

Abstract
Mitogen-activated protein kinases (MAPKs) play a critical role in regulating cardiac hypertrophy and remodeling in response to increased workload or pathological insults. The spatiotemporal activities and inactivation of MAPKs are tightly controlled by a family of dual-specificity MAPK phosphatases (DUSPs). Over the past 2 decades, we and others have determined the critical role for selected DUSP family members in controlling MAPK activity in the heart and the ensuing effects on ventricular growth and remodeling. More specifically, studies from mice deficient for individual Dusp genes as well as heart-specific inducible transgene-mediated overexpression have implicated select DUSPs as essential signaling effectors in the heart that function by dynamically regulating the level, subcellular and temporal activities of the extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs) and p38 MAPKs. This review summarizes recent literature on the physiological and pathological roles of MAPK-specific DUSPs in regulating MAPK signaling in the heart and the effect on cardiac growth and remodeling.
Keywords: cardiac hypertrophy, concentric remodeling, dual specificity phosphatases, mitogen-activated protein kinases
1. Introduction and overview
The heart can hypertrophy in response to increased workload caused by physiological or pathological stimulation [1-3]. Cardiac hypertrophy is an adaptive response that reduces wall stress in an attempt to augment or preserve cardiac function [4]. Sustained cardiac hypertrophy in response to pathologic insults is maladaptive and can eventually lead to systolic insufficiency and heart failure characterized by increased rates of myocyte apoptosis, interstitial fibrosis and ventricular dilation [4]. Over the last two decades a great deal of progress has been made in annotating the intracellular signaling pathways that are associated with cardiac remodeling and hypertrophy. Neuroendocrine receptor initiated mitogen-activated protein kinase (MAPK) signaling has been demonstrated to play a pivotal role in controlling both physiological and pathological responsiveness of the heart.
The MAPKs are central signaling intermediates that coordinate diverse physiological and pathological events such as cancer, inflammation, diabetes, memory and cardiac remodeling [5]. MAPK activation is typically achieved by membrane receptors or other ill-defined stress-sensing effectors, which then leads to activation of a highly organized sequence of successively acting protein kinases that both amplify the signal and mediate phosphorylation of diverse cytoplasmic and nuclear regulatory proteins (Figure 1). In its broadest terms the MAPK signaling cascade is divided into 3 major pathways that culminate in the activation of either the p38 MAPKs, the c-Jun N-terminal kinases (JNK), or the extracellular signal-regulated kinases (ERK) [6, 7]. Phosphorylation within their activation loop (TxY) of these three terminal MAPKs is mediated by the upstream dual-specificity MAPK kinases (MAPKKs, also named MEKs or MKKs) including MEK1/2 for ERK1/2, MKK3/MKK6 for p38, and MKK4/MKK7 for JNK1/2 [7]. Upstream of MAPKKs, multiple MAPKKKs are either directly activated by environmental stress, reactive oxygen species, or by G proteins (Ras, Rac, Rho, and others), G protein-coupled receptors, receptor tyrosine kinases or receptor serine/threonine kinases [8-11] (Figure 1). Once activated, MAPKs phosphorylate a variety of downstream substrates to regulate events such as cell proliferation, differentiation, apoptosis, growth, cellular re-organization and metabolism [12]. In end-stage heart failure or pathological cardiac hypertrophy, almost all MAPK signaling components are activated where they phosphorylate diverse effectors that drive or modify the disease process [13, 14].
Figure 1. Overview of MAPK signaling and DUSP regulation.
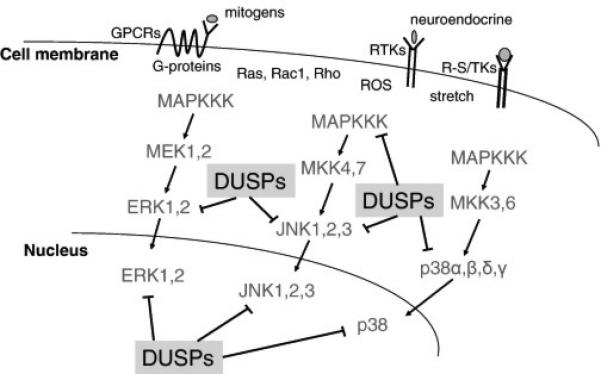
Simplified diagram of the MAPK signaling cascade showing each step in the cascade culminating in ERK1/2, JNK1/2/3 or p38α,β,δ,γ activation, which are inactivated by specific DUSP proteins in either the cytoplasm or nucleus. Other MAPK branches not shown include MEK5-ERK5 and ERK3/4, which are less well characterized for DUSPs counter-regulation.
MAPKs are phosphorylated at both threonine and tyrosine residues within their activation loop (TxY motif), and dephosphorylation is achieved by either serine/threonine phosphatases such as protein phosphatase 2A (PP2A) and protein phosphatase 2C (PP2C) [15], tyrosine phosphatases [16, 17] or more specifically by dedicated dual-specificity phosphatases (DUSPs) that can dephosphorylate both residues concurrently, previously called MAPK phosphatases (MKPs) [18, 19]. There are 25 human DUSPs genes that are roughly divided into those specifically dedicated to MAPK signaling inactivation (approximately 11 genes) and those that regulate the dephosphorylation of diverse and often unknown targets, although all 25 genes are expressed in unique patterns across cell-types and tissues [20]. For example, DUSP2 is enriched in hematopoietic cells [21] while DUSP10 is abundantly expressed in cerebellum, skeletal muscle and bone marrow [22]. The majority of the 11 MAPK-specific DUSP genes are transcriptionally induced in response to the same mitogen and stress stimuli that activate selected MAPKs, and thereafter translated leading to protein appearance within 10-30 minutes where they then inactivate the MAPKs to permit their recycling [23, 24] (Table 1).
