

Abstract
Early-onset Alzheimer’s disease (EOAD) can be familial (FAD) or sporadic (sEOAD); both have a disease onset ≤ 65 years of age. 451 sEOAD samples were screened for known causative mutations in exon 16 and 17 of the Amyloid Precursor Protein gene (APP). Four samples were shown to be heterozygous for one of three known causative mutations: p.A713T, p.V717I and p.V717G, this highlights the importance of screening EOAD patients for causative mutations. Additionally, we document an intronic 6 base pair (bp) deletion located 83 bp downstream of exon 17 (rs367709245, IVS17 83-88delAAGTAT), which has a non-significantly increased minor allele frequency in our sEOAD cohort (0.006) compared to LOAD (0.002) and controls (0.002). To assess the effect of the 6 bp deletion on splicing, COS-7 and BE(2)-C cells were transfected with a minigene vector encompassing exon 17. There was no change in splicing of exon 17 from constructs containing either wild type or deletion inserts. Sequencing of cDNA generated from cerebellum and temporal cortex of a patient harbouring the deletion found no evidence of transcripts with exon 17 removed.
Keywords: Alzheimer’s disease, early-onset, sporadic, screening, APP, rs367709245
1 Introduction
Alzheimer’s disease (AD) was estimated to cause 500,000 deaths in the U.S. in 2010 (Alzheimer’s Association, 2014), and this number is steadily increasing with the aging population. Sporadic early-onset Alzheimer’s disease (sEOAD) patients have a disease onset ≤ 65 years of age and do not harbour a known causative mutation; the remaining sporadic cases are classified as late-onset Alzheimer’s disease (LOAD). Both sporadic forms have a complex aetiology, with LOAD estimated to be 70% heritable. Patients with a causative mutation are classified as Familial Alzheimer’s disease (FAD) or autosomal dominant Alzheimer’s disease (ADAD). Since the 1990’s many causative mutations have been identified, all located in one of three genes; Amyloid Precursor Protein (APP), Presenilin 1 (PSEN1) and Presenilin 2 (PSEN2) (Goate et al., 1991; Levy-lahad et al., 1995; Sherrington et al., 1995). Studies have found that mutations in PSEN1 and APP account for the majority of FAD cases, approximately 55% and 13% respectively (Janssen et al., 2003). The remainder are caused by mutations in PSEN2 and duplications in APP. The median age of onset for all these variants is between 45 and 55 years of age, with onset presenting as early as the mid 20’s or as late as the mid 70’s (Ryman et al., 2014). Given their similar clinical presentation it is highly likely that FAD patients have been misclassified as sEOAD (or vice versa) if a proband’s family history has not been documented, or genetic screening of the proband has not been conducted.
Screening sEOAD patients for causative mutations would help prevent introduction of FAD into sEOAD cohorts and thereby increase the power to detect true genetic loci associated with sEOAD. Causative mutations in APP, PSEN1 and PSEN2 are documented in the AD&FTD mutation database (Cruts et al., 2012). Unlike PSEN1 and PSEN2, causative APP mutations are clustered into just two exons (16 and 17), both of which are less than 500 base pairs (bp) and therefore amenable to screening via Sanger sequencing.
In this paper we report on screening of 451 sEOAD samples from the ARUK DNA Resource for causative mutations in exon 16 and 17 of APP using a sequencing reaction for each exon. Additionally there is a documented 6 bp deletion (rs367709245; IVS17 83-88delAAGTAT) located downstream of exon 17 and the design of this PCR product also permitted genotyping of this variant. As rs367709245 occurs near an intron/exon boundary we have also investigated its possible effect on splicing and generation of potential alternative isoforms of APP.
2 Materials and Methods
All methods were conducted according to the manufacturer’s instructions unless otherwise stated. sEOAD samples were screened for causative mutations by Sanger sequencing. In addition, the sequencing of exon 17 permitted genotyping of rs367709245 (APP 6 bp intronic deletion, 83 bp downstream of exon 17). Further genotyping of rs367709245 in LOAD and controls was achieved via a custom designed KASP genotyping assay.
2.1 Samples
The 451 sEOAD samples had an age of disease onset (AAO) ≤ 65 years of age (Table 1A), the 584 LOAD samples had an AAO > 65 years of age (Table 1B) and the 528 controls had an age at death (AAD) > 65 years of age (Table 1C). Where AAO was not documented, it was derived assuming 8 years disease duration from age at death (Brookmeyer et al., 2002), or age at sampling (AAS) was used with the understanding it would approximate to disease onset.
