

Abstract
Fatty acid oxidation (FAO) defects often present with multi-system involvement, including several life-threatening cardiac manifestations, such as cardiomyopathy, pericardial effusion and arrhythmias.
We report herein a fatal case of cardiac dysfunction and rapid-onset tamponade following an acute illness in a neonate with molecularly proven very long chain acyl-CoA dehydrogenase (VLCAD) deficiency (harboring the known del799_802 mutation), requiring 15 days of extracorporeal membrane oxygenation (ECMO) treatment.
As data regarding the use of ECMO in FAO defects in general, and VLCAD in particular, are scarce, we review the literature and discuss insights from in vitro models and several successful reported cases.
Keywords: VLCAD, Very long chain acyl-CoA dehydrogenase deficiency, ECMO, ACADVL
1. Introduction
Fatty Acid Oxidation (FAO) defects are a heterogeneous group of disorders, affecting energy homeostasis mainly in the liver, heart and skeletal muscles [6]. Hence, cardiac manifestations are common in symptomatic cases of FAO defects, including cardiomyopathies, arrhythmias and pericardial effusion [4], [14], and can lead to cardiogenic shock [3]. Nonetheless, reports of children with FAO defects treated by extracorporeal membrane oxygenation (ECMO) are scarce. We describe a case of an infant with Very Long Chain Acyl-CoA Dehydrogenase (VLCAD) deficiency, treated in our pediatric intensive care unit with ECMO.
2. Case report
The patient was born full term by spontaneous vaginal delivery, following an uneventful pregnancy, at a birth weight of 2930 g and an Apgar score of 9/10. Born to non-consanguineous healthy parents of Jewish-Bukharian descent, the patient has five siblings, four of them healthy and a sister who died at 1 week of age, due to arrhythmia that was attributed to a FAO defect, most probably VLCAD deficiency.
At 2 days of age he presented with lethargy and apathy. Blood glucose levels were normal, however CPK was markedly elevated (13,445 IU/l) and liver transaminases were mildly elevated (ALT 58 IU/l, AST 240 IU/l). He was treated with an intravenous 10% glucose infusion and Monogen (medium-chain-triglyceride-enriched formula). At 11 days he was discharged from the hospital following clinical and laboratory improvement, reaching normal levels of CPK, ALT and AST.
Following newborn screening results, he was suspected to have VLCAD deficiency. Further lab tests showed urinary organic acids and plasma acyl carnitine profiles consistent with that diagnosis. Genetic testing of the ACADVL gene demonstrated homozygosity for the known del799_802 mutation (previously described as null mutation resulting in no residual activity of the enzyme, [2]). Echocardiography at 5 days and 1 month of age showed a small atrial septal defect (ASD) with no hypertrophy. ECG at 22 days and 1 month of age showed normal sinus rhythm. As recommended, the parents were instructed to feed their child every 3 h, to avoid fasting, and in case of prolonged febrile disease or decreased food intake to seek immediate medical care.
With regard to the patient's medical management, he was considered metabolically stable, and the parents adhered to routine checkups and were planned to arrive to the clinic a week after his presentation. At 4 months was evaluated by the nutritionist, and was fed 600 cm3/day of Pregestimil (Mead Johnson & Co, LLC, Glenview, IL) and received medium chain triglyderides (MCTs) 2.5%. His growth was overall well (Weight, length and head circumference within the 10–25th percentiles).
At 6 months of age he presented to the emergency department during an intercurrent illness, manifesting with vomiting, diarrhea, and decreased food intake beginning two days before his admission. He was clinically dehydrated, and had rales and crackles on lung auscultation. CBC and blood electrolytes levels were within normal limits, CPK was normal (121 IU/l, N 0–190), liver transaminases were slightly elevated (AST, 98 IU/l, N 0–100; ALT, 105 IU/l, N 7–45). Blood gases showed metabolic acidosis with pH 7.2, HCO3 16.5 mEq/l and lactate 30 mg/dl. ECG showed small QRS complexes and chest X-ray was suggestive of either consolidation or enlarged heart silhouette.
He was initially treated with fluid resuscitation and empirical antibiotics, and transferred to the pediatric intensive care unit. Screening tests for an infectious etiology were negative.
With regard to his metabolic management, as serum free carnitine levels were extremely low at presentation with normal total cartnitine levels, intravenous carnitine supplementation was initiated (initial loading dose of 100 mg/kg, and subsequently 50 mg/kg three times daily). Due to inability to resume enteral feedings, an intravenous infusion of 50% glucose was introduced (based on glucose disposal rate of 8 mg/kg/min), with insulin infusion as needed. Triheptanoin, which may be considered as an addition to the medical management, is unfortunately unavailable in Israel.