Table 1.
MAPK-DUSPs: names and subcellular localization
| Gene | MKP name | Other names | Localization |
|---|---|---|---|
| DUSP1 | MKP-1 | CL100, HVH1, ERP, PTPN10 | Nuclear |
| DUSP2 | PAC-1 | Nuclear | |
| DUSP4 | MKP-2 | NKP2, HVH2, TYP | Nuclear |
| DUSP5 | HVH3 | Nuclear | |
| DUSP6 | MKP-3 | PYST1 | Cytosolic |
| DUSP7 | MKP-X | PYST2 | Cytosolic |
| DUSP8 | M3/6, HVH5 | Cytosolic/nuclear | |
| DUSP9 | MKP-4 | PYST3 | Cytosolic |
| DUSP10 | MKP-5 | Cytosolic/nuclear | |
| DUSP14 | MKP-6 | Cytosolic/nuclear | |
| DUSP16 | MKP-7 | Cytosolic/nuclear |
Abbreviations: DUSP, dual-specificity phosphatase; MKP, MAPK phosphatase
The 11 MAPK-specific DUSPs are further classified into 3 subfamilies based on domain structure, sequence homology and subcellular localization [25, 26]. The first subfamily is composed of nuclear localized DUSPs: DUSP1/MKP1, DUSP2/PAC-1, DUSP4/MKP2, and DUSP5/HVH3, which more or less dephosphorylate all three MAPKs within the nucleus (Table 1). The second and cytoplasmic-localized group includes DUSP6/MKP-3, DUSP7 and DUSP9/MKP4, which are largely specific for ERK1/2 dephosphorylation [25]. The final group includes the cytoplasmic and nuclear localized DUSPs, including DUSP8 (M3/6), DUSP10/MKP5, DUSP14/MKP6, and DUSP16/MKP7, which are thought to have greater specificity for JNK and p38, although DUSP14 is also involved in inactivation of transforming growth factor beta-activated kinase 1 (TAK1) and DUSP8 appears to be more specific for ERK1/2 [27, 28]. The exact specificity of a given DUSP protein for selected MAPKs is often difficult to determine as it can vary between cell-types and depend on the type of assay empolyed. For example, DUSP8 was originally shown to be JNK-specific based on work in cultured cells [29] but in knock-out (KO) mice lacking Dusp8 we have shown a greater preference for ERK1/2 inactivation within the heart [28].
All three subfamilies of DUSPs have a common phosphatase domain as well as CDC25 homology domains and a kinase interacting motif (KIM) within the N-terminal region [20]. The DUSPs also demonstrate different subcellular localizations due to divergent N or C-terminal sequences that can contain a nuclear localization sequence [30, 31] or a nuclear export sequence [32]. DUSP8 and DUSP16 also contain PEST sequences (proline [P], glutamic acid [E], serine [S], and threonine [T] rich) within their C-terminal region that controls protein stability and turnover [20, 33]. An elegant study performed by Tanoue et al demonstrated that the interaction of MAPKs with MAPKKs or DUSPs is achieved through a domain within the MAPKs that contains two negatively charged aspartic acids and a cluster of positively charged amino acids [34]. Moreover, DUSPs are also found within signaling complexes of the cognate MAPKs and their selected scaffold proteins, which achieves efficient regulation of the MAPKs. For example, scaffold protein JNK-interacting protein-1(JIP-1) recruits MKK7, JNK, and DUSP16 to form a signaling complex for rapid proximity regulation of signaling [29, 35].
As briefly mentioned above, the DUSPs are transiently induced in response to growth factors or cellular stresses, although some are more constitutively expressed and have basal effects on MAPK activity [28, 36]. The induction and transcription of DUSPs are dependent on the activation of MAPKs, which creates a “self-limiting” feed-back loop. For example, DUSP1 was originally identified as an immediate early gene in cultured cells that is induced in response to mitogen stimulation [37]. Overexpression of calcineurin in neonatal rat cardiomyocytes led to increased level of DUSP1 through a specific regulatory site in the Dusp1 promoter, which then reduced p38 activity [23]. Analysis of the Dusp5 promoter showed two CArG boxes that bound to the transcription factor, serum response factor (SRF), which was part of a regulatory mechanism whereby the serum response factor interacting factor Elk-1 was phosphorylated by ERK1/2 to induce gene transcription [38]. DUSP6 expression was also induced in response to elevated ERK1/2 signaling as a feedback regulator [39]. Further analysis of Dusp6 promoter discovered two binding sites for Ets2 protein, a known target of ERK2 [40].
2. MAPK signaling in cardiac remodeling and diseases
Studies in KO and transgenic (Tg) mice with altered p38, JNK or ERK activity have annotated the physiological or pathological function of these MAPK effectors in the heart. The first reported study on the functional role of ERK1/2 in the heart utilized Tg mice with cardiac-specific expression of an activated MEK1 mutant protein [41]. MEK1 Tg mice developed mild concentric hypertrophy by 2 months of age without signs of cardiomyopathy or lethality up to 12 months of age. These mice were also resistant to apoptotic stimulation and were protected after ischemia-reperfusion (I/R) injury [42]. Moreover, mice with cardiac-specific overexpression of DN-Raf (a dominant negative form of Raf-1) were resistant to pressure overload-induced cardiac hypertrophy in conjunction with reduced ERK1/2 activation, suggesting the prohypertrophic effects of MEK1/2-ERK1/2 signaling in the heart [43]. Loss-of-function approaches were also pursued to address the necessity of MEK1/2-ERK1/2 signaling in cardiac hypertrophy and remodeling. Cardiac-specific Mapk1/Mapk3 (ERK1/2) KO mice developed cardiac dilation and eccentric growth of the heart, suggesting that ERK1/2 regulate the dimensional growth of the heart, which was linked to individual cardiomyocytes growing in either width (more ERK1/2 activity) or length (loss of ERK1/2 activity) [44]. Consistent with these results, cardiac-specific Tg mice overexpressing DUSP6 in the heart, which showed specific and complete loss of all ERK1/2 signaling, developed eccentric or dilatory growth with a loss of ventricular performance upon stress stimulation [45]. Based on these findings, it is clear that ERK1/2 signaling regulates the balance between eccentric and concentric cardiac growth by regulating how individual cardiomyocytes grow in either length or width [2, 44, 46].
