

Abstract
Cardiovascular dysfunction as a result of tumor burden is becoming a recognized complication; however, the mechanisms remain unknown. A murine model of cancer cachexia has shown marked increases of matrix metalloproteinases (MMPs), known mediators of cardiac remodeling, in the left ventricle. The extent to which MMPs were involved in remodeling remained obscured. To this end a common antibiotic, minocycline, with MMP inhibitory properties was used to elucidate MMP involvement in tumor induced cardiovascular dysfunction. Tumor-bearing mice showed decreased cardiac function with reduced posterior wall thickness (PWTs) during systole, increased MMP and collagen expression consistent with fibrotic remodeling. Administration of minocycline preserved cardiac function in tumor bearing mice and decreased collagen RNA expression in the left ventricle. MMP protein levels were unaffected by minocycline administration, with the exception of MMP-9, indicating minocycline inhibition mechanisms are directly affecting MMP activity. Cancer induced cardiovascular dysfunction is an increasing concern, novel therapeutics are needed to prevent cardiac complications. Minocycline is a well-known antibiotic and recently has been shown to possess MMP inhibitory properties. Our findings presented here show that minocycline could represent a novel use for a long established drug in the prevention and treatment of cancer induced cardiovascular dysfunction.
Keywords: Matrix metalloproteinase, tissue inhibitors of matrix metalloproteinase, extracellular matrix, cancer cachexia, cardiomyocyte, minocycline
1. Introduction
Cancer is a pervasive disease that was the leading cause of deaths world-wide in 2014 [1], and is predicted to be the leading cause of death in 2015 [2, 3]. Of those who develop cancer, approximately half of these patients will develop cachexia. Cachexia is a wasting syndrome, characterized by marked loss of adipose tissue and skeletal muscle mass [4]. Cancer-induced cachexia will be the primary cause of death in a quarter of those affected [5]. While primarily thought of as a syndrome affecting adipose tissue and skeletal muscle, recent investigations have determined that cancer-induced cachexia also negatively affects heart function. In animal models of cancer-induced cachexia, there is a significant decrease in fractional shortening (FS%) and Ejection Fraction (EF) via echocardiography, consistent with heart failure [6]. While no human studies have determined the cardiac functional alterations caused by cancer cachexia, there is histological evidence from patients who died from cancer cachexia that there was marked fibrotic remodeling [7]. Remodeling in heart failure is an accumulation of multiple factors, but is strongly dependent on the activity of matrix metalloproteinases [8–10].
Matrix metalloproteinases (MMPs) are zinc-peptidases that alter the structure of the extracellular matrix (ECM) by hydrolyzing the peptide bonds holding proteins together [11, 12]. MMPs are a normal component of cardiac homeostasis but uncontrolled MMP activity leads to detrimental remodeling that can negatively affect cardiac function [13]. MMPs are endogenously controlled by Tissue Inhibitors of Metalloproteinase (TIMPs) [12]; however, previous research in the c26 mouse model of cancer cachexia showed an insufficient TIMP response to compensate for increased MMPs. This insufficient response leads to MMP-mediated collagen deposition in the heart presenting as fibrosis which can compromise heart structure and negatively impact function. While MMP levels were increased in the c26 cachectic mouse model [14], the degree in which they participate in remodeling remain unknown. In order to determine how increased MMP activity could lead to altered function, we utilized a known inhibitor of MMPs, minocycline.
Minocycline is a semisynthetic tetracycline derivative first synthesized in 1967 by Ledele Laboratories. It quickly gained FDA approval in 1970 as a broad spectrum antibiotic and is still used today for both its antibacterial effects and recently discovered neuroprotective effects. It has been used as an MMP inhibitor in diseases primarily caused through MMP activity in both animal and human studies [15–17]. The mechanism of inhibition remains unknown, however, two theories have been hypothesized. One is that minocycline binds the zinc in the active site of the MMP since minocycline has been shown to have chelating properties, and the other is that it binds allosterically and causes an active site conformational change preventing enzymatic action. Minocycline has been shown to inhibit collagenases as well as gelatinases but seems to have an unexplained increased specificity for MMP-9 [16]. The effects of minocycline on TIMPs are difficult to discern because of different effects based on location. Minocycline has been shown to increase TIMP expression but only in the brain and spinal cord [18]. Minocycline and other tetracyclines have been shown to have no effect on TIMP expression in other tissues. This could be due to changes to minocycline when crossing the blood-brain-barrier, but further work is required. Despite the mechanism of inhibition not being fully characterized, minocycline and other tetracyclines, have been shown to be potent MMP inhibitors. Because of its current status as an FDA approved drug and its well documented use and tolerability in humans, minocycline represents apossible treatment strategy to limit the effects of MMPs in cancer-induced cardiac dysfunction.
In the present study, we injected the c26 adenocarcinoma cell line into CD2F1 mice treated with or without minocycline, which was delivered through their drinking water. We hypothesize that minocycline treatment in tumor burdened animals will improve cardiac function by inhibiting MMP activity.
2. Materials and Methods
2.1 Animal Husbandry
Adult ~10 week old female mice weighing 20–22 grams were obtained from Charles River Laboratories (Charles River, Willmington, MA). Mice were housed 1–3 per cage and were maintained on a 12 hour light/dark cycle at 25°C and were provided ad libitum access to standard rodent chow as well as water. Treated mice were administered minocycline in their drinking water and provided ad libitum access as well. All animal care and procedures were approved by the Ohio State University Institutional Animal Care and Use Committee.
2.2 Minocycline Administration
Mice were administered minocycline orally through supplemented water at the same dosage in previous studies [19]. Water bottles provided were supplemented with 1 mg/ml minocycline for a dose of 100 mg/kg/day (Sigma, St. Louis, MO). Water bottles were changed every other day throughout the duration of the study. No fluid intake differences were observed between any of the experimental groups (data not shown).
