

Abstract
AIM
To investigate the role of the complement 5a (C5a)/C5a receptor (C5aR) pathway in the pathogenesis of acute liver failure (ALF) in a mouse model.
METHODS
BALB/c mice were randomly assigned to different groups, and intraperitoneal injections of lipopolysaccharide (LPS)/D-galactosamine (D-GalN) (600 mg/kg and 10 μg/kg) were used to induce ALF. The Kaplan-Meier method was used for survival analysis. Serum alanine aminotransferase (ALT) levels, at different time points within a 1-wk period, were detected with a biochemistry analyzer. Pathological examination of liver tissue was performed 36 h after ALF induction. Serum complement 5 (C5), C5a, tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, high-mobility group protein B1 (HMGB1) and sphingosine-1-phosphate levels were detected by enzyme-linked immunosorbant assay. Hepatic morphological changes at 36 h after ALF induction were assessed by hematoxylin and eosin staining. Expression of C5aR, sphingosine kinase 1 (SphK1), p38-MAPK and p-p38-MAPK in liver tissue, peripheral blood mononuclear cells (PBMCs) and peritoneal exudative macrophages (PEMs) of mice or RAW 264.7 cells was analyzed by western blotting. C5aR mRNA levels were detected by quantitative real-time PCR.
RESULTS
Activation of C5 and up-regulation of C5aR were observed in liver tissue and PBMCs of mice with ALF. Blockade of C5aR with a C5aR antagonist (C5aRa C5aRa) significantly reduced the levels of serum ALT, inflammatory cytokines (TNF-α, IL-1β and IL-6) and HMGB1, as well as the liver tissue damage, but increased the survival rates (P < 0.01 for all). Blockade of C5aR decreased SphK1 expression in both liver tissue and PBMCs significantly at 0.5 h after ALF induction. C5aRa pretreatment significantly down-regulated the phosphorylation of p38-MAPK in liver tissues of ALF mice and C5a stimulated PEMs or RAW 264.7 cells. Moreover, inhibition of p38-MAPK activity with SB203580 reduced SphK1 protein production significantly in PEMs after C5a stimulation.
CONCLUSION
The C5a/C5aR pathway is essential for up-regulating SphK1 expression through p38 MAPK activation in ALF in mice, which provides a potential immunotherapeutic strategy for ALF in patients.
Keywords: Acute liver failure, C5a/C5aR, p38-MAPK, Sphingosine kinase 1
Core tip: Recent studies and our work show that SphK1 and complement activation play an important role in systemic inflammation in acute liver failure (ALF). It has been shown that C5a activates sphingosine kinase 1 (SphK1) in macrophages. However, the mechanism of C5a-induced SphK1 activation is unknown. In this study we found that excessive activation of C5 and up-regulation of C5aR in liver tissue, and the C5a/C5aR pathway is essential for potentiating SphK1 expression through p38 MAPK activation in ALF. To our knowledge, this is the first report of the mechanism of C5a-induced SphK1 activation, which provides a potential immunotherapeutic strategy for ALF in patients.
INTRODUCTION
Despite availability of efficient antiviral drugs and artificial liver support system, acute liver failure (ALF) remains a largely intractable clinical problem, with high mortality rates (about 80%)[1]. It often requires urgent liver transplantation due to the limited therapeutic options[1-3]. Growing evidence suggests that ALF can trigger systemic inflammation through release of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6[4,5].
Sphingosine kinase 1 (SphK1) is an intracellular signaling enzyme that generates the lipid mediator sphingosine-1-phosphate (S1P)[6]. Several pro-inflammatory stimuli, including complement 5a (C5a), activate SphK1 on macrophages, and blockade of SphK1 attenuates inflammatory responses[7-9]. Previous studies have shown that SphK1 plays a critical role in sepsis-induced inflammatory responses[10], and our recent work showed that expression and activation of SphK1 play an important role in ALF in a mouse model[11,12].
The C5a fragment is the most powerful pro-inflammatory anaphylatoxin generated during complement activation[13]. Increasing evidence suggests that excessive C5a can cause deleterious exaggeration of the innate immune responses during bacterial infections[14-16]. In animal models of sepsis, blockade of the C5a/C5aR pathway attenuates organ injury and increases survival rates in mice[16]. Recent studies indicate that complement activation plays an important role in lipopolysaccharide (LPS)/D-galactosamine (D-GalN)- and acetaminophen (APAP)-induced ALF in mice[17,18]. Moreover, C5a is over-produced during ALF, and inhibition of C5aR signaling alleviates liver injury in an animal model of ALF[17].
Based on the above results, we speculated that activation of the C5a/C5aR pathway plays an important role in SphK1 activation in ALF. Here, we report that SphK1 activation relies on C5a/C5aR interactions, which involve the mitogen-activated protein kinase (MAPK) signaling pathway. Blocking C5a/C5aR interactions effectively prevents LPS/D-GalN-induced ALF in mice, indicating that intervention of complement activation may be a useful immunotherapeutic strategy for ALF in patients.