Table 1. Sample Demographics.
Sample demographics of each cohort; sporadic early-onset Alzheimer’s disease (sEOAD) (A), late-onset Alzheimer’s disease (LOAD) (B), and control (C). Each cohort contains samples from multiple centres with each centre represented one per row. The number of samples from each centre (N) is given along with the mean age of onset with standard deviation (Mean age at onset (±SD)), the number and percentage of female samples per centre (Females (%)), the number and percentage of samples harbouring at least one APOE ε4 allele (APOE ε4 + (%)), the number and percentage of samples with APOE ε4ε4 genotype (APOE ε4ε4 (%)), the minor allele frequency of the APOE ε4 allele (APOE ε4 MAF), and finally the number of samples classed as post mortem confirmed Alzheimer’s disease (Definite) or probable Alzheimer’s disease (Probable). Key: N, number of samples; SD, standard deviation.
| A. Sporadic early-onset Alzheimer’s disease (sEOAD)
| ||||||||
|---|---|---|---|---|---|---|---|---|
| N | Mean age at onset (±SD) | Females (%) | APOE ε4+ (%) | APOE ε 4ε4 (%) | APOE ε4 MAF | Definite | Probable | |
| Bristol | 24 | 53.5 (5.3) | 11 (45.8) | 12 (50.0) | 3 (12.5) | 0.31 | 24 | 0 |
| Manchester | 356 | 57.3 (5.4) | 171 (48.0) | 211 (59.3) | 49 (13.8) | 0.56 | 61 | 295 |
| Nottingham | 37 | 58.5 (6.1) | 18 (48.6) | 17 (45.9) | 2 (5.4) | 0.26 | 7 | 30 |
| Oxford | 34 | 55.5 (4.2) | 19 (55.9) | 20 (58.8) | 4 (11.8) | 0.35 | 24 | 10 |
| All | 451 | 57.0 (5.5) | 219 (48.6) | 260 (57.6) | 58 (12.9) | 0.51 | 116 | 335 |
| B. Late-onset Alzheimer’s disease (LOAD)
| |||||||
|---|---|---|---|---|---|---|---|
| N | Mean age at onset (±SD) | Females (%) | APOE ε4+ (%) | APOE ε 4ε4 (%) | APOE ε4 MAF | Definite | |
| Belfast | 295 | 78.1 (6.3) | 187 (63.4) | 162 (54.6) | 24 (8.1) | 0.31 | 295 |
| Bonn | 115 | 75.6 (6.4) | 84 (73.0) | 69 (60.0) | 19 (16.5) | 0.38 | 115 |
| Leeds | 103 | 79.4 (6.1) | 55 (53.4) | 67 (65.0) | 0 (0.0) | 0.40 | 103 |
| Nottingham | 71 | 78.1 (6.0) | 48 (67.6) | 40 (56.3) | 3 (4.2) | 0.30 | 71 |
| All | 584 | 77.8 (6.4) | 374 (63.9) | 338 (57.8) | 61 (10.4) | 0.34 | 584 |
| C. Control
| ||||||
|---|---|---|---|---|---|---|
| N | Mean age at death (±SD) | Females (%) | APOE ε4+ (%) | APOE ε 4ε4 (%) | APOE ε4 MAF | |
| Belfast | 47 | 73.8 (5.3) | 33 (70.2) | 9 (19.1) | 0 (0.0) | 0.10 |
| Bonn | 134 | 74.9 (7.6) | 74 (55.2) | 41 (30.6) | 3 (2.2) | 0.16 |
| Leeds | 191 | 78.4 (5.7) | 98 (51.3) | 39 (20.4) | 4 (2.1) | 0.11 |
| Manchester | 3 | 83.7 (6.1) | 2 (66.7) | 0 (0.0) | 0 (0.0) | 0.00 |
| Nottingham | 111 | 77.4 (7.5) | 41 (36.9) | 22 (19.8) | 0 (0.0) | 0.10 |
| Oxford | 15 | 81.5 (6.3) | 11 (73.3) | 3 (20.0) | 0 (0.0) | 0.10 |
| Southampton | 27 | 77.1 (5.5) | 12 (44.4) | 6 (22.2) | 0 (0.0) | 0.11 |
| All | 528 | 76.9 (6.8) | 271 (51.3) | 120 (22.7) | 7 (1.3) | 0.12 |
All case samples were diagnosed as either definite (post mortem confirmed) or probable Alzheimer’s disease (AD) according to National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA), and the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) guidelines. All samples used in this study were received with informed consent and were approved by the local Ethics Committee.