2.3 Model of Tumor Growth
The c26 line was maintained and injected as previously described [6]. Upon arrival, mice were allowed to acclimate to their environment for one week before being randomly assigned into experimental groups. After acclimatization and group assignment, 5×105 c26 cells suspended in PBS were injected subcutaneously between the scapulae region as described previously [6]. Non-tumor bearing mice were subjected to a sham procedure and injected subcutaneously with the same volume of PBS. Female mice are used exclusively in this study since they have been shown to guard their body weight better than male mice and are less prone to an anorexic state than male mice. Cachectic effects in females are more representative of a true cachectic state than their male counterparts [6]. Tumor growth was observed as early as day twelve and mice typically were cachectic by day 21 post injection as determined by body weight as well as body score. Food intake has been shown previously to be reduced in this model; however, this is not the main cause of the wasting condition as pair fed animals did not show as significant body mass loss [20]. The c26 model shows weight loss beginning at approximately the second week and continues as body score worsens, reaching a wasted state. Minocycline as already been shown in our lab to reverse this wasting condition and the results will not be repeated here [19]. At the time of euthanasia, approximately day 21 post-injection, mice were anesthetized with a ketamine/xylazine cocktail (10/1 mg/ml respectively) at a volume of 0.01 ml per gram of body weight or approximately 0.2 ml. A cardiectomy was performed, the left ventricle was dissected, snap frozen in liquid nitrogen and stored at −80°C until biochemical analyses. The tumor was also removed and weighed at the end of the study, 21 days, to determine if treatment had any effect on tumor growth. Previous study by our lab has shown minocycline did not have any effect on final tumor weight and will not be repeated here [19].
2.4 Echocardiography
Cardiac function was determined using echocardiography with a 40 mHz VEVO 2100 Ultrasound System (Visual Sonics, Toronto, Ontario, Canada). Mice were anesthetized using isoflurane; 3% in 100% oxygen for induction and 1% in 100% oxygen for maintenance. Animals were placed on a warm table, fur around the chest was removed using a depilatory agent, and temperature was measured via a rectal probe. Pre-warmed ultrasound gel was applied to a 15 mHz transducer probe optimized for mouse echocardiography. The transducer was placed on the parasternal short axis to obtain a view of the left ventricle (LV) at the mid-papillary level for image capture and measurements. LV dimensions; LV End Diastolic Dimension, LV End Systolic Dimension (LVEDd and LVEDs) as well as posterior wall thickness during systole and diastole (PWTs, PWTd) were acquired using the leading edge method as recommended by the American Society for Echocardiography [21]. Percent fractional shortening (FS%) was calculated using the following formula; . Ejection Fraction (EF) was also measured and calculated using the following formula; . For echocardiography results an n=6 was used for each experimental group.
2.5 Cardiomyocyte Isolation
Cardiomyocytes were isolated as previously described [22]. In brief, hearts were removed and digested using liberase and trypsin. Once digested, the cardiomyocytes were plated on laminin coated glass inserts. Inserts were loaded into a perfusion chamber mounted on an Olympus IX-71 microscope. Cells were stimulated (1 Hz, 3-ms duration) with a Myopacer Field-Stimulator system and function was determined using Sarclen Sarcomere Length Acquisition Module (IonOptix, Milton, MA). Approximately 10–15 cells were measured from each mouse to provide adequate measurements for peak shortening (PS%), cellular equivalent to FS%. Sarcomere contractile velocity was also measured as ±dL/dt and time to 90% shortening and relengthening as TPS90 and TR90, respectively. For Ca+2 measurements, a calcium sensitive fluorimetric compound, FURA-2AM, was loaded onto the cells at a concentration of 5 μM for 20 minutes. Fluorometric measurements were acquired using a dual excitation single emission system. These transients were analyzed for transient calcium amplitude (Δ340/380), and calcium reuptake (T). For cardiomyocyte studies, separate mice were used independent of those used for biochemical studies to ensure that there were no artifacts in the biochemical studies caused by digestion of the heart for cardiomyocyte isolation. Cardiomyocyte results use an n=3 for each experimental group, for each mouse 15 cells were used to measure cardiomyocyte function.
2.6 RNA Isolation and qPCR
RNA was isolated from frozen tissue using the Trizol/Chloroform method. Briefly, the tissue was homogenized with a TissueLyser (Qiagen, Boston, MA) with Trizol buffer, transferred to a chloroform containing tube and centrifuged. The supernatant was mixed with alcohol and loaded onto a Qiagen RNA processing column and the Qiagen RNA isolation protocol was followed from the Qiagen RNeasy Min-Kit (Qiagen, Boston, MA). RNA quality and concentration was checked using a NanoDrop 2000c (ThermoScientific, Wilmington, DE), then a known amount of RNA was reverse transcribed to generate cDNA using the iScript Supermix kit (Bio-Rad, Hercules, CA). The cDNA was diluted to an appropriate concentration and qPCR was performed as previously described [6]. Briefly, the cDNA was mixed with SYBR Green Master Mix (Bio-Rad, Hercules, CA), and forward and reverse primer sequences. The resulting mix was run through a CFX96 (Bio-Rad, Hercules, CA) Three Step Amplification protocol and each cycle was repeated forty times. The Ct values of an internal control, GAPDH, were then used against the target Ct values and analyzed with the Livak 2ˆ(−ΔΔCt) method. QPCR results used an n=6 for each experimental group.