MATERIALS AND METHODS
Animal model of ALF and treatment
Male BALB/c mice, weighing 20 ± 0.5 g, were obtained from the Experimental Animal Center of Nanchang University (Nanchang, China). Specific pathogen-free male mice around 6-wk-old were used for all experiments. Mice were handled and treated in accordance with the strict guiding principles of the National Institution of Health for experimental care and use of animals and approved by the animal care and use committee of Zhejiang Hospital. After ALF was induced, mice were sacrificed at the indicated time points as described previously[12]. Mice were randomly assigned to five groups (12 mice per group): PBS group, ALF group, C5aR antagonist (C5aRa) or cobra venom factor (CVF) pretreatment group, and C5aRa + CVF pretreatment group. All of the groups were observed for 1 wk.
Blockade of C5aR and complement depletion
For blockade of C5aR, mice were intraperitoneally injected with 1 mg/kg C5aRa (GL Biochem Ltd., Shanghai, China) 30 min before D-GalN/LPS challenge or treatment with PBS as a mock control. Complement was depleted by two intraperitoneal injections (7.5 U each in 200 μL of saline/mouse) of CVF (Quidel Corporation, San Diego, CA, United States). The first injection was given 24 h before D-GalN/LPS or saline administration, and the second injection was given 5 h after the first CVF injection. This was done to prevent any adverse effects of rapid loss of complement.
Quantification of alanine aminotransferase and detection of serum cytokines, high-mobility group protein B1, C5, C5a and S1P
Serum alanine aminotransferase (ALT) levels were measured using an Olympus AU5400 automatic biochemistry analyzer, and the levels of serum cytokines (TNF-α, IL-1β and IL-6), high-mobility group protein B1 (HMGB1) and S1P were measured by enzyme linked-immunosorbant assay (ELISA) as described previously[12]. Serum C5a and C5 levels were measured with commercial ELISA kits according to the manufacturer’s instructions.
Western blot analysis and histological study of liver tissue
Western blot analysis was performed as previously described[11,12]. Briefly, 40 μg of protein from total tissue or cell lysate were used, and the blots were probed using polyclonal anti-mouse SphK1, C5aR, p38-MAPK and phospho-p38-MAPK antibodies (Santa Cruz Biotechnology, Dallas, TX, United States), with β-actin detected with anti-β-actin antibody (Santa Cruz Biotechnology) as a loading control. For histological study, liver tissue was fixed in 40 g/L phosphate-buffered formalin, and 3-5 μm tissue sections were cut and stained with hematoxylin and eosin (HE) before microscopic evaluation at × 200 or × 400 magnification.
Quantitative real-time polymerase chain reaction
Liver tissues were obtained from mice at the indicated time points and total RNA was extracted using TRIZOL (Invitrogen, Carlsbad, CA, United States) and then reverse transcribed into cDNA using ReverTra Ace qPCR RT kit (Toyobo, Osaka, Japan). The cDNA was then amplified by PCR. Quantitative PCR with SYBR Green Realtime PCR Master Mix-Plus (Toyobo) was performed using a Prism 7000 (Applied Biosystems Inc, Foster City, CA, United States) sequence detection system, and mRNA levels were normalized to that of the housekeeping gene β-actin. Primer sequences for PCR amplification were as follows: C5aR forward, 5′-TGGACCCCATAGATAACAGCAG-3’ and C5aR reverse, 5′-GGAACACCACCGAGTAGATGAT-3′; β-actin forward, 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ and β-actin reverse, 5-AAAACGCAGCTCAGTAACAGTCCG-3′.
Peritoneal exudative macrophage isolation and cell culture
Peritoneal exudative macrophages (PEMs) were harvested from BALB/c mice. Cells were re-suspended in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) at 5 × 106 cells/mL in a 6-well plate and incubated for 4 h; 500 ng/mL of C5a (Biovision, Milpitas, CA, United States) and 10 nmol/L of C5aRa (60-min pre-incubation) were used. PEMs were harvested and lysed for western blot analysis of SphK1. RAW 264.7 cells were obtained from the Institute of Cell Biology of the Chinese Academy of Sciences (Shanghai, China) and were cultured in 6-well plates and propagated in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were stimulated with C5a (500 ng/mL) or C5a + C5aRa (pre-incubated for 60 min with 10 nmol/L of C5aRa in the presence of C5a). Inhibition of p38-MAPK activity was achieved with SB203580 (10 mmol/L).
Statistical analysis
Data are expressed as the mean ± standard error of the mean. Statistical significance was determined by a two-tailed Student’s t-test or one-way analysis of variance (ANOVA), and, specifically, a log-rank test for survival analysis. A P value < 0.05 was considered statistically significant. Statistical image analysis was performed after determining that the data fit a normal distribution. A two-tailed Student’s t-test was employed after the exclusion of outliers that were less or greater than two standard deviations away from the median. All statistical analyses were performed using SPSS 13.0 for Windows.