Cardiac-specific expression of activated mutants of MKK3 and MKK6 (specific to p38 activation) did not promote cardiac hypertrophy but instead led to profound disease with interstitial fibrosis, ventricular dysfunction, ventricular dilation and early death [47]. Consistent with these observations, dominant-negative p38α, MKK3, or MKK6 Tg mice each showed enhanced cardiac hypertrophy following aortic banding or infusion of different neuroendocrine factors for 14 days, whereas Mapk14 (p38) heart-specific null mice were more prone to heart failure and pathological remodeling [48, 49]. Similarly, genetic inhibition of JNK1/2 rendered the heart more susceptible to cardiac hypertrophy, similar to the phenotype of mice with cardiac-specific deletion of the genes encoding MKK4 or MKK7 protein [50-52]. Thus, studies in genetically modified mice generally suggest that greater p38 or JNK signaling in the heart promotes profound disease with ventricular remodeling and dilation, while inhibition of p38 or JNK appears to promote hypertrophy and a different type of disease phenotype.
3. DUSPs in cardiac remodeling through MAPK regulation
3.1 Nuclear localized DUSPs: DUSP1, DUSP4, and DUSP5
DUSP1 was the first identified family member, which is implicated in diverse biological events such as inflammation, cancer, metabolism, and diabetes [53]. DUSP1 is expressed in a variety of tissues with the highest levels being observed in the lung [37], and it is induced in the heart upon disease stimulation [54]. Both in vitro and in vivo studies have demonstrated that DUSP1 dephosphorylates all three MAPKs in a cell-type dependent manner [55]. Treatment of cardiac myocytes with an endogenous peptide hormone angiotensin-(1-7) decreased the phosphorylation of ERK1/2 in association with DUSP1 induction [56]. The first study of DUSP1 in the adult heart was from in Tg mice with cardiac-specific expression [57] (Table 2). High expressing Tg mice had reduced activity of all 3 MAPKs and severe ventricular dilation with early postnatal lethality [57]. Lower expressing DUSP1 Tg mice were also resistant to pressure overload induced cardiac hypertrophy collectively suggesting that a physiologic “mix” of all 3 MAPK signaling branches is necessary to support adaptive growth of the heart [57].
Table 2.
Role of DUSPs in the heart from knockout and transgenic mouse studies
| DUSP | MAPK activity in KO mice | Phenotype of KO mice | MAPK activity in Tg mice | Phenotype of Tg mice | References |
|---|---|---|---|---|---|
| DUSP1 | No change in ERK1/2, JNK, and p38 in fibroblasts | Greater infarction injury following I/R; hypertension following endotoxin treatment | Reduced activity of ERK1/2, JNK, and p38 | Dilation, attenuated hypertrophy, increased apoptosis | [57, 58, 60, 62, 63] |
| DUSP4 | Increased p38 activity | Cardiomyopathy in Dusp1/4−/− mice | Reduced ERK1/2 activity | Cardiac hypertrophy and impaired function | [59, 71] |
| DUSP6 | Increased ERK1/2 activity at baseline | Increased myocyte proliferation; Increased heart weight; protected from decompensation following prolonged TAC | Reduced ERK1/2 activity | Decompensation, increased fibrosis and apoptosis following prolonged TAC | [79, 45] |
| DUSP8 | Increased ERK1/2 activity at baseline and following stimulation | Concentric remodeling; increased contractility; protected from injury | Reduced activity of ERK1/2, p38 and JNK | Dilation, fibrosis and cardiac dysfunction | [28] |
| DUSP14 | Increased activity of p38 and JNK following TAC | Hypertrophy, fibrosis, dilation, dysfunction following TAC | Decreased activity of p38 and JNK following TAC | Attenuated TAC-induced cardiac dysfunction and remodeling | [27] |
Abbreviations: DUSP, dual-specificity phosphatase; ERK, extracellular signal-regulated kinase; I/R, ischemia-reperfusion; JNK, c-Jun N-terminal kinase; KO, knockout; MAPK, mitogen-activated protein kinase; TAC, transverse aortic constriction; Tg, transgenic.
Dusp1−/− mice were normal and fertile and did not show overt phenotypical or histological abnormalities or changes in MAPK phosphorylation [58] (Table 2). However, when both Dusp1 and Dusp4 were deleted together, two DUSPs that appear to selectively inactivate p38 in the heart, mice demonstrated increased baseline p38 phosphorylation and cardiomyopathy in response to pathological insults [59] (Table 2). This later result is likely because Dusp4 induction can compensate for loss of Dusp1 in mediating p38 inactivation in response to cardiac pathological conditions [20]. Despite this argument of compensation, Dusp1−/− mice did show greater infarction injury compared to controls after I/R insult [60], and p38 over-activation is generally known to promote cell death [61]. Mice deficient for Dusp1 also exhibited increased hypertension following endotoxin treatment or a hypoxia procedure [62, 63]. Finally, DUSP1 was also involved in regulation of cardiac fibroblast proliferation and migration through p38 and ERK1/2 signaling [64]. Taken together, these data suggest that DUSP1 is a critical signaling effector in the heart, most likely due to its preference of p38 inactivation.