DNA was extracted from blood or brain tissue using a standard phenol chloroform extraction method. DNA quality and quantity was assessed via gel electrophoresis and NanoDrop™ 3300 spectrometer respectively.
2.2 Sequencing of APP
APP exon 16 (485bp) was amplified using primers E16F 5′ CAG-GTT-TCC-CTT-ACC-CTT-TC 3′ and E16R 5′ GCG-CTC-AGC-CTA-GCC-TAT-TT 3′ (Eurogenomics). APP exon 17 (482 bp) was amplified using primers E17F 5′ CAA-CCA-GTT-GGG-CAG-AGA-AT 3′ and E17R 5′ CAC-GGT-AAG-TTG-CAA-TGA-ATG 3′ (Eurogenomics). Both amplicons were sequenced in the forward direction using primer E17F or E16F, those found to harbour a causative mutation were validated by sequencing in the reverse direction using reverse primer E16R or E17R.
Genomic DNA was amplified using 2 ng/μl gDNA, 1 pM forward primer, 1 pM reverse primer, 1x Buffer (BioLabs), 0.2 mM dNTPs (Thermo Scientific), 0.1 U/μl LongAmp Taq DNA polymerase (New England Biolabs). The reaction was subjected to the following conditions: initial denaturation step of 94°C for 2 min, followed by 35 cycles of 94°C for 30 sec, 58°C (exon 16) or 61°C (exon 17) for 15 sec, and 72°C for 45 sec, with a final extension step at 72°C for 7 min. PCR products were cleaned using ExoSAP-IT (Affymetrix) and Sanger sequenced using BigDye Terminator v3.1 (ThermoFisher Scientic). The reactions were cleaned using Performa DTR Gel filtration Cartridges (Edge Biosystems) and sequenced on the ABI 3130.
2.3 Exontrap Minigene Assay
2.3.1 TA Cloning
gDNA was amplified for exon 17 using the method described in section 2.2, but with primer E17SF 5′ CAA-ATA-GTC-GAC-CAA-CCA-GTT-GGG-CAG-AGA-AT 3′ (Eurogenomics) which has a SalI restriction site at position 7 to 12 of the primer, and E17SR 5′ GAG-CAG-TCT-AGA-CAC-GGT-AAG-TTG-CAA-TGA-ATG 3′ (Eurogenomics) which has a Xbal restriction site at position 7 to 12 of the primer. The product was 494 bp in length and contained exon 17 (147 bp) along with 167 bp upstream and 192 bp downstream intronic sequence.
Amplicon DNA ligated with the pCR 2.1-TOPO vector (Invitrogen) was transformed into One Shot TOP10 Chemically Competent E. coli cells (Invitrogen). Colonies harbouring insert were selected and DNA extracted from liquid bacterial cultures using the QIAprep Spin Miniprep Kit (QIAGEN) and sequenced to confirm the presence/absence of the deletion. The APP insert for each allele was then subcloned into the Exon-trap vector pET01 (MoBiTech) using SalI and XbaI digestion followed by ligation with T4 Ligase.
2.3.2 pET01 Cloning and COS-7 and BE(2)-C Cell Culture
Plasmids were sequenced to confirm successful cloning using primer pET01S 5′ GAT-CGA-TCC-GCT-TCC-TG 3′ or pET01AS 5′ GTC-ATA-GCT-GTT-TCC-TG 3′ (Eurofin Genomics). The NucleoBond® Xtra Midi EF/Maxi EF kit (Macherey-Nagel) was used to extract pET01 plasmids free of endotoxins and suitable for transfection.
COS-7 and BE(2)-C cells were obtained from the European Collection of Cell Cultures (ECACC). COS-7 cells were cultured in Dulbecco’s modified eagle medium (Sigma) with 10% foetal bovine serum (Gibco), 2mM L-Glutamine, 100 U/ml penicillin (Gibco), 100 μg/ml streptomycin (Gibco) and 2.5 μg/ml fungizone. BE(2)-C were cultured with 50% of eagle’s minimum essential medium and 50% Ham’s F12 supplement with additional components 1% of non-essential amino acids, 2 mM L-Glutamine, 1 mM Sodium Pyruvate, 10% foetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 100 U/ml fungizone. Cells were incubated at 37°C in a humidified atmosphere containing 5% carbon dioxide and split when they reached 90% confluence. Cells were detached by trypsinisation at 37°C for 2–5 minutes and once detached complete medium was added. Cells were plated for transfection using 3.5 × 105 COS-7 cells or 6 × 105 BE(2)-C cells per 6cm dish.