RESULTS
Excessive activation of C5 and up-regulation of C5aR in liver tissue of mice with ALF
To address the relevance of C5 activation in ALF in mice, we first challenged BALB/c mice with D-GalN/LPS and examined C5 activation over a time course. Upon D-GalN/LPS challenge, C5a levels in serum rapidly increased within 6 h and peaked at 12 h (Figure 1A). Although serum C5a levels began to decrease after 24 h of ALF induction (Figure 1B), the serum C5a level at 36 h was still higher than that of the unchallenged mice (Figure 1B). Compared to the controls, expression of C5aR mRNA (> 15-fold) and protein was elevated significantly in liver tissue of ALF mice (Figure 1C and D). These results suggest that excessive complement activation occurs during D-GalN/LPS-induced ALF in mice.
Figure 1.
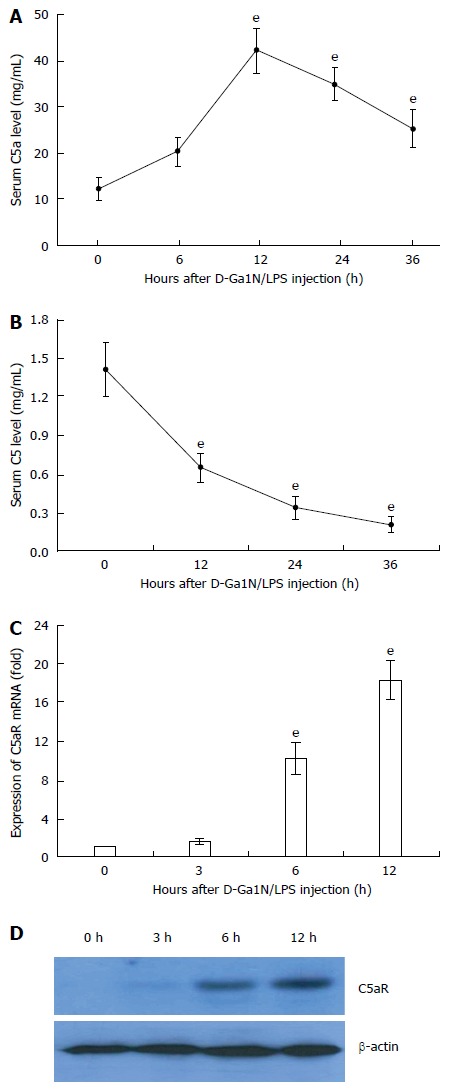
Excessive activation of C5 and up-regulation of C5aR in liver tissue of mice with acute liver failure. Acute liver failure was induced in BALB/c mice using D-GalN (600 mg/kg) and LPS (10 μg/kg). A: Serum levels of C5a at 12, 24 and 36 h increased significantly compared with that at 0 h (42.8 ng/mL ± 4.77 ng/mL, 35.22 ng/mL ± 3.62 ng/mL, 25.52 ng/mL ± 4.02 ng/mL vs 12.23 ng/mL ± 2.55 ng/mL, t = 19.31, 17.2, and 9.67, respectively, eP < 0.001); B: Serum levels of C5 at 12, 24 and 36 h increased significantly compared with that at 0 h (0.65 mg/mL ± 0.117 mg/mL, 0.343 mg/mL ± 0.09 mg/mL, 0.211 mg/mL ± 0.06 mg/mL vs 1.413 mg/mL ± 0.209 mg/mL, t = 11.03, 16.29, and 19.15, respectively, eP < 0.001); C and D: Expression of C5aR mRNA and protein in liver tissue. The mean ± SE of three independent experiments is shown (error bar indicates standard error).
Blockade of the C5a/C5aR pathway attenuates D-GalN/LPS-induced ALF in mice
To achieve a blockade of C5aR signaling during ALF, we chose to use a C5aRa. Blockade of C5aR apparently attenuated ALF, as demonstrated by a significant reduction in serum levels of ALT (Figure 2A), inflammatory cytokines (Figure 2B and C) and the liver tissue damage (Figure 2D), as well as an increase in survival rates (Figure 2E) in mice after D-GalN/LPS challenge. In addition, depleting complement by CVF pretreatment also resulted in a significant decrease in susceptibility to D-GalN/LPS challenge, as evidenced by a reduction in serum ALT levels (Figure 3A) along with an increase in survival rates (Figure 3B) compared with the control. Furthermore, treatment with C5aRa further lessened the pathogenic effect on D-GalN/LPS challenged mice receiving CVF pretreatment, as evidenced by lower levels of serum ALT (Figure 3C), inflammatory cytokines (TNF-α, IL-1β and IL-6) and HMGB1 (Figure 3D and E). These data clearly demonstrate that abrogating the C5a/C5aR pathway can alleviate the severity of ALF.
Figure 2.