2.7 Western Blotting
Proteins were homogenized from snap frozen tissue using a tissue lysis buffer described previously [23]. The protein concentration was then quantified using a Bicinchinonic Acid Assay (BCA) (ThermoScientific, Wilmington, DE). The samples plus standards were incubated at 37°C and then read using a PowerWave plate reader (BioTek, Winooski, VT) at 562 nm. The sample concentration was extrapolated using the slope of the standard curve and was used to assure equal loading. The protein homogenate was mixed with a sample buffer and loaded onto a polyacrylamide gel (5% stacking, 12% resolving) and run at constant voltage (100v). The gel was transferred onto a PVDF membrane using a wet transfer system (Bio-Rad, Hercules, CA) that was run at room temperature for 90 minutes at 100 V. The membranes were blocked for 1 hour at room temperature with a TBS-T solution containing 5% Bovine Serum Albumin (BSA). The primary antibody was suspended at the same concentration as previously [14] used and incubated overnight at 4°C. The membrane was washed in TBST in triplicate for five minutes, then secondary antibody was applied at a concentration of 1:5000 in TBST containing 2.5% BSA for an hour at room temperature. The membrane was then washed again in TBST in triplicate for five minutes. The membrane was developed using the LiCor (Li-Cor, Lincoln, NE) imaging system.
2.8 Protein Quantification
Immunoblots were quantified using the Li-Cor Odyssey system (Li-Cor, Lincoln, NE). In brief, secondary antibodies with an infrared conjugate were excited using a laser system in the Li-Core Odyssey Infrared Imaging System. The emission was quantified as an emission signal with automatic background subtraction. Additionally, a separate gel was run and stained with Coomassie Blue, where the actin region was used to ensure equal loading and normalization. The Coomassie stained gel was placed into the Odyssey system (Li-Core, Lincoln, NE) and a signal was obtained using the 700 nm channel, as previously described [24, 25]. This signal was used to normalize the immunoblots; the average of the normalized signal from the control group was used to determine the fold change of the protein of interest. Three samples from each group were exposed on one immunoblot and the fold change was determined by normalization of the signal with the loading control signal and then divided by the average from the normalized control signal. Fold change calculations utilized an n=6 for each experimental group.
2.9 Statistical Analyses
All data are reported as the mean ±SEM. The data were analyzed using a two-way analysis of variance (ANOVA) to determine any significance caused by the tumor, drug, or both using Prism 6 (Graphpad Software, Inc., La Jolla, CA). A Bonferroni post hoc test was used to determine significant differences between experimental groups; untreated control, treated control, untreated tumor-bearing, and treated tumor-bearing. Using these statistical methods we were not only able to investigate tumor, drug, and interaction effects, but further determine significant differences between each group. Differences were considered statistically significant if p<0.05.
3. Results
3.1 Minocycline treatment attenuated cardiac dysfunction
Mice bearing c26 tumors had significantly reduced posterior wall thickness during systole (PWTs), reduced Fractional Shortening and Ejection Fraction (FS%, and EF respectively) when compared to untreated controls (Figure 2a–b). Treatment with minocycline in tumor-bearing mice significantly improved both FS% and EF when compared to untreated tumor-bearing mice. When compared to untreated controls, regardless of tumor-burden, minocycline treatments did not have any significant effect in FS% or EF. PWTs in tumor-bearing untreated mice was significantly reduced when compared to untreated control mice. Treated tumor-bearing mice did not show significant increases in PWTs when compared to untreated tumor-bearing mice; however, they also did not show significant decreases when compared to untreated non tumor-bearing mice. (Figure 2c).
Figure 2.
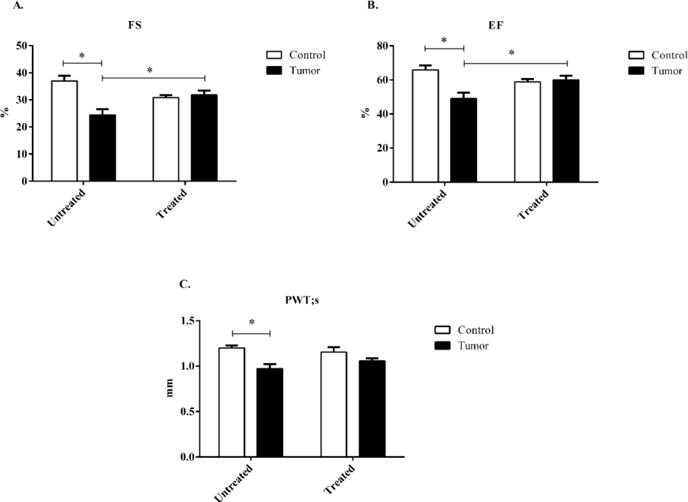
Systolic echocardiography data taken at the parasternal short axis from control and c26 tumor-bearing mice with or without treatment. A) Fractional shortening (FS%), B) Ejection Fraction (EF), and C) Posterior wall thickness (PWT;s) at systole were determined at day 19 post injection. Bars are used to demonstrate significant effects between groups. p<0.05 was considered statistically significant.
3.2 Minocycline restored contractile time in isolated cardiomyocytes with no effect on calcium transient amplitude or reuptake
Baseline sarcomere length was not different between any of the groups (Figure 3a). Cardiomyocytes from tumor-bearing mice with or without minocycline treatment showed no change in peak shortening when compared to untreated control mice. (PS%) (Figure 3b). When contractile velocity was measured there were no significant effects regardless of tumor-burden or minocycline treatment (Figure 3c–d). Both contraction and relaxation times were significantly reduced in untreated tumor-bearing mice when compared to untreated control mice (Figure 3d–e). Treated tumor-bearing mice had significantly increased contraction and relaxation times when compared to untreated tumor-bearing mice (Figure 3e–f.). Treated control mice also had significantly increased contraction and relaxation times when compared to untreated control mice, indicating a significant minocycline effect regardless of tumor burden (Figure 3e–f). There was a significant tumor effect showing reduced calcium transient amplitude of treated and untreated tumor-bearing mice, Δ340/380, when compared to untreated control mice. Treated control mice had no significant differences in calcium transient compared to untreated control mice, indicating no minocycline effect (Figure 4a). Calcium reuptake (T) was not significantly affected regardless of treatment or tumor burden (Figure 4b)
Figure 3.