DUSP4 was first cloned from PC-12 cells and characterized to inactivate all three MAPKs [65-69]. Overexpression of DUSP4 in human umbilical vein endothelial cells abolished JNK activity and protected cells against tumor necrosis factor α-mediated apoptosis [70]. Genetic ablation of Dusp4 in neurons enhanced ERK1/2 activity during neuronal differentiation [67]. In terms of the functional role of DUSP4 in the heart, we showed that deletion of either Dusp1 or Dusp4 alone had little effect but that the double deletion of these genes promoted greater p38 activity in the heart with no effect on JNK or ERK1/2, which produced a disease susceptible phenotype [59] (Table 2). The Dusp1/4 double null mice showed induction of cardiomyopathy with aging that was reversed by pharmacological inhibition of p38 MAPK with SB731445 suggesting that these 2 DUSPs function primarily through p38 [59]. In a parallel study, Choi et al discovered that DUSP4 mRNA was selectively upregulated in the hearts of a mouse model of lamin A/C cardiomyopathy (Lmna H222P/H222P mice), and generation and characterization of DUSP4 cardiac-specific Tg mice showed cardiomyopathy [71]. Such results are consistent with the disease predisposing phenotype observed in DUSP1 heart-specific Tg mice [57].
Considered as a regulator and anchor for nuclear ERK2, DUSP5 has yet to be extensively investigated for effects on the heart. However, in cultured neonatal rat ventricular myocytes selective inhibitors for class I histone deacetylases (HDACs) led to decreased DUSP5 expression, and overexpression of DUSP5 promoted ERK1/2 inactivation and the reduction in agonist-induced hypertrophy [72]. DUSP5 was also shown to dephosphorylate ERK1/2 during cardiac fibroblast proliferation [73]. Finally, while Dusp5 KO mice are viable, the effect of gene deletion on cardiac growth and remodeling has yet to be investigated [74].
3.2 Cytosolic DUSPs in the heart: DUSP6, DUSP7, and DUSP9
DUSP6 is exclusively cytosolic and highly specific for ERK1/2 dephosphorylation in a diverse array of cell-types and tissues [75-78]. Dusp6 KO mice are viable, fertile and overtly normal [79]. Interestingly, loss of Dusp6 resulted in increased baseline ERK1/2 activity in multiple tissues without any effect on phosphorylation of JNK, p38, or ERK5 [79] (Table 2). However, loss of Dusp6 did not further enhance or prolong ERK1/2 signaling after stimulation, suggesting that DUSP6 is more dedicated to baseline ERK1/2 activity. This increase in ERK1/2 activity at baseline in Dusp6 KO mice produced an increase in heart weight at every age investigated due to increased rate of myocyte proliferation during development, which protected the heart from decompensation and cardiomyopathy in response to pressure overload and myocardial infarction injuries [79]. Cardiac-specific DUSP6 Tg mice exhibited inactivation of ERK1/2 without reduction in p38 or JNK phosphorylation at baseline or following stimulation, demonstrating the highly specific nature of DUSP6 for ERK1/2 [45]. As discussed earlier, DUSP6 Tg mice showed alterations in cardiac ventricular remodeling with pressure overload, such as greater dilation and loss of ventricular performance [45]. This is consistent with the phenotype of Mapk1/3 (ERK1/2) heart-specific null mice that also showed failure with ventricular dilation and increase in myocyte growth in the long axis [28, 44]. Taken together, manipulation of ERK1/2 signaling by DUSP6 suggests that ERK1/2 are not required for mediating hypertrophy per se, but rather that DUSP6-ERK1/2 influence the balance between concentric and eccentric growth of the heart with myocytes growing in either their long or short axis.
Given that DUSP6, DUSP7 and DUSP9 each regulate ERK1/2; we might expect that loss of these later 2 DUSPs might impact the heart with either greater myocyte proliferation during development or greater concentric remodeling with stimulation, all due to greater ERK1/2 activity. However, deletion of Dusp9 in the mouse resulted in embryonic lethality due to placental abnormalities, hence this gene would have to be LoxP targeted to allow for heart-specific deletion along with the appropriate Cre expressing Tg lines [80].
3.3 Dually localized DUSPs in the heart: DUSP8, DUSP10, DUSP14, and DUSP16
DUSP8 was initially identified as a JNK-specific phosphatase and implicated in diabetes and alcohol dependence [81-83]. Recently, both Dusp8 KO mice and heart-specific inducible Tg mice were generated and analyzed [28] (Table 2). DUSP8 was found to be predominantly expressed in cardiomyocytes over other non-myocyte populations in the heart, and induced in response to pressure overload or myocardial infarction injury [28]. Dusp8 KO mice were viable and fertile and while they maintained normal cardiac function, the morphology of the left ventricle was altered with a mild form of concentric hypertrophy [28]. Specifically, the left ventricular chamber dimension was smaller and septal thickness was increased compared to the wild type controls. Isolation of adult myocytes from hearts of Dusp8 KO mice showed myocyte thickening [28], similar to heart-specific MEK1 Tg mice [41]. This alteration in ventricular geometry in null mice was protective and resulted in less cardiomyopathy following ischemic injury and TAC stimulation, while Tg overexpression of DUSP8 in the heart promoted cardiomyopathy and cardiomyocyte lengthening [28]. Mechanistically, Dusp8 KO mice had increased activity of ERK1/2 at baseline and following acute stimulation, which is the first study to suggest a preference for ERK1/2 inactivation, although a partial effect on JNK inactivation in the heart was also noted [28]. However, the results observed in Dusp8 null mice is highly consistent with many past studies in mice with altered ERK1/2 activity, which collectively underscores the biology of ERK1/2 as master regulators of cardiac remodeling by directly controlling how individual cardiomyocytes grow in either their long or short axis.