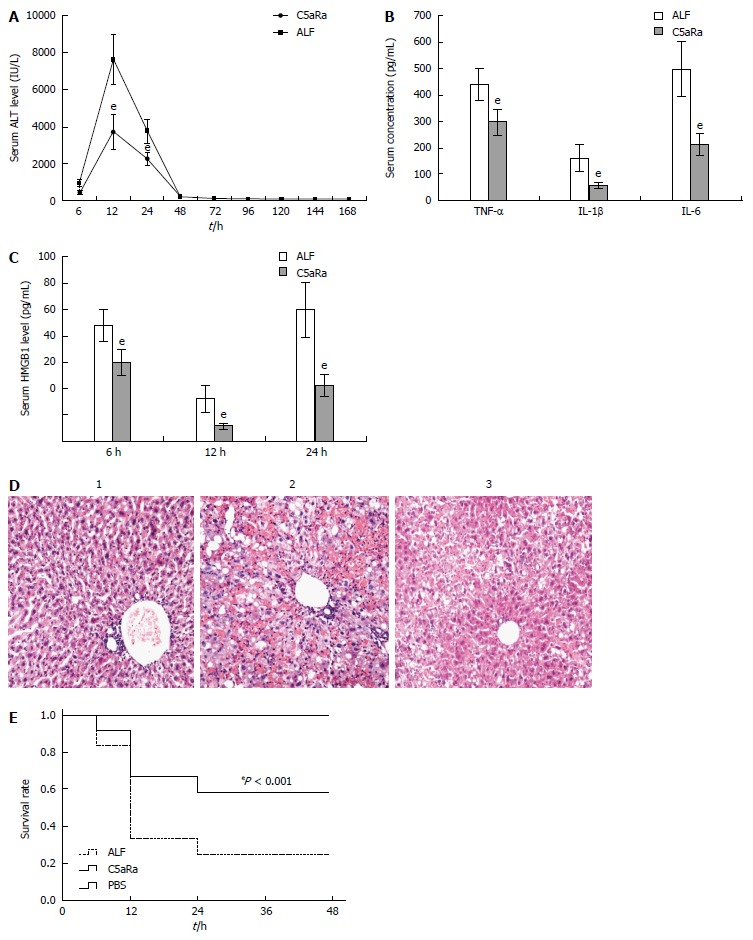
C5aRa attenuates D-GalN/LPS induced acute liver failure in mice. A: C5aRa decreased serum levels of ALT at 12 h and 24 h significantly (3736.12 IU/L ± 937.98 IU/L vs 7612.78 IU/L ± 1379.21 IU/L, 2225.07 IU/L ± 381.99 IU/L vs 3741.74 IU/L ± 637.53 IU/L, t = 8.05 and 7.07, respectively, eP < 0.001); B: C5aRa reduced serum levels of TNF-α, IL-1β and IL-6 at 12 h (299.35 pg/mL ± 50.61 pg/mL vs 439.33 pg/mL ± 63.59 pg/mL, 57.42 pg/mL ± 12.98 pg/mL vs 106.69 pg/mL ± 49.87 pg/mL, 213.52 pg/mL ± 42.69 pg/mL vs 500.87 pg/mL ± 104.14 pg/mL, t = 5.96, 6.94, and 8.84 respectively, eP < 0.001); C: C5aRa reduced HMGB1 levels at 6, 12 and 24 h (18.14 ng/mL ± 4.08 ng/mL vs 60.23 ng/mL ± 5.47 ng/mL; 16.21 ng/mL ± 5.11 ng/mL vs 67.14 ng/mL ± 14.27 ng/mL; 15.42 ng/mL ± 6.23 ng/mL vs 48.71 ng/mL ± 15.6 ng/mL, t = 9.13, 11.64, and 6.85, respectively, eP < 0.001); D: Immune cell infiltration and tissue damage were detected by HE staining at 36 h after onset of ALF (1 = normal mice; 2 = ALF mice; 3 = C5aRa treated mice; magnification, × 100); E: Kaplan-Meier analysis of the effect of C5aRa on survival rates of animals (eP < 0.001, log-rank test, F = 14.06). The mean ± SE of three independent experiments is shown (error bar indicates standard error). ALF: Acute liver failure. TNF-α: Tumor necrosis factor-α; IL: Interleukin.
Figure 3.