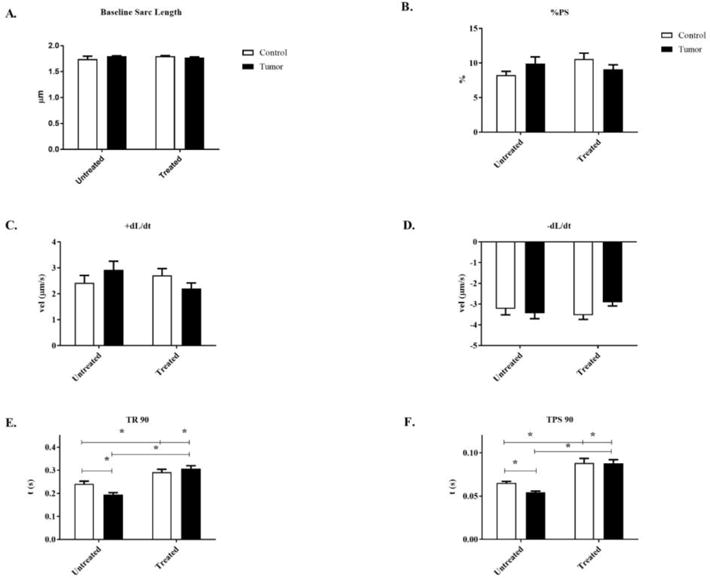
Sarcomere length and contractile properties of isolated cardiomyocytes from control and c26 tumor-bearing mice with or without treatment. A) Sarcomere length (μm), B) Peak shortening (PS%), C) Negative velocity (−dL/dt), D) Positive velocity (+dL/dt), E) Time to peak shortening 90% (TPS 90), and F) Time to relaxation 90% (TR 90) were determined at day 21 post injection. Bars are used to demonstrate significant effects between groups. p<0.05 was considered statistically significant.
Figure 4.
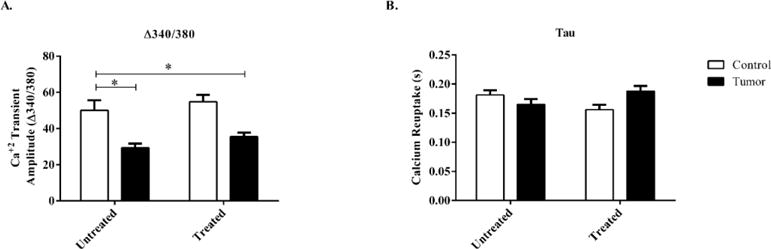
Calcium transient amplitude and calcium reuptake of isolated cardiomyocytes from control and c26 tumor-bearing mice with or without treatment. A) Calcium transient amplitude (Δ360/380), and B) Calcium reuptake (T) were measured day 21 post injection. Bars are used to demonstrate significant. p<0.05 was considered statistically significant.
3.3 Minocycline treatment reduced Collagen I and III RNA expression in tumor-bearing mice
Collagen I and III RNA expression were significantly increased in untreated tumor-bearing mice when compared to untreated control mice. Minocycline administration significantly reduced the amount of collagen I and III RNA expression in tumor-bearing mice when compared to untreated tumor-bearing mice (Figure 5.)
Figure 5.
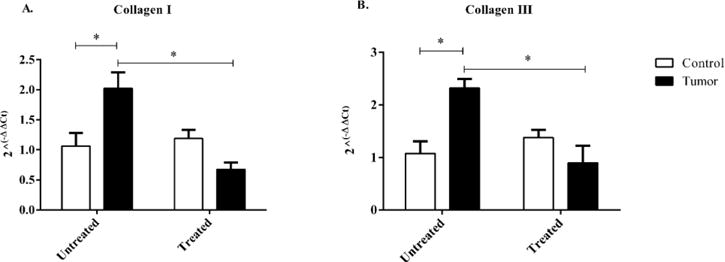
RNA expression of Collagen I and III in control and c26 tumor-bearing mice with or without treatment. A) Collagen I, and B) Collagen III RNA expression were measured from hearts collected at day 21 post injection. Bars are used to indicate significance between groups. p<0.05 was considered statistically significant
3.4 RNA expression of MMPs are unaffected by minocycline administration, protein expression of MMP-9 is significantly decreased compared to untreated tumor-bearing mice
The RNA expression of MMP-2, -3, and -14 were significantly increased in tumor-bearing mice and administration of minocycline did not show any significant effects. The RNA expression of MMP-9 in tumor-bearing groups regardless of treatment was not significantly increased when compared to untreated control mice (Figure 6a). The protein expression of MMP-2, -3, and -14 of tumor-bearing mice, untreated or treated, were significantly increased when compared to untreated control mice indicating a significant tumor effect. Protein expression of MMP-9 was significantly increased in tumor-bearing mice regardless of treatment, when compared to untreated control mice indicating a significant tumor effect. In treated tumor-bearing mice the protein expression of MMP-9 was significantly decreased when compared to untreated tumor-bearing mice indicating a significant interaction effect (Figure 6b).
Figure 6.
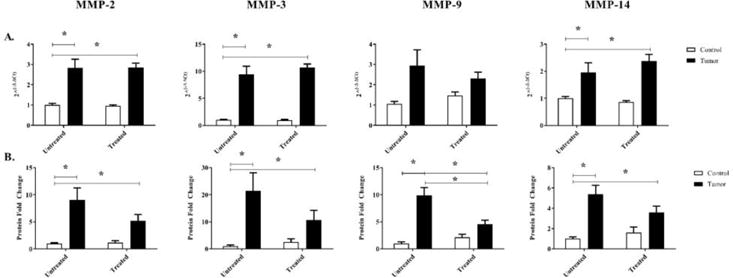
RNA and protein expression of MMP-2, -3, -9, and -14 in control and c26 tumor-bearing mice with or without treatment. A) RNA expression, and B) protein expression were measured from hearts collected at day 21 post injection. Bars are used to indicate significance between groups. p<0.05 was considered statistically significant.