Echocardiography at day 1 demonstrated a large pericardial effusion (13 mm in the dorsal part and 20 mm in the lateral part, Fig. 1), with mild right atrial collapse and reduced flow across the mitral valve, requiring pericardiocentesis. Post-pericardiocentesis echocardiographs showed left ventricle hypertrophy with normal function (LVSF 33%, norm 28–44%), with no residual pericardial effusion. Over the next 48 h his cardiac function deteriorated – LVSF decreased to 20% and lower, and he developed mitral regurgitation. The effusion recurred and a second pericardiocentesis was conducted. He also suffered multiple organ failure including cardiac arrest, and after a successful resuscitation extracorporeal membrane oxygenation (ECMO) was initiated. During the next two weeks, despite maximal supportive treatment, his cardiac function worsened. After 15 days under ECMO, head CT demonstrated subarachnoid and intraventricular hemorrhages with parenchymal edema. ECMO was withdrawn, following which the patient had asystole and died.
Fig. 1.
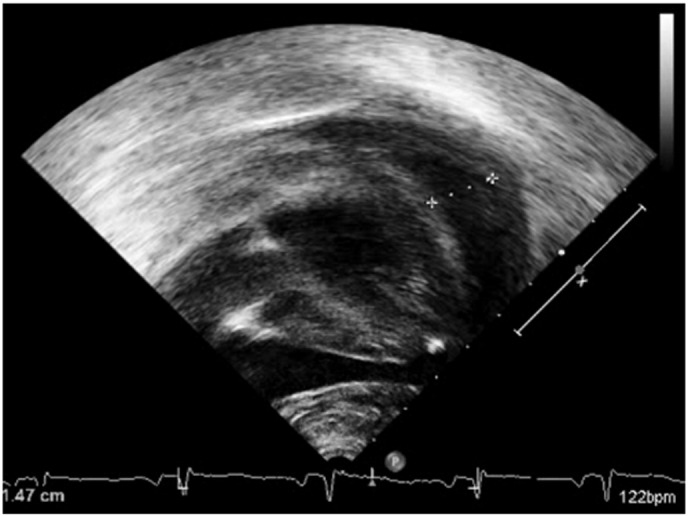
Echcardiograph of a patient with VLCAD deficiency upon admission, demonstrating severe pericardial effusion.
3. Discussion
Cardiac manifestations may occur in most FAO disorders, including VLCAD deficiency. These comprise of cardiomyopathies, arrhythmias, conduction abnormalities and pericardial effusion [4], [14], and used to be common before the era of newborn screening programs [16].
VLCAD deficiency is associated with a variety of phenotypes, the most severe of which is characterized by early-onset cardiomyopathy, leading to high mortality rates [2]. Spiekerkoetter et al. described a case of an infant presenting at 4 months of age with hypertrophic cardiomyopathy and a large pericardial effusion. After pericardiocentesis and under Monogen diet and regular feedings, the pericardial effusion resolved in two weeks, and cardiomyopathy was reversed in two months [17]. Other cases in which cardiac symptoms resolved under sufficient energy supply were also reported [3], [15]. In the case reported herein, perinatal cardiac evaluation showed no hypertrophy and normal cardiac functioning. Nevertheless, at 6 months of age upon admission, the patient had a large pericardial effusion and echo signs of cardiac tamponade, with no improvement despite repeated pericardiocentesis.
ECMO serves as a temporary life support measure in a variety of cardiac and respiratory indications. Review of the literature reveals only a few cases of FAO defects treated with ECMO. Kumar et al. described a neonate with MCAD (Medium Chain Acyl-CoA Dehydrogenase) deficiency, who was successfully resuscitated with ECMO, following an episode of VT progressing to VF. The authors concluded that ECMO support is not necessarily contraindicated in cases of inborn errors of metabolism, and can lead to excellent result [8]. In case series of 18 patients with VLCAD deficiency, out of 12 who presented with cardiomyopathy, one patient was treated with ECMO with good outcome (described shortly as survival during follow up period, [9]). Our patient was placed on ECMO following multi-organ failure and cardiac deterioration. However, during the 15 days he was on ECMO there was no amelioration of his cardiac function. This lack of improvement can be attributed to a number of reasons. First, although outcomes of ECMO treatment are improving, mortality and morbidity are still high. Recent reports show survival rates of less than 50% in pediatric cardiac patients treated with ECMO [13], with worse outcome under prolonged treatment [10]. Moreover, ECMO patients suffer from many complications, most commonly cardiac, but also renal, neurological, metabolic and more [5].
Second, ECMO promotes a number of metabolic disturbances. Previous studies have shown that ECMO induces an inflammatory reaction, which increases insulin resistance, reduces glucose utilization and promotes a general catabolic state [12]. Attempts to administer high dose insulin had minimal influence on protein turnover [1]. Interestingly, Kajimoto et al. modeled ECMO procedure in immature swine and examined the metabolic changes in the myocardium. They found that the myocardium under unloading by ECMO increases long chain fatty acid (LCFA) oxidation, and concluded that LCFA may be an alternative energy source to carbohydrates in this unique situation [7]. In children with VLCAD deficiency, part of the recommended treatment during acute illness with high fever, low intake or recurrent vomiting or diarrhea, is intravenous high dose glucose infusion [11]. It is possible that in these patients their basic disorder prevents them from increasing LCFA oxidation, while insulin resistance induced by ECMO prevents efficient utilization of glucose.