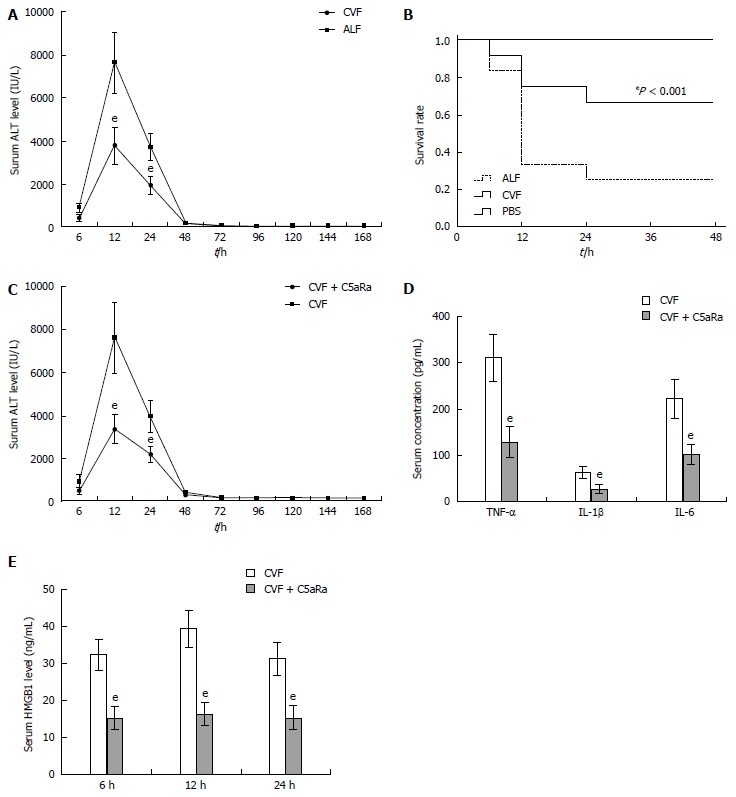
C5aRa further lessens the pathogenic effect on D-GalN/LPS challenged mice receiving cobra venom factor pretreatment. A: Cobra venom factor (CVF) treatment decreased serum levels of ALT at 12 h and 24 h significantly (3798.28 IU/L ± 839.68 IU/L vs 7612.78 IU/L ± 1379.21 IU/L, 1965.93 IU/L ± 371.74 IU/L vs 3798.28 IU/L ± 839.68 IU/L, t = 8.34 and 8.18, respectively, eP < 0.001); B: Kaplan-Meier analysis of the effect of CVF on survival rates of animals (eP < 0.001, log-rank test, F = 14.84); C: C5aRa further decreased serum levels of ALT at 12 h and 24 h significantly compared with CVF (1668.4 IU/L ± 339.68 IU/L vs 3798.28 IU/L ± 839.68 IU/L, 1069.69 IU/L ± 171.74 IU/L vs 1965.93 IU/L ± 371.74 IU/L, t = 8.14 and 7.58, respectively, eP < 0.001); D: C5aRa further reduced serum levels of TNF-α, IL-1β and IL-6 at 12 h significantly compared with CVF (129.67 pg/mL ± 32.79 pg/mL vs 312.19 pg/mL ± 51.25 pg/mL; 27.73 pg/mL ± 8.78 pg/mL vs 63.28 pg/mL ± 13.27 pg/mL; 103.66 pg/mL ± 22.33 pg/mL vs 223.67 pg/mL ± 41.77 pg/mL, t = 10.39, 7.74, and 8.78, respectively, eP < 0.001); E: C5aRa further reduced HMGB1 levels at 6, 12 and 24 h in ALF mice (15.14 ng/mL ± 3.08 ng/mL vs 33.23 ng/mL ± 4.17 ng/mL; 16.21 ng/mL ± 3.11 ng/mL vs 39.44 ng/mL ± 5.07 ng/mL; 15.42 ng/mL ± 3.23 ng/mL vs 31.33 ng/mL ± 4.36 ng/mL, t = 11.49, 13.53, and 10.16, respectively, eP < 0.001). ALF: Acute liver failure. TNF: Tumor necrosis factor; IL: Interleukin; ALT: Alanine aminotransferase; HMGB1: High-mobility group protein B1.
C5a/C5aR pathway is required for the expression of SphK1 during ALF
Given the critical role of SphK1 in D-GalN/LPS-induced ALF[11,12], we measured SphK1 expression in C5aRa pretreated mice. These mice exhibited a significant reduction in SphK1 expression in both liver tissue and peripheral blood mononuclear cells (PBMCs) at 0.5 h after ALF induction (Figure 4A and B). Blockade of SphK1 activity in vivo was confirmed by reduced S1P level (Figure 4C). Moreover, treatment with CVF appeared to be capable of further reducing SphK1 expression in ALF mice receiving C5aRa (Figure 4A and B).
Figure 4.

C5a/C5aR signaling is required for the expression of SphK1 during acute liver failure. A and B: Blocking of C5aR with C5aRa reduced SphK1 expression in liver tissue and peripheral blood mononuclear cells (PBMCs) of acute liver failure (ALF) mice; C: Blocking of C5aR by C5aRa reduced S1P level in liver tissue significantly than that of control (ALF group), eP < 0.01; D, E: Recombinant murine C5a (500 ng/mL) induced SphK1 expression in peritoneal exudative macrophages (PEMs) and RAW 264.7 cells, and was abolished by incubation for 60 min with 10 nmol/L C5aRa. The data shown are representative of three separate experiments.