3.5 RNA expression of TIMP-1 is significantly increased in tumor-bearing mice regardless of treatment. Protein expression of TIMP-2 in treated tumor-bearing is significantly decreased compared to untreated tumor-bearing mice
The RNA expression of TIMP-1 was significantly increased in tumor bearing mice regardless of treatment when compared to untreated control mice, the RNA expression of TIMP-2 was not significantly different regardless of tumor burden or treatment (Figure 7a). Protein expression of TIMP-1 and TIMP-2 in both untreated and treated tumor-bearing mice were significantly increased indicating a primary tumor effect. There was a significant decrease in TIMP-2 protein expression between untreated tumor-bearing mice and treated tumor-bearing mice, indicating a significant interaction effect. (Figure 7b).
Figure 7.
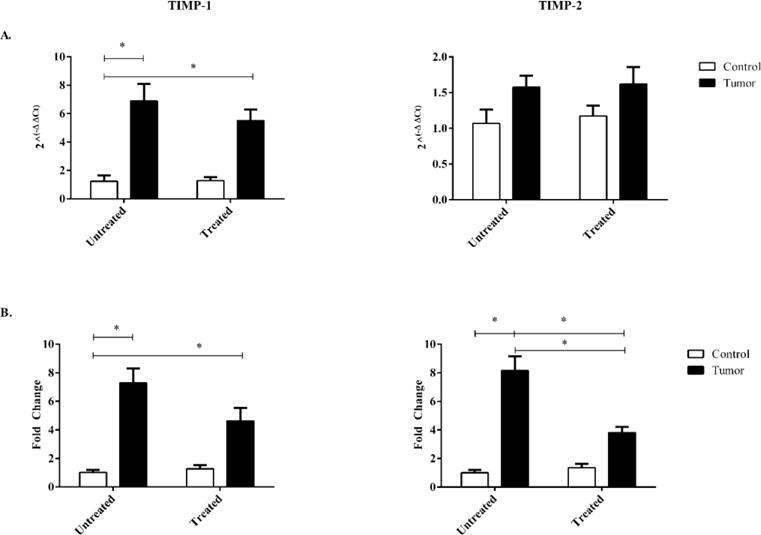
RNA and protein expression of TIMP-1 and TIMP-2 in control and c26 tumor-bearing mice with or without treatment. A) RNA expression, and B) protein expression were measured from hearts collected at day 21 post injection. Bars are used to indicate significance between groups. p<0.05 was considered statistically significant.
4. Discussion
In our current study, we expanded upon previous research showing collagen deposition and increased MMP levels in the failing hearts of tumor-burdened mice. MMPs are increased in other models of heart failure and are correlated with morbidity in human heart failure patients [8, 13]. Minocycline, a member of the tetracycline family, represents an attractive means to determine MMP contribution to the failing heart as an inhibitor of MMP activity. Minocycline has been used clinically and experimentally in disease models to inhibit MMPs, demonstrating its efficacy in inhibiting MMP activity [15, 17, 26]. Based on our previous findings and the known effects of minocycline we utilized this drug to understand the contributions of MMPs to depressed cardiac function. A previous study demonstrated that minocycline administration to c26 tumor-bearing mice had no effect on systemic inflammation, measured as plasma concentrations of IL-6 [19]. Minocycline was also shown to have no effect on tumor mass, meaning the probable mechanism of minocycline is direct MMP inhibition and not due to anti-tumor effects [19]. Since minocycline does not reduce systemic inflammation or overall tumor mass in the tumor-bearing mouse, we can infer that its cardioprotective effects are due to MMP inhibition rather than anti-tumor or anti-inflammatory effects.
Heart function is notably depressed in this cancer model with reduced FS% and EF [6, 20, 22, 27]. This reduction in the FS% and EF is the result of thinning of the posterior wall during systole, causing chamber expansion and reducing blood ejection. The posterior wall thickness during diastole is unchanged in tumor-bearing mice resulting in a normal diastolic chamber dimension [6]. The increased compliance of the left ventricular wall is most likely due to the actions of MMPs. By degrading components of the ECM, the ventricle becomes increasingly compliant such that during a high pressure system, systole, causes a thinning of the left ventricular wall that is not present during diastole, a lower pressure system. The administration of minocycline to tumor-bearing mice showed significantly increased FS% and EF when compared to untreated tumor-bearing mice, indicating an improvement in cardiac function. PWTs is significantly decreased in untreated tumor-bearing mice, in agreement with previous studies. Treated tumor-bearing mice did not show significantly improved PWTs compared to untreated tumor-bearing mice; however, it was not significantly decreased from untreated control levels. Diastolic function was not measured in this study since previous findings have not shown diastolic dysfunction in this model. While there is significantly increased collagen deposition, it may indicate that the collagen deposition has not reached a point to impact diastolic function. There may be post-translational modifications occurring to collagen adjusting its mechanical properties, further study is necessary to understand collagen contribution in this model.