DUSP14 is not considered a traditional MAPK-DUSP compared with the 10 other members of this subfamily, but functional studies with DUSP14 have shown the ability to directly regulate the MAPKs [84]. For example, Li et al characterized the role of DUSP14 in the heart in the hypertrophic response using both Dusp14−/− mice as well as Tg mice with cardiac overexpression of this gene [27] (Table 2). In cardiomyopathic human hearts, as well as in TAC-operated mouse hearts, DUSP14 expression was decreased. Indeed, Dusp14−/− mice exhibited cardiac hypertrophy, fibrosis, ventricular dilation and dysfunction in response to 4 weeks of pressure overload injury. In contrast, DUSP14 Tg mice had attenuated cardiac hypertrophy and less dysfunction following stress [27]. Interestingly, loss of Dusp14 led to increased activity of TAK1, p38 and JNK1/2 without alterations in ERK1/2 phosphorylation in TAC operated mice, whereas hearts from DUSP14 Tg mice had reduced activity of TAK1, p38 and JNK1/2.
Both DUSP10 and DUSP16 were initially identified as phosphatases for JNK and p38, although studies in mice deficient for these genes also suggest a role in regulating ERK1/2 signaling [85-90]. However, the role that these two genes play in the heart has yet to be examined, although KO mice have been reported [91, 92].
4 Conclusions and future directions
One critical point to consider in DUSP biology is specificity validation for ERK, JNK and p38 kinases. Earlier studies were largely based on cultured cells with overexpression approaches (transfection of expression plasmids) or with some sort of partial knock-down technique. Such studies gave a profile of DUSP regulation towards selected MAPKs that sometimes were inconsistent with studies from KO mice or null cells. For example, DUSP1 was originally shown to be specific for ERK1/2, yet our analysis in Dusp1 gene-deleted hearts clearly shows a strong preference for p38 MAPK. Other factors that might affect observed profiles of specificity between DUSPs and the MAPK terminal effector proteins is the presence or absence of select MAPK docking proteins that could vary between tissues and cell-types, or more importantly, the repertoire of other expressed DUSPs with similar specificity, thus more or less masking the effects of the DUSP factor under investigation. For example, high levels of Dusp4 expression appear to mask the effects of Dusp1 deletion in the heart, such that only in the absence of both genes is an alteration in p38 MAPK observed [59]. By extension, it is possible that Dusp4 is not highly expressed in other cell-types or tissues, such that deletion of Dusp1 now has an easily observable effect on MAPK signaling and associated biologic processes. A final issue to consider is that overexpression approaches, even in Tg mice with tissue specific promoters, is not an ideal means of annotating specificity compared with loss-of-function approaches. For example, overexpression of DUSP1 in the heart generated promiscuity and inactivation of all three MAPK terminal effectors groups [57], a finding that is inconsistent with the loss-of-function approach discussed above (specific for p38 MAPK).
In conclusion, studies over more than 2 decades have provided considerable insight into the function and importance of the MAPK dedicated Dusp genes in the heart. This review focused mostly on studies conducted in genetically modified mice so we apologize for not covering all aspects of the field that were based on studies in cultured cells. Table 2 summarizes the most prominent of these studies conducted in genetically modified mice, including gene-deleted mice. Further characterization is needed to investigate the role of additional Dusp genes to collectively understand the dynamic regulation of MAPK signaling in the heart and how the DUSPs are involved.
Highlights.
MAPK signaling critically regulates cardiac adaptive and maladaptive cardiac hypertrophy
Inactivation of the MAPKs are regulated by dedicated dual-specificity phosphatases (DUSPs)
DUSP-dependent regulation of the MAPKs dynamically alters cardiac responses to physiological and pathological stimuli
Acknowledgments
Funding sources
This work was supported by grants from the National Institutes of Health (to J.D. Molkentin). J.D. Molkentin was also supported by the Howard Hughes Medical Institute. R.L., was supported by a training grant from the National Heart Lung and Blood Institute of the NIH (T32HL125204) and a start-up grant from Grand Valley State University, Michigan.
Abbreviations
- DUSP
dual-specificity phosphatase
- ERK
extracellular signal-regulated kinase
- I/R
ischemia reperfusion
- JNK
c-Jun N-terminal kinase
- KO
knockout
- MAPK
mitogen-activated protein kinase
- MAPKKs
MAPK kinases
- MKPs
MAPK phosphatases
- TAC
transverse aortic constriction
- Tg
transgenic
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
No financial or other conflicts of interest exist with either of the authors.