Previous evidence has suggested that macrophages and PBMCs are the major source of SphK1 expression during D-GalN/LPS-induced ALF in mice[11,12] and C5aR is highly expressed in macrophages and the macrophages-derived cell line RAW 264.7[19]. To further explore whether C5a could directly induce SphK1 expression in macrophage, we stimulated murine PEMs with recombinant C5a. Compared to the controls, the C5a stimulated PEMs exhibited a significant increase in SphK1 expression, and this effect was abolished by C5aRa pretreatment (Figure 4C), indicating that C5a/C5aR interactions directly modulate SphK1 production in macrophages. Furthermore, C5a treatment led to an increase in SphK1 expression in RAW 264.7 cells and this effect was also abolished by C5aRa pretreatment (Figure 4D). These results indicate that the C5a/C5aR pathway associates with SphK1 expression and the pathogenesis of ALF in mice.
C5aR signaling induces SphK1 expression through activation of p38-MAPK
Recent studies show that C5a has a critical role in regulating macrophage functions and p38-MAPK is activated in macrophages during C5a stimulation[20]. Whether C5a induced SphK1 expression through p38-MAPK activation needs further investigation. To explore the relation of C5a/C5aR interactions with p38-MAPK phosphorylation in vivo, we first examined the tyrosine phosphorylation status of p38-MAPK after C5aRa pretreatment. As expected, the liver tissues presented with remarkably lower levels of p38-MAPK phosphorylation at 6 h and 12 h in C5aRa pretreated mice compared to controls (Figure 5A). In line with the in vivo result, a faster and persistent phosphorylation of p38-MAPK was clearly observed in PEMs stimulated with C5a, and C5aRa abolished this effect (Figure 5B). These results indicated that the C5a/C5aR pathway is involved in mediating C5a-induced p38-MAPK phosphorylation.
Figure 5.

C5aR signaling induces SphK1 expression through activation of p38-MAPK. A: C5a induced p38-MAPK activation in liver tissue of acute liver failure (ALF) mice; B: PEMs were stimulated with C5a (500 ng/mL) or C5a + C5aRa (pre-incubated 60 min with 10 nmol/L of C5aRa in the presence of C5a); C: Inhibition of p38-MAPK activity with SB203580 down-regulated SphK1 expression in PEMs stimulated with C5a.
To investigate whether p38-MAPK activation mediates C5aR-dependent SphK1 induction, we used SB203580 to directly inhibit p38-MAPK activity. Pre-incubation with SB20380 significantly reduced SphK1 production in PEMs upon C5a stimulation (Figure 5C). Under our experimental condition, SB203580 showed no cytotoxicity, as confirmed by > 95% of the cells with trypan blue exclusion after incubation for 6 h and 24 h (data not shown). These data indicate that the p38-MAPK signaling pathway mediates C5a/C5aR-induced SphK1 production in macrophages.
DISCUSSION
D-GalN/LPS-induced ALF in mice efficiently reproduces the clinical syndrome of ALF in humans. Although it has been noticed that complement activation plays an important role in LPS/D-GalN- and APAP-induced ALF[17,18], the possible role of complement activation in promoting ALF has not been investigated previously. Here, we found that ALF strongly activates the complement system, leading to a C5a increase. Attenuation in wild-type mice treated with C5aRa upon LPS/D-GalN-induction clearly validated the role of the C5a/C5aR pathway in the pathogenesis of ALF. In vivo and in vitro experiments also suggest that C5a/C5aR interactions up-regulate the expression and activation of SphK1 in macrophages through p38-MAPK activation.
Extensive expression and activation of SphK1 are a hallmark for LPS/D-GalN-induced ALF[11,12]. Our data show that upon LPS/D-GalN challenge, inhibition of C5aR signaling with C5aRa can substantially reduce SphK1 production, thus protecting the animals from ALF. These results demonstrate that the C5a/C5aR pathway, via induction of SphK1 expression, plays a key role in ALF. It also implies that the excessive activation of complement, particularly C5a levels, may serve as a clinical criterion for disease diagnosis and prediction of severity in patients with ALF.
It has been shown that bacterial infections and LPS can quickly activate the complement system[16,21]. In accordance with the previous results[17,18], the complement system is rapidly activated in ALF. This is confirmed by the observation of serum C5a level upsurge at the early stage of ALF development, indicating that C5a is an early mediator of ALF. This excessive complement activation and consumption at the early stage appear to explain the drop in serum C5 levels and C5 exhaustion in LPS/D-GalN-induced ALF. Although serum C5a levels in these mice began to decrease at 24 h after challenge, likely due to C5 exhaustion, the serum C5a level at 36 h was still higher than that of control, implying that C5a may also play a role in pathogenesis at the late stage of disease.
Excessive expression and activation of SphK1 are critical to the pathogenesis of ALF and inhibition of SphK1 with a specific inhibitor (N,N-dimethylsphingosine, DMS) attenuates mouse liver injury and increases the survival rate, supporting a critical role for SphK1 in ALF[17]. Our study clearly showed that if C5aR signaling was inhibited, LPS/D-GalN failed to induce massive expression of SphK1 in the affected liver, indicating that C5a/C5aR interactions participate in the expression and activation of SphK1 for causing the disease.