2.3.3 Transfection and cDNA Sequencing
COS-7 and BE(2)-C cells were transfected following overnight incubation at 37ΰC using 1 ug of DNA, 9 μl of transfast (Promega), and 2ml of serum free medium. The mixture was added to the cells and incubated for 1 hour. Finally, 4 ml of complete medium was added and the cells were incubated for a further 24 hours before cells were harvested by trypsinisation. The experiment was repeated a minimum of three times for each cell line.
Total RNA was extracted using an RNeasy mini kit (Qiagen). RNA was treated with DNase (Ambion® TURBO DNA-free Kit) and cDNA was synthesised in duplicate using the AffinityScript Multi Temperature cDNA Synthesis Kit (Agilent) with either 0.3 μg random primer (Agilent) or 0.5 μg Oligo(dT) primers (Agilent), along with 2 μg of RNA. Reactions containing no enzyme were included as a negative control.
cDNA was amplified in a final volume of 30 μl using 500ng of cDNA, 0.5 pM primer pET01S (Eurofins Genomics), 0.5 pM primer pET01AS (Eurofins Genomics), 1x Buffer (Roche), 1.5 mM MgCl2 (Roche), 0.2 mM dNTPs (Thermo Scientific), 5 U/μl High Fidelity Taq DNA polymerase (Roche), and molecular grade water to the required volume. The product was predicted to be 389 bp in length if exon 17 was retained or 242 bp if exon 17 was removed. The reaction was subjected to the following conditions: initial extension at 94°C for 2 min followed by 35 cycles of 94°C for 30 sec, 58°C for 30 sec and 72°C for 1 min, with a final extension step of 72°C for 7 min. PCR product (5 μl) was cleaned and sequenced using primer pET01S/pET01AS.
2.4 Patient tissue cDNA analysis
Brain tissue from the cerebellum and temporal cortex from a patient who harboured the APP 6bp deletion was obtained from our collaborators (Oxford OPTIMA Study); tissue was homogenised using liquid nitrogen prior to RNA extraction and cDNA synthesis.
cDNA was amplified using 1ul cDNA, forward primer E16SF 5′ TGG-ATG-CAG-AAT-TCC-GAC-ATG 3′ (Eurofins Genomics) and reverse primer E18SR 5′ TTC-TGC-TGC-ATC-TTG-GAC-AG 3′ (Eurofins Genomics). The product was predicted to be 253 bp in length if exon 17 was retained or 106 bp if exon 17 was removed and this was verified by sequencing. Reactions containing no RNA or enzyme (RT minus) were included as negative controls.
2.5 Genotyping assay
Reactions were conducted in a final volume of 8ul using 1x MasterMix (KASP), 1x Assay (KASP), 2.5 ng/μl gDNA and molecular grade water to the required volume. Reactions were thermally cycled with an initial step of 94°C for 15 min, followed by 10 cycles of 94°C for 20 sec, 61°C (decreasing 0.6°C every cycle) for 60 sec, 32 cycles of 94°C for 20 sec, 55°C for 60 sec, and a final step of 30°C for 60 sec. A further 6 cycles of 94°C for 20 sec and 55°C for 60 sec was conducted to generate tighter clusters where appropriate. All samples positive for the deletion were confirmed by Sanger sequencing.
2.6 Statistical analyses
Power calculations were completed in Quanto using a ‘gene-only’ hypothesis and ‘log-additive’ model.
3 Results and Discussion
3.1 Screening exon 16 and exon 17 of APP in sEOAD
From our cohort of 451 sEOAD samples, all samples were successfully Sanger sequenced with good coverage of all causative mutations in exon 16 and 17. None of the samples carried a causative mutation in exon 16; however we did identify four samples (9%) (Table 2) heterozygous for one of three known causative mutations in exon 17; p.A713T (two samples), p.V717I (one sample) and p.V717G (one sample), all were verified by sequencing in both directions as shown in Figure 1. In addition to the above; unpublished data has successfully genotyped 408 of the 451 sEOAD samples using the NeuroChip (NeuroX) version 1 (Nalls et al., 2015). Included on the NeuroX are 82 of the 222 causative mutations from the AD&FTD mutation database (www.molgen.vib-ua.be/ADMutations/) (Cruts et al., 2012), this includes 24 in APP, 185 in PSEN1 and 13 in PSEN2. In our data, 80 of these mutations passed quality control; 6 in APP, 68 in PSEN1 and 6 in PSEN2. The NeuroX data confirmed 2/3 APP causative mutations found during Sanger sequencing, the third was not present on the NeuroX. All 408 samples were wild type for the 68 PSEN1 and 6 PSEN2 causative mutations genotyped on the NeuroX.