Our cardiomyocyte data agrees with previous findings, indicating no change in the sarcomere percent peak shortening in the c26 adenocarcinoma model of cancer cachexia [6, 22]. There were significant decreases in TPS90 and TR90 in tumor-bearing mice compared to control, as the velocity was unchanged. This effect was ameliorated when minocycline was administrated, implicating MMP activity in the sarcomere. MMP-2 has recently been shown to localize to the sarcomere in the heart and degrade cardiac sarcomeric proteins such as; titin, myosin light chain, apha-actinin, and TnI among others [28]. It is possible that minocycline-treated mice had restored shortening and re-lengthening times due to inhibition of MMP-2 actions on the sarcomere [28]. This effect does not seem to be due to sarcomere length, though it does not discount possible MMP-mediated sarcomere effects. Another possibility is that minocycline has been shown to act as a calcium ionophore possibly transporting calcium from the bloodstream into the cardiac myocytes and fibroblasts. Further work will need to be performed to determine whether MMPs are causing changes to proteins in the sarcomere. Additional work will need to be done to understand the effects of minocycline on the heart, while it has been established that cardiomyocytes and fibroblasts take up minocycline readily, more information is needed on what effects minocycline may have on these cell types.
Calcium transient amplitude, or the release of calcium from the sarcoplasmic reticulum (SR), was decreased in tumor-bearing mice, similar to our previous results in this model [22]. The release of calcium was not improved in tumor-bearing mice with the administration of minocycline. The depressed calcium release might be indicative of posttranslational alterations to calcium handling proteins in an MMP-independent manner since this was not recovered with the administration of minocycline. Calcium release dysfunction is usually caused by alterations to the ryanodine receptor, which is typically altered in heart failure [29]. Another possibility would be calstabin, a protein that stabilizes the pores of the ryanodine receptor, is being degraded which has been shown in the skeletal muscle of another tumor model [30]. Together changes to these proteins may explain the calcium transient changes in tumor-bearing mice, but further work is needed to understand the mechanism. The time to calcium uptake, or tau, in tumor-bearing mice was unchanged from control mice. In the context of calcium handling this indicates that phospholamban and SERCA are unaffected by the presence of tumor-burden in this model. Minocycline treatment, in control or tumor-bearing, had no effect on tau. Collectively, this indicates that MMPs could be altering sarcomeric function through proteolytic activity but the calcium dysfunction present is MMP-independent and warrants further investigation to determine how the proteins are being altered in the tumor-bearing myocardium.
In previous studies it has been shown that there is increased collagen deposition in the hearts of c26 tumor-bearing mice through Picrosirius red staining [14]. The study did not examine the RNA expression of collagen, which we examined in this study. We examined gene expression of both collagen I and III to determine if the fibrotic response remains. In tumor-bearing mice, we found significant increases in both collagen I and III, agreeing with previous findings of increased collagen deposition. In treated tumor-bearing mice we found significantly less when compared to untreated tumor-bearing mice. MMPs can provoke an increased collagen deposition. Increased MMP activity begins a cycle of collagen breakdown causing increased collagen deposition as a compensatory response eventually leading to fibrosis [31, 32]. While it may seem counter-intuitive that MMPs, as enzymes that degrade extracellular matrix, would provoke increased collagen response, it is due to the balancing act of the normal physiological system. In the normal heart as collagen is worn down, MMPs remove this old or damaged collagen in order for it to be replaced by newly synthesized collagen. This leads to a carefully regulated balance of MMPs to collagen expression so that overall collagen content is not disturbed. In a pathological setting, increased MMP expression causes increased collagen expression but the collagen is deposited randomly throughout the heart leading to a fibrotic phenotype [33, 34]. In minocycline treated tumor-bearing mice, the RNA expression of collagen I and III was decreased, implicating decreased collagen deposition in the heart. Inhibition of MMP enzymatic activity by minocycline possibly prevented the pathological fibrotic response from occurring. Further work is required to determine if MMPs are directly provoking this response or acting as or on a signaling factor promoting collagen synthesis in the c26 model.
The protein expression in tumor-bearing mice of matrix metalloproteinases were largely unaffected with the administration of minocycline, with the exception of MMP-9. This was most likely due to the influence of MMP-2 which has been shown to regulate MMP-9 [35]. With the inhibitory effect of minocycline, MMP-2 is no longer able to provoke an MMP-9 up-regulation [36]. The lack of change in MMP expression was expected and experimentally beneficial as the action of minocycline could not be misinterpreted as a result of reduced MMP expression. The protein expression of TIMP-2 was significantly decreased in treated tumor-bearing mice compared to untreated tumor-bearing mice. The decrease in TIMP-2 in treated tumor-bearing mice may be due to the lack of MMP activity, as MMPs may cleave certain transcription factors to increase TIMPs in order to rebalance the MMP to TIMP ratio. By rebalancing this MMP/TIMP ratio, MMP mediated remodeling would be attenuated or prevented altogether. This indicates minocycline is acting in a direct fashion on MMPs as opposed to secondary signaling.
Our results have demonstrated that minocycline is capable of restoring cardiac function in a tumor-bearing mouse without altering tumor burden or inflammatory state. We anticipate that the improved cardiac function of tumor burdened minocycline mice is due to the inhibition of MMP activity by minocycline, though this current study does not explicitly examine MMP activity. Minocycline has been shown to suppress MMP activity in experimental models and clinical studies in MMP-mediated diseases such as Fragile X Syndrome and Marfan Syndrome [17, 37, 38]. The mechanism of inhibition of MMP activity is still unknown with spectroscopic studies indicating either that minocycline is binding to the active site as a competitive inhibitor or binding allosterically and modifying the active site area preventing enzymatic activity [39, 40]. While the cardiomyocytes were studied extensively in this work along with whole heart function, the contribution of other cells in the heart warrant further investigation. Previous studies have demonstrated that minocycline is taken up into cardiomyocytes and fibroblasts at approximately the same rate reaching similar concentrations [41]. Fibroblasts are important in remodeling, therefore further work will need to be done to isolate fibroblasts from the hearts of tumor-burdened mice to better understand their contribution to the impaired function. Minocycline could represent a therapeutic that can be administered before or alongside chemotherapeutics in cancer patients to prevent cardiac complications. Administration may not only prevent cardiac complications during treatment but also preserve quality of life post treatment by ameliorating any cardiac dysfunction and improving mood. More work is still needed to further elucidate the roles that MMPs play in cardiac dysfunction in a tumor-bearing mice. Minocycline restored contractile time in the cardiomyocyte, indicating that MMPs may function within the sarcomeric complex. Regardless, minocycline administration has demonstrated marked cardiac functional improvement and further elucidated the roles that MMPs play in the tumor-burdened murine myocardium.