Several proinflammatory stimuli, including anaphylatoxin C5a and TNF-α, activate SphK1 on human macrophages, and blockade of SphK1 inhibits several pro-inflammatory responses triggered by these stimuli[7-9]. As a result, we speculated that C5a/C5aR interactions are essential for SphK1 expression and activation. It has been shown that C5a is able to activate the three major MAPK pathways in most of inflammatory cells, including macrophages[22,23], and a recent study indicated that the C5a/C5aR pathway is necessary for p38-MAPK phosphorylation in macrophages[20]. Furthermore, MAPK activation plays a key role in C5a-induced production of inflammatory cytokines[15,22]. In accordance with these results, we found that in vitro, C5a-induced SphK1 expression is largely dependent on activation of the p38-MAPK pathways. Our result identified that C5 activation and C5a/C5aR interactions play an important role in mediating p38-MAPK activation in liver tissue that leads to the pathogenesis of ALF.
Although the specific mechanism of complement activation during ALF is not very clear, the observation that C5a/C5aR signaling play an important role in the development of experimental ALF sheds light on the new strategies for treating ALF patients. Further research is needed to investigate the potential role of C5a in ALF in humans, and C5aR antagonist or C5a-neutralizing antibodies appear to have existing advantages for their usage in clinical treatment of ALF.
In conclusion, our results provide direct evidence that C5a/C5aR signaling plays a critical role in the pathogenesis of LPS/D-GalN-induced ALF in mice. Moreover, it is the first time we found that the C5a/C5aR pathway participates in the expression and activation of SphK1 through p38-MAPK activation. We have reasons to believe that interfering in the C5a/C5aR signaling pathway can become a promising immunotherapeutic strategy for ALF in patients.
COMMENTS
Background
Recent studies and our work show that sphingosine kinase 1 (SphK1) and complement activation play an important role in systemic inflammation in acute liver failure (ALF). It has been shown that complement 5a (C5a) activates SphK1 in macrophages. However, the mechanism of C5a-induced SphK1 activation is unknown.
Research frontiers
Systemic inflammation is an important feature of ALF, in which macrophages and inflammatory cytokines released by macrophages play a critical role. C5a is the most powerful pro-inflammatory mediator, and excessive C5a can cause exaggeration of the inflammatory responses while blockade of C5a/complement 5a receptor (C5aR) interactions increases survival rates in mice with sepsis. Moreover, complement activation also plays an important role in ALF.
Innovations and breakthroughs
In this study the authors found excessive activation of C5 and up-regulation of C5aR in liver tissue of ALF mice. The C5a/C5aR pathway is essential for potentiating SphK1 expression through p38-MAPK activation in ALF in mice. This is the first report of the mechanism of C5a-induced SphK1 activation.
Applications
Blockade of the C5a/C5aR pathway may represent a valuable immunotherapeutic strategy for ALF in patients.
Peer-review
Very interesting study. The authors investigated the role of the C5a/C5aR pathway in ALF in a mouse model. BALB/c mice were randomly assigned to different groups, intraperitoneal injection of D-galactosamine/lipopolysaccharide were used to induce ALF. Serum C5, C5a, tumor necrosis factor-α, interleukin (IL)-1β, IL-6, high-mobility group protein B1 and sphingosine-1-phosphate were detected by enzyme-linked immunosorbent assay. Hepatic morphological changes at 36 h were assessed, and the expression of C5aR, SphK1, p38-MAPK and p-p38-MAPK in liver tissue, peripheral blood monocytes and peritoneal exudative macrophages of mice or RAW 264.7 cells were analyzed. The authors found that the C5a/C5aR pathway is essential for potentiating SphK1 expression through p38-MAPK activation in ALF, providing a potential immunotherapeutic strategy for ALF in patents.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Zhejiang Hospital Institutional Review Board.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Zhejiang Hospital.
Conflict-of-interest statement: We declare that there are no conflicts of interest to disclose.
Data sharing statement: No additional data are available.