Table 2.
Information of 13 individuals harbouring a causative mutation or the variant rs367709245
Sample ID is given (Sample) along with the sample origin (Origin), gender (Gender), diagnosis (Diagnosis), age at onset of disease (AAO), APOE ε status (APOE ε), the cohort the sample is from (Cohort), and finally the variant present (Variant) given as the protein change or reference SNP ID. Key: AAO, age at onset; sEOAD, sporadic early onset Alzheimer’s disease; LOAD, late onset Alzheimer’s disease.
| Sample | Origin | Gender | Diagnosis | AAO | APOE ε | Cohort | Variant |
|---|---|---|---|---|---|---|---|
| M562 | Manchester, UK | Female | Definite AD | 62 | 33 | sEOAD | p.A713T |
| MRC045 | Nottingham, UK | Female | Probable AD | 61 | 34 | sEOAD | p.A713T |
| O0691 | Oxford, UK | Male | Probable AD | 50 | 44 | sEOAD | p.V717I |
| M758 | Manchester, UK | Female | Probable AD | 61 | 33 | sEOAD | p.V717G |
| O0483 | Oxford, UK | Male | Definite AD | 51 | 34 | sEOAD | rs367709245 |
| M111 | Manchester, UK | Female | Probable AD | 54 | 33 | sEOAD | rs367709245 |
| M200 | Manchester, UK | Female | Probable AD | 63 | 33 | sEOAD | rs367709245 |
| M400 | Manchester, UK | Female | Probable AD | 50 | 34 | sEOAD | rs367709245 |
| M461 | Manchester, UK | Female | Probable AD | 64 | 44 | sEOAD | rs367709245 |
| LE328 | Leeds, UK | Male | Definite AD | 70 | 34 | LOAD | rs367709245 |
| LE431 | Leeds, UK | Female | Definite AD | 76 | 34 | LOAD | rs367709245 |
| BN1842 | Bonn, Germany | Female | Non-diseased | 68 | 33 | Control | rs367709245 |
| BN0424 | Bonn, Germany | Female | Non-diseased | 84 | 33 | Control | rs367709245 |
Figure 1. Sequence chromatograms from individuals harbouring known causative APP mutations.
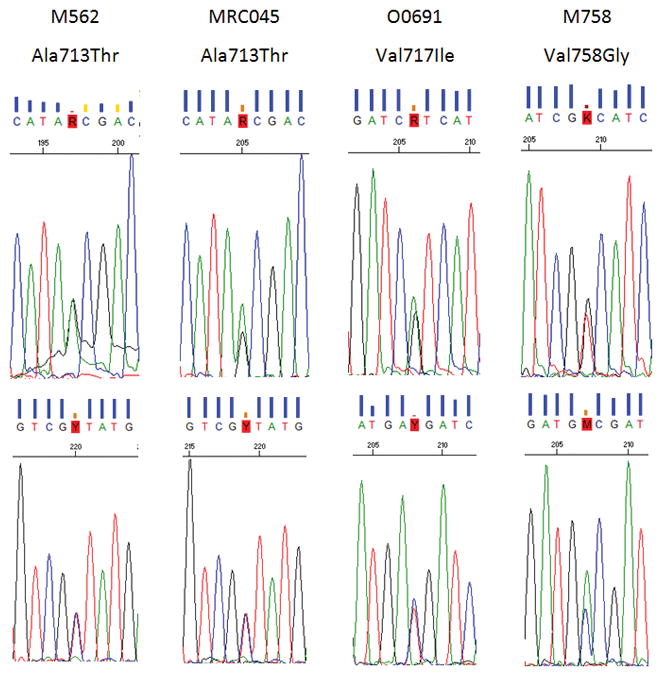
Each sample is presented one per column, with sample ID and mutation ID stated at the top and sequence chromatographs underneath. The chromatographs were generated using Sequence Scanner version 2 (Applied Biosystems) and show the heterozygous causative mutation at the centre with 8 surrounding bases. The top image in each column shows the sequence in the forward direction using primer E17F and the bottom image sequence in the reverse direction using primer E17R.