Figure 1.
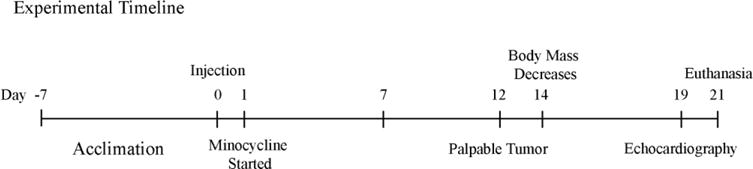
Experimental timeline of the c26 model including when drug treatment was started, along with echocardiography, and euthanasia.
Figure 8.
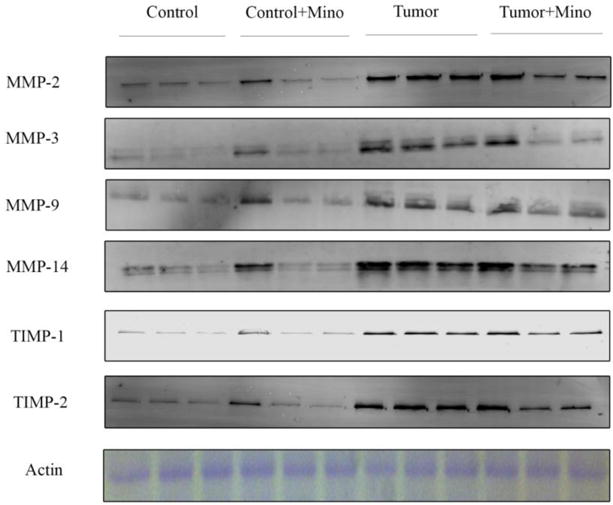
Representative immunoblots of MMP and TIMP expression in the left ventricle of control and c26 tumor-bearing mice with or without treatment.
Highlights.
Minocycline treatment improves cardiac function in tumor-burdened mice.
Contractile time is improved in minocycline-treated tumor-burdened mice.
MMP protein expression is reduced in treated tumor-bearing mice.
Collagen RNA expression is reduced in treated tumor-bearing mice.
Acknowledgments
The authors would like to thank Ms. Diana Norden for assistance with animal husbandry.
Funding
The work was supported by grants from the National Institutes of Health [NR012618 and ES019923to LEW].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Hughes BB, Kuhn R, Peterson CM, Rothman DS, Solorzano JR, Mathers CD, et al. Projections of global health outcomes from 2005 to 2060 using the International Futures integrated forecasting model. Bulletin of the World Health Organization. 2011;89:478–86. doi: 10.2471/BLT.10.083766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS medicine. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeWys WD. Nutritional care of the cancer patient. Jama. 1980;244:374–6. doi: 10.1001/jama.1980.03310040056033. [DOI] [PubMed] [Google Scholar]
- 5.Tisdale MJ. Cachexia in cancer patients. Nature reviews Cancer. 2002;2:862–71. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 6.Xu H, Crawford D, Hutchinson KR, Youtz DJ, Lucchesi PA, Velten M, et al. Myocardial dysfunction in an animal model of cancer cachexia. Life sciences. 2011;88:406–10. doi: 10.1016/j.lfs.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. European heart journal. 2014;35:932–41. doi: 10.1093/eurheartj/eht302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiological reviews. 2007;87:1285–342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 9.Lindsey ML, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti-Morrow L, et al. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002;105:753–8. doi: 10.1161/hc0602.103674. [DOI] [PubMed] [Google Scholar]
- 10.Iwanaga Y, Aoyama T, Kihara Y, Onozawa Y, Yoneda T, Sasayama S. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. Journal of the American College of Cardiology. 2002;39:1384–91. doi: 10.1016/s0735-1097(02)01756-4. [DOI] [PubMed] [Google Scholar]
- 11.Lemaitre V, D’Armiento J. Matrix metalloproteinases in development and disease. Birth defects research Part C, Embryo today: reviews. 2006;78:1–10. doi: 10.1002/bdrc.20065. [DOI] [PubMed] [Google Scholar]
- 12.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular research. 2006;69:562–73. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Shirakabe A, Asai K, Hata N, Yokoyama S, Shinada T, Kobayashi N, et al. Clinical significance of matrix metalloproteinase (MMP)-2 in patients with acute heart failure. International heart journal. 2010;51:404–10. doi: 10.1536/ihj.51.404. [DOI] [PubMed] [Google Scholar]
- 14.Devine RD, Bicer S, Reiser PJ, Velten M, Wold LE. Metalloproteinase Expression is Altered in Cardiac and Skeletal Muscle of Cancer Cachexia. American journal of physiology Heart and circulatory physiology. 2015 doi: 10.1152/ajpheart.00106.2015. ajpheart 00106 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang TY, Chu HC, Lin YL, Lin CK, Hsieh TY, Chang WK, et al. Minocycline attenuates experimental colitis in mice by blocking expression of inducible nitric oxide synthase and matrix metalloproteinases. Toxicology and applied pharmacology. 2009;237:69–82. doi: 10.1016/j.taap.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 16.Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC neuroscience. 2006;7:56. doi: 10.1186/1471-2202-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siller SS, Broadie K. Matrix metalloproteinases and minocycline: therapeutic avenues for fragile X syndrome. Neural plasticity. 2012;2012:124548. doi: 10.1155/2012/124548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behavioural brain research. 2009;196:168–79. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 19.Norden DM, Bicer S, Clark Y, Jing R, Henry CJ, Wold LE, et al. Tumor growth increases neuroinflammation, fatigue and depressive-like behavior prior to alterations in muscle function. Brain, behavior, and immunity. 2015;43:76–85. doi: 10.1016/j.bbi.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian M, Nishijima Y, Asp ML, Stout MB, Reiser PJ, Belury MA. Cardiac alterations in cancer-induced cachexia in mice. International journal of oncology. 2010;37:347–53. doi: 10.3892/ijo_00000683. [DOI] [PubMed] [Google Scholar]