To conclude, we describe a case of an infant with VLCAD deficiency presenting with cardiac deterioration, who was treated with ECMO, unfortunately without success. Considering physiological models showing that ECMO induces metabolic changes in the myocardium, we suggest that ECMO treatment may be even more complicated in these children. Further physiological research and clinical evidence are needed to determine whether treatment by ECMO earlier in the course of cardiac and general deterioration is effective, and whether specific supportive measures can improve outcomes of such treatment in this vulnerable patient population.
References
- 1.Agus M.S., Javid P.J., Piper H.G. The effect of insulin infusion upon protein metabolism in neonates on extracorporeal life support. Ann. Surg. 2006;244(4):536. doi: 10.1097/01.sla.0000237758.93186.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andresen B.S., Olpin S., Poorthuis B.J. Clear correlation of genotype with disease phenotype in very–long-chain Acyl-CoA dehydrogenase deficiency. Am. J. Hum. Genet. 1999;64(2):479–494. doi: 10.1086/302261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dereddy N.R., Kronn D., Krishnan U. Defects in long chain fatty acid oxidation presenting as severe cardiomyopathy and cardiogenic shock in infancy. Cardiol. Young. 2009;19(05):540–542. doi: 10.1017/S104795110999134X. [DOI] [PubMed] [Google Scholar]
- 4.Exil V.J., Summar M., Boles M.A. Metabolic basis of pediatric heart disease. Prog. Pediatr. Cardiol. 2005;20(2):143–159. [Google Scholar]
- 5.Haines N.M., Rycus P.T., Zwischenberger J.B., Bartlett R.H., Ündar A. Extracorporeal Life Support Registry Report 2008: neonatal and pediatric cardiac cases. ASAIO J. 2009;55(1):111–116. doi: 10.1097/MAT.0b013e318190b6f7. [DOI] [PubMed] [Google Scholar]
- 6.Houten S.M., Wanders R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010;33(5):469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kajimoto M., O'Kelly Priddy C.M., Ledee D.R. Extracorporeal membrane oxygenation promotes long chain fatty acid oxidation in the immature swine heart in vivo. J. Mol. Cell. Cardiol. 2013;62:144–152. doi: 10.1016/j.yjmcc.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar G., Mattke A.C., Bowling F., McWhinney A., Alphonso N., Karl T.R. Resuscitation of a neonate with medium chain acyl-coenzyme A dehydrogenase deficiency using extracorporeal life support. World Journal for Pediatric and Congenital Heart Surgery. 2014;5(1):118–120. doi: 10.1177/2150135113501900. [DOI] [PubMed] [Google Scholar]
- 9.Mathur A., Sims H.F., Gopalakrishnan D. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99(10):1337–1343. doi: 10.1161/01.cir.99.10.1337. [DOI] [PubMed] [Google Scholar]
- 10.Merrill E.D., Schoeneberg L., Sandesara P. Outcomes after prolonged extracorporeal membrane oxygenation support in children with cardiac disease—Extracorporeal Life Support Organization registry study. J. Thorac. Cardiovasc. Surg. 2014;148(2):582–588. doi: 10.1016/j.jtcvs.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 11.New England Consortium of Metabolic Programs, acute illness protocols, available on http://newenglandconsortium.org/for-professionals/acute-illness-protocols.
- 12.O'Kelly Priddy C.M., Kajimoto M., Ledee D.R. Myocardial oxidative metabolism and protein synthesis during mechanical circulatory support by extracorporeal membrane oxygenation. Am. J. Phys. Heart Circ. Phys. 2013;304(3):H406–H414. doi: 10.1152/ajpheart.00672.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paden M.L., Conrad S.A., Rycus P.T., Thiagarajan R.R. Extracorporeal life support organization registry report 2012. ASAIO J. 2013;59(3):202–210. doi: 10.1097/MAT.0b013e3182904a52. [DOI] [PubMed] [Google Scholar]
- 14.Saudubray J.M., Martin D., De Lonlay P. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J. Inherit. Metab. Dis. 1999;22(4):487–502. doi: 10.1023/a:1005556207210. [DOI] [PubMed] [Google Scholar]
- 15.Sharef S.W., Al-Senaidi K., Joshi S.N. Successful treatment of cardiomyopathy due to very long-chain acyl-CoA dehydrogenase deficiency: first case report from Oman with literature review. Oman Medical Journal. 2013;28(5):354. doi: 10.5001/omj.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J. Inherit. Metab. Dis. 2010;33(5):527–532. doi: 10.1007/s10545-010-9090-x. [DOI] [PubMed] [Google Scholar]
- 17.Spiekerkoetter U., Tenenbaum T., Heusch A., Wendel U. Cardiomyopathy and pericardial effusion in infancy point to a fatty acid b-oxidation defect after exclusion of an underlying infection. Pediatr. Cardiol. 2003;24(3):295–297. doi: 10.1007/s00246-002-0277-2. [DOI] [PubMed] [Google Scholar]