Sassi et al (Sassi et al., 2014) previously screened 47 EOAD samples and found one sample (2%) heterozygous for a causative APP mutation and four samples (8.5%) heterozygous for one of four PSEN1 mutations, two were causative and the remaining two were novel variants which are probably pathogenic. Nicolas et al (Nicolas et al., 2015) screened 264 EOAD samples and found three samples (1%) heterozygous for one of three PSEN1 variants that are possibly or probably pathogenic; they also found one sample (0.4%) heterozygous for a causative PSEN2 mutation. None of the patients in this study harboured APP mutations. In comparison to these studies we have identified a higher frequency of samples harbouring a causative APP mutation (9% compared with 2% and 0%), a lower frequency of PSEN1 mutations (0% compared to 8.5% and 1%) with a similar frequency for PSEN2 mutations. Our screening for PSEN1/PSEN2 mutations was not exhaustive, as the NeuroX genotyped under half of the causative PSEN1/PSEN2 mutations. Given this data and the fact causative PSEN1 mutations account for the majority of EOAD our cohort are likely to harbour causative PSEN1 mutations and possibly a causative PSEN2 mutation.
74% of our cohort (335 samples) were clinically diagnosed as probable AD, thus it is possible that a proportion of these samples have mixed dementia or a different type of dementia, further screening of our sEOAD samples for mutations that cause or are associated with other dementias would shed some light on this, and would also investigate genetic crossover between different types of neurodegeneration.
This data demonstrates the need to screen sEOAD cohorts for causative mutations as there are likely to be a low frequency of such changes present due to their similar clinical presentation. There is currently no rapid method to screen for all causative mutations simultaneously, typically this is done via Sanger or targeted next generation sequencing. The NeuroX (Nalls et al., 2015) is a customised Illumina HumanExome genotyping chip and the first version genotypes just under half of the currently known APP/PSEN1/PSEN2 causative mutations (Cruts et al., 2012). In the light of emerging data demonstrating the role of rare variants a successful genotyping array needs to contain all documented mutations. Updated versions of NeuroX will need to be more exhaustive before they can be used as a screening tool in both the research and clinical setting.
3.2 Genotyping rs367709245 in LOAD and controls
Screening of exon 16 and exon 17 of APP in 451 sEOAD identified five samples (Table 2) heterozygous for a 6 base pair (bp) intronic deletion (rs367709245, IVS17 83-88delAAGTAT) located downstream of exon 17 with a minor allele frequency (MAF) of 0.006. To assess the function of rs637709245 in other cohorts we genotyped 584 LOAD and 528 control samples, and identified four samples heterozygous for rs367709245, 2 in LOAD and 2 in control, both with a MAF of 0.002. Interestingly, this variant is 83 bp downstream of exon 17 and the Aβ peptide is coded by exon 16 and 17 of APP; which are always retained during mRNA splicing. The deletion has only ever been seen in a heterozygous form and was first reported in 1992 (Kamino et al., 1992) in 3/98 LOAD families (MAF 0.02) where it co-segregated with AD in 2 of the LOAD families. It was also found in 1/207 age matched controls (MAF 0.002); however, it was not seen in 29 FAD families or 153 sporadic cases. It has since been identified by phase 3 of the 1000 genome project (1000 Genomes Project Consortium et al., 2012), where it was seen in 1/661 individuals of African ancestry (MAF 0.002) and in 2/346 individuals of Puerto Rican ancestry (MAF 0.006). rs367709245 has recently been seen in an exome sequencing project (n=11) conducted on individuals with schizophrenia who are of South African ancestry (MAF 0.05) (Drogemoller et al., 2014). It is clear that this is a rare variant (MAF < 0.01) and in order to have power to detect any association with disease or potentially age of onset, thousands of samples would need to be screened. Our study has 13% and 14% power for sEOAD and LOAD respectively, to achieve a power of 80%, a total of over 10,000 samples would be needed (assuming a MAF of 0.002 and an odds ratio of 2).