References
- 1.Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102:470–9. doi: 10.1161/01.cir.102.4.470. [DOI] [PubMed] [Google Scholar]
- 2.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–9. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 5.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–46. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 7.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122–42. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- 9.Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–5. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- 10.Goupil E, Wisehart V, Khoury E, Zimmerman B, Jaffal S, Hebert TE, et al. Biasing the prostaglandin F2alpha receptor responses toward EGFR-dependent transactivation of MAPK. Mol Endocrinol. 2012;26:1189–202. doi: 10.1210/me.2011-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Fan D, Wang C, Wang JY, Cui XB, Wu D, et al. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-beta1 pathways in cardiac fibroblasts. Cardiovasc Res. 2011;91:80–9. doi: 10.1093/cvr/cvr067. [DOI] [PubMed] [Google Scholar]
- 12.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–12. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 13.Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–7. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 14.Toischer K, Rokita AG, Unsold B, Zhu W, Kararigas G, Sossalla S, et al. Differential cardiac remodeling in preload versus afterload. Circulation. 2010;122:993–1003. doi: 10.1161/CIRCULATIONAHA.110.943431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol. 1995;5:283–95. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- 16.Pulido R, Zuniga A, Ullrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998;17:7337–50. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–65. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 18.Patterson KI, Brummer T, O'Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–89. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- 19.Kondoh K, Nishida E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim Biophys Acta. 2007;1773:1227–37. doi: 10.1016/j.bbamcr.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Huang CY, Tan TH. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012;2:24. doi: 10.1186/2045-3701-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeffrey KL, Brummer T, Rolph MS, Liu SM, Callejas NA, Grumont RJ, et al. Positive regulation of immune cell function and inflammatory responses by phosphatase PAC-1. Nat Immunol. 2006;7:274–83. doi: 10.1038/ni1310. [DOI] [PubMed] [Google Scholar]
- 22.Tanoue T, Moriguchi T, Nishida E. Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J Biol Chem. 1999;274:19949–56. doi: 10.1074/jbc.274.28.19949. [DOI] [PubMed] [Google Scholar]
- 23.Lim HW, New L, Han J, Molkentin JD. Calcineurin enhances MAPK phosphatase-1 expression and p38 MAPK inactivation in cardiac myocytes. J Biol Chem. 2001;276:15913–9. doi: 10.1074/jbc.M100452200. [DOI] [PubMed] [Google Scholar]
- 24.Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119:4607–15. doi: 10.1242/jcs.03266. [DOI] [PubMed] [Google Scholar]
- 25.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salojin K, Oravecz T. Regulation of innate immunity by MAPK dual-specificity phosphatases: knockout models reveal new tricks of old genes. J Leukoc Biol. 2007;81:860–9. doi: 10.1189/jlb.1006639. [DOI] [PubMed] [Google Scholar]
- 27.Li CY, Zhou Q, Yang LC, Chen YH, Hou JW, Guo K, et al. Dual-specificity phosphatase 14 protects the heart from aortic banding-induced cardiac hypertrophy and dysfunction through inactivation of TAK1-P38MAPK/-JNK1/2 signaling pathway. Basic Res Cardiol. 2016;111:19. doi: 10.1007/s00395-016-0536-7. [DOI] [PubMed] [Google Scholar]
- 28.Liu R, van Berlo JH, York AJ, Vagnozzi RJ, Maillet M, Molkentin JD. DUSP8 Regulates Cardiac Ventricular Remodeling by Altering ERK1/2 Signaling. Circ Res. 2016;119:249–60. doi: 10.1161/CIRCRESAHA.115.308238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willoughby EA, Perkins GR, Collins MK, Whitmarsh AJ. The JNK-interacting protein-1 scaffold protein targets MAPK phosphatase-7 to dephosphorylate JNK. J Biol Chem. 2003;278:10731–6. doi: 10.1074/jbc.M207324200. [DOI] [PubMed] [Google Scholar]
- 30.Mandl M, Slack DN, Keyse SM. Specific inactivation and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual-specificity protein phosphatase DUSP5. Mol Cell Biol. 2005;25:1830–45. doi: 10.1128/MCB.25.5.1830-1845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu JJ, Zhang L, Bennett AM. The noncatalytic amino terminus of mitogen-activated protein kinase phosphatase 1 directs nuclear targeting and serum response element transcriptional regulation. Mol Cell Biol. 2005;25:4792–803. doi: 10.1128/MCB.25.11.4792-4803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlsson M, Mathers J, Dickinson RJ, Mandl M, Keyse SM. Both nuclear-cytoplasmic shuttling of the dual specificity phosphatase MKP-3 and its ability to anchor MAP kinase in the cytoplasm are mediated by a conserved nuclear export signal. J Biol Chem. 2004;279:41882–91. doi: 10.1074/jbc.M406720200. [DOI] [PubMed] [Google Scholar]
- 33.Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–71. [PubMed] [Google Scholar]
- 34.Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2:110–6. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 35.Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. Scaffold proteins of MAP-kinase modules. Oncogene. 2007;26:3185–202. doi: 10.1038/sj.onc.1210411. [DOI] [PubMed] [Google Scholar]
- 36.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- 37.Charles CH, Abler AS, Lau LF. cDNA sequence of a growth factor-inducible immediate early gene and characterization of its encoded protein. Oncogene. 1992;7:187–90. [PubMed] [Google Scholar]
- 38.Buffet C, Catelli MG, Hecale-Perlemoine K, Bricaire L, Garcia C, Gallet-Dierick A, et al. Dual Specificity Phosphatase 5, a Specific Negative Regulator of ERK Signaling, Is Induced by Serum Response Factor and Elk-1 Transcription Factor. PLoS One. 2015;10:e0145484. doi: 10.1371/journal.pone.0145484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bermudez O, Pages G, Gimond C. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am J Physiol Cell Physiol. 2010;299:C189–202. doi: 10.1152/ajpcell.00347.2009. [DOI] [PubMed] [Google Scholar]