- 21.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. Journal of the American Society of Echocardiography: official publication of the American Society of Echocardiography. 2005;18:1440–63. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Stevens SC, Velten M, Youtz DJ, Clark Y, Jing R, Reiser PJ, et al. Losartan treatment attenuates tumor-induced myocardial dysfunction. Journal of molecular and cellular cardiology. 2015;85:37–47. doi: 10.1016/j.yjmcc.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velten M, Gorr MW, Youtz DJ, Velten C, Rogers LK, Wold LE. Adverse perinatal environment contributes to altered cardiac development and function. American journal of physiology Heart and circulatory physiology. 2014;306:H1334–40. doi: 10.1152/ajpheart.00056.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris LR, Churchward MA, Butt RH, Coorssen JR. Assessing detection methods for gel-based proteomic analyses. Journal of proteome research. 2007;6:1418–25. doi: 10.1021/pr0700246. [DOI] [PubMed] [Google Scholar]
- 25.Luo S, Wehr NB, Levine RL. Quantitation of protein on gels and blots by infrared fluorescence of Coomassie blue and Fast Green. Analytical biochemistry. 2006;350:233–8. doi: 10.1016/j.ab.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 26.Lee SM, Yune TY, Kim SJ, Kim YC, Oh YJ, Markelonis GJ, et al. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J Neurochem. 2004;91:568–78. doi: 10.1111/j.1471-4159.2004.02780.x. [DOI] [PubMed] [Google Scholar]
- 27.Tian M, Asp ML, Nishijima Y, Belury MA. Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. International journal of oncology. 2011;39:1321–6. doi: 10.3892/ijo.2011.1150. [DOI] [PubMed] [Google Scholar]
- 28.Ali MA, Fan X, Schulz R. Cardiac sarcomeric proteins: novel intracellular targets of matrix metalloproteinase-2 in heart disease. Trends in cardiovascular medicine. 2011;21:112–8. doi: 10.1016/j.tcm.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Scoote M, Williams AJ. The cardiac ryanodine receptor (calcium release channel): emerging role in heart failure and arrhythmia pathogenesis. Cardiovascular research. 2002;56:359–72. doi: 10.1016/s0008-6363(02)00574-6. [DOI] [PubMed] [Google Scholar]
- 30.Waning DL, Guise TA. Cancer-associated muscle weakness: What’s bone got to do with it? BoneKEy reports. 2015;4:691. doi: 10.1038/bonekey.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix biology: journal of the International Society for Matrix Biology. 2015;44–46C:247–54. doi: 10.1016/j.matbio.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Polyakova V, Loeffler I, Hein S, Miyagawa S, Piotrowska I, Dammer S, et al. Fibrosis in endstage human heart failure: severe changes in collagen metabolism and MMP/TIMP profiles. International journal of cardiology. 2011;151:18–33. doi: 10.1016/j.ijcard.2010.04.053. [DOI] [PubMed] [Google Scholar]
- 33.Quan T, Little E, Quan H, Qin Z, Voorhees JJ, Fisher GJ. Elevated matrix metalloproteinases and collagen fragmentation in photodamaged human skin: impact of altered extracellular matrix microenvironment on dermal fibroblast function. The Journal of investigative dermatology. 2013;133:1362–6. doi: 10.1038/jid.2012.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu B, Li C, Liu Z, Dai Z, Tao Y. Increasing extracellular matrix collagen level and MMP activity induces cyst development in polycystic kidney disease. BMC nephrology. 2012;13:109. doi: 10.1186/1471-2369-13-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toth M, Chvyrkova I, Bernardo MM, Hernandez-Barrantes S, Fridman R. Pro-MMP-9 activation by the MT1-MMP/MMP-2 axis and MMP-3: role of TIMP-2 and plasma membranes. Biochemical and biophysical research communications. 2003;308:386–95. doi: 10.1016/s0006-291x(03)01405-0. [DOI] [PubMed] [Google Scholar]
- 36.Ogata Y, Enghild JJ, Nagase H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. The Journal of biological chemistry. 1992;267:3581–4. [PubMed] [Google Scholar]
- 37.Hartog AW, Franken R, Zwinderman AH, Groenink M, Mulder BJ. Current and future pharmacological treatment strategies with regard to aortic disease in Marfan syndrome. Expert opinion on pharmacotherapy. 2012;13:647–62. doi: 10.1517/14656566.2012.665446. [DOI] [PubMed] [Google Scholar]
- 38.Williams A, Davies S, Stuart AG, Wilson DG, Fraser AG. Medical treatment of Marfan syndrome: a time for change. Heart. 2008;94:414–21. doi: 10.1136/hrt.2006.109454. [DOI] [PubMed] [Google Scholar]
- 39.Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: far beyond an antibiotic. British journal of pharmacology. 2013;169:337–52. doi: 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garrido-Mesa N, Zarzuelo A, Galvez J. What is behind the non-antibiotic properties of minocycline? Pharmacological research. 2013;67:18–30. doi: 10.1016/j.phrs.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Romero-Perez D, Fricovsky E, Yamasaki KG, Griffin M, Barraza-Hidalgo M, Dillmann W, et al. Cardiac uptake of minocycline and mechanisms for in vivo cardioprotection. Journal of the American College of Cardiology. 2008;52:1086–94. doi: 10.1016/j.jacc.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]