Peer-review started: August 3, 2016
First decision: August 19, 2016
Article in press: October 10, 2016
P- Reviewer: Sharma P, Yamada A S- Editor: Qi Y L- Editor: Filipodia E- Editor: Wang CH
References
- 1.Stravitz RT, Kramer DJ. Management of acute liver failure. Nat Rev Gastroenterol Hepatol. 2009;6:542–553. doi: 10.1038/nrgastro.2009.127. [DOI] [PubMed] [Google Scholar]
- 2.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atillasoy E, Berk PD. Fulminant hepatic failure: pathophysiology, treatment, and survival. Annu Rev Med. 1995;46:181–191. doi: 10.1146/annurev.med.46.1.181. [DOI] [PubMed] [Google Scholar]
- 4.Muto Y, Nouri-Aria KT, Meager A, Alexander GJ, Eddleston AL, Williams R. Enhanced tumour necrosis factor and interleukin-1 in fulminant hepatic failure. Lancet. 1988;2:72–74. doi: 10.1016/s0140-6736(88)90006-2. [DOI] [PubMed] [Google Scholar]
- 5.Wu Z, Han M, Chen T, Yan W, Ning Q. Acute liver failure: mechanisms of immune-mediated liver injury. Liver Int. 2010;30:782–794. doi: 10.1111/j.1478-3231.2010.02262.x. [DOI] [PubMed] [Google Scholar]
- 6.Melendez AJ. Sphingosine kinase signalling in immune cells: potential as novel therapeutic targets. Biochim Biophys Acta. 2008;1784:66–75. doi: 10.1016/j.bbapap.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Abdin AA. Targeting sphingosine kinase 1 (SphK1) and apoptosis by colon-specific delivery formula of resveratrol in treatment of experimental ulcerative colitis in rats. Eur J Pharmacol. 2013;718:145–153. doi: 10.1016/j.ejphar.2013.08.040. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W, Mottillo EP, Zhao J, Gartung A, VanHecke GC, Lee JF, Maddipati KR, Xu H, Ahn YH, Proia RL, et al. Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. J Biol Chem. 2014;289:32178–32185. doi: 10.1074/jbc.M114.601096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melendez AJ, Harnett MM, Pushparaj PN, Wong WS, Tay HK, McSharry CP, Harnett W. Inhibition of Fc epsilon RI-mediated mast cell responses by ES-62, a product of parasitic filarial nematodes. Nat Med. 2007;13:1375–1381. doi: 10.1038/nm1654. [DOI] [PubMed] [Google Scholar]
- 10.Lufrano M, Jacob A, Zhou M, Wang P. Sphingosine kinase1 mediates endotoxemiainduced hyperinflammation in aged animals. Mol Med Rep. 2013;8:645–649. doi: 10.3892/mmr.2013.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei YC, Yang LL, Li W, Luo P. Sphingosine kinase 1 dependent protein kinase C-δ activation plays an important role in acute liver failure in mice. World J Gastroenterol. 2015;21:13438–13446. doi: 10.3748/wjg.v21.i48.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lei YC, Yang LL, Li W, Luo P, Zheng PF. Inhibition of sphingosine kinase 1 ameliorates acute liver failure by reducing high-mobility group box 1 cytoplasmic translocation in liver cells. World J Gastroenterol. 2015;21:13055–13063. doi: 10.3748/wjg.v21.i46.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 14.Ward PA. The harmful role of c5a on innate immunity in sepsis. J Innate Immun. 2010;2:439–445. doi: 10.1159/000317194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, Zetoune FS, Gerard NP, Cianflone K, Köhl J, et al. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun S, Guo Y, Zhao G, Zhou X, Li J, Hu J, Yu H, Chen Y, Song H, Qiao F, et al. Complement and the alternative pathway play an important role in LPS/D-GalN-induced fulminant hepatic failure. PLoS One. 2011;6:e26838. doi: 10.1371/journal.pone.0026838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singhal R, Ganey PE, Roth RA. Complement activation in acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther. 2012;341:377–385. doi: 10.1124/jpet.111.189837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu GL, Chen J, Yang F, Li GQ, Zheng LX, Wu YZ. C5a/C5aR pathway is essential for the pathogenesis of murine viral fulminant hepatitis by way of potentiating Fgl2/fibroleukin expression. Hepatology. 2014;60:114–124. doi: 10.1002/hep.27114. [DOI] [PubMed] [Google Scholar]
- 20.Issuree PD, Maretzky T, McIlwain DR, Monette S, Qing X, Lang PA, Swendeman SL, Park-Min KH, Binder N, Kalliolias GD, et al. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J Clin Invest. 2013;123:928–932. doi: 10.1172/JCI66168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patel SN, Berghout J, Lovegrove FE, Ayi K, Conroy A, Serghides L, Min-oo G, Gowda DC, Sarma JV, Rittirsch D, et al. C5 deficiency and C5a or C5aR blockade protects against cerebral malaria. J Exp Med. 2008;205:1133–1143. doi: 10.1084/jem.20072248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaeffer V, Cuschieri J, Garcia I, Knoll M, Billgren J, Jelacic S, Bulger E, Maier R. The priming effect of C5a on monocytes is predominantly mediated by the p38 MAPK pathway. Shock. 2007;27:623–630. doi: 10.1097/SHK.0b013e31802fa0bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiou WF, Tsai HR, Yang LM, Tsai WJ. C5a differentially stimulates the ERK1/2 and p38 MAPK phosphorylation through independent signaling pathways to induced chemotactic migration in RAW264.7 macrophages. Int Immunopharmacol. 2004;4:1329–1341. doi: 10.1016/j.intimp.2004.05.017. [DOI] [PubMed] [Google Scholar]