3.3 Effect of rs367709245 on APP transcripts in COS7 and Be(2)-C cell lines
It is interesting to note that Human Splicing Finder 2.4.1 (Desmet et al., 2009) predicts rs367709245 to almost completely overlap (5 out of 6 bp) a consensus binding site for the splicing protein SRp55 (SFRS6), and that this protein is thought to promote exclusion of exon 10 in the gene MAPT (Yu et al., 2004), although its exact binding site has not yet been determined. It is well documented that deletions and single nucleotide polymorphisms (SNPs) at exon 10’s acceptor or donor site in MAPT are sufficient to alter the splicing ratio of exon 10 and cause Frontotemporal Dementia with Parkinsonism linked to chromosome 17 (FTDP-17) (Liu and Gong, 2008), though none of these variants are located as far downstream as rs367709245 (83 bp). Given this we wanted to test if rs367709245 would affect splicing of exon 17 in APP. COS7 cells and Be(2)-C cells were transfected with minigene constructs with either rs367709245 or wild type insert. PCR of cDNA from these cells showed no alternative isoforms; a single amplicon was obtained of expected size in all instances (389 bp - Figure 2), and sequencing of all constructs confirmed exon 17 was retained. Additionally, PCR of cDNA from cerebellum and temporal cortex from a patient harbouring this deletion (rs367709245) showed no alternative isoforms; a single amplicon was obtained of expected size in both instances (253 bp – Figure 3) and sequencing of both cDNAs verified exon 17 was retained. Both approaches indicate a lack of alternative splicing in the presence of rs367709245; given its location from the exon intron boundary (83 bp), it could be too far away to influence splicing.
Figure 2. RT-PCR products from cells transfected with vector containing wild type or variant insert (rs367709245).

RT-PCR products from COS7 cells (A) and Be(2)-C cells (B) transfected with pET01 vector containing wild type or variant insert (rs367709245). Lane 1 contains the DNA ladder GeneRuler 1 kb Plus (Life Technologies) which has two reference bands; 1000 bp (top) and 500 bp (bottom). Lanes 2 contains the RT-PCR product from a colony harbouring vector with wild type insert, lane 3 being its respective RT-PCR negative control (no enzyme). Lanes 4 contains the RT-PCR product from a colony harbouring a variant insert (rs367709245) with lane 5 being its respective RT-PCR negative control (no enzyme). Lane 6 contains the RT-PCR negative control (no RNA).
Figure 3. RT-PCR products from individual O0483 who harbours rs367709245.

Reverse transcriptase polymerase chain reaction (RT-PCR) products generated using Oligo(dT) or random primers and RNA from individual (O0483) cerebellum (C) and Temporal cortex (TC). Lanes 1 and 12 contain GeneRuler 100 bp Plus DNA ladder (ThemoFisher Scientific) containing one reference band marked at 500bp. Lanes 2 and 3 contain the RT-PCR products generated using Oligo(dT) primers. Lanes 4 and 5 contain the RT-PCR product generated using random primers. Lanes 6 to 9 are the same as lanes 2 to 5 but with no reverse transcriptase enzyme (cDNA –ve). Lane 10 contains an RNA negative control (-ve) and lastly lane 11 contains a PCR positive control (+ve), which was cDNA generated from Lymphoblastoid cell lines from two individuals obtained from the NHGRI sample repository.
4 Conclusion
We have highlighted the need to screen sEOAD cohorts for FAD patients who harbour exon 16/17 APP causative mutations; this holds especially true for sEOAD cohorts over LOAD; due to similar clinical presentation there is a greater chance of misclassification. Screening will help increase power to detect true genetic loci associated with the sporadic form of the disease. In addition, we identified a 6 bp deletion (rs367709245; IVS17 83-88delAAGTAT) in our sEOAD cohort with a non-significant increased MAF when compared to LOAD and controls. Due to lack of power we cannot say if rs367709245 is associated with disease; a much larger study with samples in the thousands would be needed to establish this. Minigene assays provided no evidence to suggest rs367709245 affects exon 17 splicing; however, we cannot rule out that it may have alternative functional effects on APP e.g. via gene regulation.
Acknowledgments
The University of Nottingham lab is funded by Alzheimer’s Research UK and the Big Lottery Fund. IB’s PhD studentship is jointly funded by Alzheimer’s Research UK and the School of Life Sciences at The University of Nottingham. JB and RG’s Fellowships are funded by The Alzheimer’s Society.