- 40.Ekerot M, Stavridis MP, Delavaine L, Mitchell MP, Staples C, Owens DM, et al. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008;412:287–98. doi: 10.1042/BJ20071512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–50. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lips DJ, Bueno OF, Wilkins BJ, Purcell NH, Kaiser RA, Lorenz JN, et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004;109:1938–41. doi: 10.1161/01.CIR.0000127126.73759.23. [DOI] [PubMed] [Google Scholar]
- 43.Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C, Muslin AJ. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation. 2004;110:718–23. doi: 10.1161/01.CIR.0000138190.50127.6A. [DOI] [PubMed] [Google Scholar]
- 44.Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res. 2011;108:176–83. doi: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Purcell NH, Wilkins BJ, York A, Saba-El-Leil MK, Meloche S, Robbins J, et al. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci U S A. 2007;104:14074–9. doi: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mutlak M, Kehat I. Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front Pharmacol. 2015;6:149. doi: 10.3389/fphar.2015.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–8. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–86. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, et al. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–20. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J. 2003;22:5079–89. doi: 10.1093/emboj/cdg474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu W, Zi M, Jin J, Prehar S, Oceandy D, Kimura TE, et al. Cardiac-specific deletion of mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ Res. 2009;104:905–14. doi: 10.1161/CIRCRESAHA.108.188292. [DOI] [PubMed] [Google Scholar]
- 52.Liu W, Zi M, Chi H, Jin J, Prehar S, Neyses L, et al. Deprivation of MKK7 in cardiomyocytes provokes heart failure in mice when exposed to pressure overload. J Mol Cell Cardiol. 2011;50:702–11. doi: 10.1016/j.yjmcc.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 53.Low HB, Zhang Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016;16:85–98. doi: 10.4110/in.2016.16.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohki R, Yamamoto K, Ueno S, Mano H, Misawa Y, Fuse K, et al. Transcriptional profile of genes induced in human atrial myocardium with pressure overload. Int J Cardiol. 2004;96:381–7. doi: 10.1016/j.ijcard.2003.07.025. [DOI] [PubMed] [Google Scholar]
- 55.Lawan A, Shi H, Gatzke F, Bennett AM. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell Mol Life Sci. 2013;70:223–37. doi: 10.1007/s00018-012-1041-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCollum LT, Gallagher PE, Ann Tallant E. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. Am J Physiol Heart Circ Physiol. 2012;302:H801–10. doi: 10.1152/ajpheart.00908.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bueno OF, De Windt LJ, Lim HW, Tymitz KM, Witt SA, Kimball TR, et al. The dual-specificity phosphatase MKP-1 limits the cardiac hypertrophic response in vitro and in vivo. Circ Res. 2001;88:88–96. doi: 10.1161/01.res.88.1.88. [DOI] [PubMed] [Google Scholar]
- 58.Dorfman K, Carrasco D, Gruda M, Ryan C, Lira SA, Bravo R. Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene. 1996;13:925–31. [PubMed] [Google Scholar]
- 59.Auger-Messier M, Accornero F, Goonasekera SA, Bueno OF, Lorenz JN, van Berlo JH, et al. Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ Res. 2013;112:48–56. doi: 10.1161/CIRCRESAHA.112.272963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, et al. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J Biol Chem. 2004;279:15524–30. doi: 10.1074/jbc.M313717200. [DOI] [PubMed] [Google Scholar]
- 61.Yokota T, Wang Y. p38 MAP kinases in the heart. Gene. 2016;575:369–76. doi: 10.1016/j.gene.2015.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calvert TJ, Chicoine LG, Liu Y, Nelin LD. Deficiency of mitogen-activated protein kinase phosphatase-1 results in iNOS-mediated hypotension in response to low-dose endotoxin. Am J Physiol Heart Circ Physiol. 2008;294:H1621–9. doi: 10.1152/ajpheart.01008.2007. [DOI] [PubMed] [Google Scholar]
- 63.Jin Y, Calvert TJ, Chen B, Chicoine LG, Joshi M, Bauer JA, et al. Mice deficient in Mkp-1 develop more severe pulmonary hypertension and greater lung protein levels of arginase in response to chronic hypoxia. Am J Physiol Heart Circ Physiol. 2010;298:H1518–28. doi: 10.1152/ajpheart.00813.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valente AJ, Yoshida T, Gardner JD, Somanna N, Delafontaine P, Chandrasekar B. Interleukin-17A stimulates cardiac fibroblast proliferation and migration via negative regulation of the dual-specificity phosphatase MKP-1/DUSP-1. Cell Signal. 2012;24:560–8. doi: 10.1016/j.cellsig.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Misra-Press A, Rim CS, Yao H, Roberson MS, Stork PJ. A novel mitogen-activated protein kinase phosphatase. Structure, expression, and regulation. J Biol Chem. 1995;270:14587–96. doi: 10.1074/jbc.270.24.14587. [DOI] [PubMed] [Google Scholar]
- 66.Robinson CJ, Sloss CM, Plevin R. Inactivation of JNK activity by mitogen-activated protein kinase phosphatase-2 in EAhy926 endothelial cells is dependent upon agonist-specific JNK translocation to the nucleus. Cell Signal. 2001;13:29–41. doi: 10.1016/s0898-6568(00)00121-2. [DOI] [PubMed] [Google Scholar]
- 67.Kim SY, Han YM, Oh M, Kim WK, Oh KJ, Lee SC, et al. DUSP4 regulates neuronal differentiation and calcium homeostasis by modulating ERK1/2 phosphorylation. Stem Cells Dev. 2015;24:686–700. doi: 10.1089/scd.2014.0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lawan A, Torrance E, Al-Harthi S, Shweash M, Alnasser S, Neamatallah T, et al. MKP-2: out of the DUSP-bin and back into the limelight. Biochem Soc Trans. 2012;40:235–9. doi: 10.1042/BST20110648. [DOI] [PubMed] [Google Scholar]