References
- 1000 Genomes Project Consortium. Abecasis G, Auton A, Brooks L, DePristo M, Durbin R, Handsaker R, Kang R, Marth G, McVean G. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association. 2014 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2014;10:e47–92. doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Brookmeyer R, Corrada MM, Curriero FC, Kawas C. Survival following a diagnosis of Alzheimer disease. Arch Neurol. 2002;59:1764–1767. doi: 10.1001/archneur.59.11.1764. [DOI] [PubMed] [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33:1340–1344. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet F, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogemoller B, Niehause D, Chiliza B, Merwe L, Asmal L, Malhotra A, Emsley R, Warnich L. Pattern of variation influencing antipychotic treatment outcomes in South African first-episode schizophrenia patients. Pharmacogenomics. 2014 doi: 10.2217/pgs. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin M-C, Mullan M, Brown J, Crawford F, Fidani L, Guiffra L, Haynest A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rosser M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nat Lett. 1991 doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Janssen JC, Beck JA, Campbell TA, Dickinson A, Fox NC, Harvey RJ, Houlden H, Rossor MN, Collinge J. Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology. 2003;60:235–239. doi: 10.1212/01.wnl.0000042088.22694.e3. [DOI] [PubMed] [Google Scholar]
- Kamino K, Orr H, Payami H, Wijsman M, Olonson M, Pulst S, Anderson L, O’dahl S, Nemens E, White J. Linkage and Mutational Analysis of Familial Alzheimer Disease Kindreds for the APP Gene Region. Am J Hum Genet. 1992;51:998–1014. [PMC free article] [PubMed] [Google Scholar]
- Levy-lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu C, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate Gene for the Chromosome 1 Familial Alzheimer’s Disease Locus. Science (80-.) 1995 doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. 2008;3:8. doi: 10.1186/1750-1326-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Bras J, Hernandez DG, Keller MF, Majounie E, Renton AE, Saad M, Jansen I, Guerreiro R, Lubbe S, Plagnol V, Gibbs JR, Schulte C, Pankratz N, Sutherland M, Bertram L, Lill CM, DeStefano AL, Faroud T, Eriksson N, Tung JY, Edsall C, Nichols N, Brooks J, Arepalli S, Pliner H, Letson C, Heutink P, Martinez M, Gasser T, Traynor BJ, Wood N, Hardy J, Singleton AB. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging. 2015;36:1605e7–1605.e12. doi: 10.1016/j.neurobiolaging.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas G, Wallon D, Charbonnier C, Quenez O, Rousseau S, Richard A, Rovelet-Lecrux A, Coutant S, Le Guennec K, Bacq D, Garnier J, Olaso R, Boland A, Meyer V, Deleuze J, Munter H, Bourque G, Auld D, Montpetit A, Lathrop M, Guyant-Maréchal L, Martinaud O, Pariente J, Rollin-Sillaire A, Pasquier F, Le Ber I, Sarazin M, Croisile B, Boutoleau-Bretonnière C, Thomas-Antérion C, Paquet C, Sauvée M, Moreaud O, Gabelle A, Sellal F, Ceccaldi M, Chamard L, Blanc F, Frebourg T, Campion D, Hannequin D. Screening of dementia genes by whole-exome sequencing in early-onset Alzheimer’s disease: input and lessons. Eur J Hum Genet. 2015 doi: 10.1038/ejhg.2015.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, Goate A, Frommelt P, Ghetti B, Langbaum JBS, Lopera F, Martins R, Masters CL, Mayeux RP, McDade E, Moreno S, Reiman EM, Ringman JM, Salloway S, Schofield PR, Sperling R, Tariot PN, Xiong C, Morris JC, Bateman RJ. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–60. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi C, Guerreiro R, Gibbs R, Ding J, Lupton MK, Troakes C, Lunnon K, Al-Sarraj S, Brown KS, Medway C, Lord J, Turton J, Mann D, Snowden J, Neary D, Harris J, Bras J, Morgan K, Powell JF, Singleton A, Hardy J. Exome sequencing identifies 2 novel presenilin 1 mutations (p L166V and p S230R) in British early-onset Alzheimer’s disease. Neurobiol Aging. 2014;35:2422e13–2422.e16. doi: 10.1016/j.neurobiolaging.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev E, Liang Y, Rogaeva E, Levesque G, Ikeda M, Chi H, Li G, Holman K, Tsuda T, Mar LJF, Bruni A, Mentesi M, Sorbi S, Raino I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky R, Wasco W, Silva H, Haines J, Pericak-Vance M, Tanzi R, Roses A, Fraser P, Rommens J, St George-Hyslop P. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995 doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Yu Q, Guo J, Zhou J. A minimal length between tau exon 10 and 11 is required for correct splicing of exon 10. J Neurochem. 2004;90:164–172. doi: 10.1111/j.1471-4159.2004.02477.x. [DOI] [PubMed] [Google Scholar]