- 69.Berasi SP, Huard C, Li D, Shih HH, Sun Y, Zhong W, et al. Inhibition of gluconeogenesis through transcriptional activation of EGR1 and DUSP4 by AMP-activated kinase. J Biol Chem. 2006;281:27167–77. doi: 10.1074/jbc.M602416200. [DOI] [PubMed] [Google Scholar]
- 70.Al-Mutairi M, Al-Harthi S, Cadalbert L, Plevin R. Over-expression of mitogen-activated protein kinase phosphatase-2 enhances adhesion molecule expression and protects against apoptosis in human endothelial cells. Br J Pharmacol. 2010;161:782–98. doi: 10.1111/j.1476-5381.2010.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choi JC, Wu W, Muchir A, Iwata S, Homma S, Worman HJ. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J Biol Chem. 2012;287:40513–24. doi: 10.1074/jbc.M112.404541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, et al. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2013;110:9806–11. doi: 10.1073/pnas.1301509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tao H, Cao W, Yang JJ, Shi KH, Zhou X, Liu LP, et al. Long noncoding RNA H19 controls DUSP5/ERK1/2 axis in cardiac fibroblast proliferation and fibrosis. Cardiovasc Pathol. 2016;25:381–9. doi: 10.1016/j.carpath.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 74.Rushworth LK, Kidger AM, Delavaine L, Stewart G, van Schelven S, Davidson J, et al. Dual-specificity phosphatase 5 regulates nuclear ERK activity and suppresses skin cancer by inhibiting mutant Harvey-Ras (HRasQ61L)-driven SerpinB2 expression. Proc Natl Acad Sci U S A. 2014;111:18267–72. doi: 10.1073/pnas.1420159112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kidger AM, Keyse SM. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin Cell Dev Biol. 2016;50:125–32. doi: 10.1016/j.semcdb.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Muda M, Boschert U, Dickinson R, Martinou JC, Martinou I, Camps M, et al. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J Biol Chem. 1996;271:4319–26. doi: 10.1074/jbc.271.8.4319. [DOI] [PubMed] [Google Scholar]
- 77.Shojaee S, Caeser R, Buchner M, Park E, Swaminathan S, Hurtz C, et al. Erk Negative Feedback Control Enables Pre-B Cell Transformation and Represents a Therapeutic Target in Acute Lymphoblastic Leukemia. Cancer Cell. 2015;28:114–28. doi: 10.1016/j.ccell.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li GY, Zhou Y, Ying RS, Shi L, Cheng YQ, Ren JP, et al. Hepatitis C virus-induced reduction in miR-181a impairs CD4(+) T-cell responses through overexpression of DUSP6. Hepatology. 2015;61:1163–73. doi: 10.1002/hep.27634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maillet M, Purcell NH, Sargent MA, York AJ, Bueno OF, Molkentin JD. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem. 2008;283:31246–55. doi: 10.1074/jbc.M806085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Christie GR, Williams DJ, Macisaac F, Dickinson RJ, Rosewell I, Keyse SM. The dual-specificity protein phosphatase DUSP9/MKP-4 is essential for placental function but is not required for normal embryonic development. Mol Cell Biol. 2005;25:8323–33. doi: 10.1128/MCB.25.18.8323-8333.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dayeh TA, Olsson AH, Volkov P, Almgren P, Ronn T, Ling C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia. 2013;56:1036–46. doi: 10.1007/s00125-012-2815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hink RL, Hokanson JE, Shah I, Long JC, Goldman D, Sikela JM. Investigation of DUSP8 and CALCA in alcohol dependence. Addict Biol. 2003;8:305–12. doi: 10.1080/13556210310001602211. [DOI] [PubMed] [Google Scholar]
- 83.Schwenk RW, Vogel H, Schurmann A. Genetic and epigenetic control of metabolic health. Mol Metab. 2013;2:337–47. doi: 10.1016/j.molmet.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng H, Li Q, Chen R, Zhang J, Ran Y, He X, et al. The dual-specificity phosphatase DUSP14 negatively regulates tumor necrosis factor- and interleukin-1-induced nuclear factor-kappaB activation by dephosphorylating the protein kinase TAK1. J Biol Chem. 2013;288:819–25. doi: 10.1074/jbc.M112.412643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Theodosiou A, Smith A, Gillieron C, Arkinstall S, Ashworth A. MKP5, a new member of the MAP kinase phosphatase family, which selectively dephosphorylates stress-activated kinases. Oncogene. 1999;18:6981–8. doi: 10.1038/sj.onc.1203185. [DOI] [PubMed] [Google Scholar]
- 86.James SJ, Jiao H, Teh HY, Takahashi H, Png CW, Phoon MC, et al. MAPK Phosphatase 5 Expression Induced by Influenza and Other RNA Virus Infection Negatively Regulates IRF3 Activation and Type I Interferon Response. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.030. [DOI] [PubMed] [Google Scholar]
- 87.Png CW, Weerasooriya M, Guo J, James SJ, Poh HM, Osato M, et al. DUSP10 regulates intestinal epithelial cell growth and colorectal tumorigenesis. Oncogene. 2016;35:206–17. doi: 10.1038/onc.2015.74. [DOI] [PubMed] [Google Scholar]
- 88.Tanoue T, Yamamoto T, Maeda R, Nishida E. A Novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38 alpha and beta MAPKs. J Biol Chem. 2001;276:26629–39. doi: 10.1074/jbc.M101981200. [DOI] [PubMed] [Google Scholar]
- 89.Masuda K, Shima H, Watanabe M, Kikuchi K. MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J Biol Chem. 2001;276:39002–11. doi: 10.1074/jbc.M104600200. [DOI] [PubMed] [Google Scholar]
- 90.Zhang Y, Nallaparaju KC, Liu X, Jiao H, Reynolds JM, Wang ZX, et al. MAPK phosphatase 7 regulates T cell differentiation via inhibiting ERK-mediated IL-2 expression. J Immunol. 2015;194:3088–95. doi: 10.4049/jimmunol.1402638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Blattman JN, Kennedy NJ, Duong J, Nguyen T, Wang Y, et al. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature. 2004;430:793–7. doi: 10.1038/nature02764. [DOI] [PubMed] [Google Scholar]
- 92.Niedzielska M, Bodendorfer B, Munch S, Eichner A, Derigs M, da Costa O, et al. Gene trap mice reveal an essential function of dual specificity phosphatase Dusp16/MKP-7 in perinatal survival and regulation of Toll-like receptor (TLR)-induced cytokine production. J Biol Chem. 2014;289:2112–26. doi: 10.1074/jbc.M113.535245. [DOI] [PMC free article] [PubMed] [Google Scholar]