

Abstract
Previously, our study showed that prohibitin interacts with phospholipids, including phosphatidylinositide and cardiolipin. Under stress conditions, prohibitin interacts with cardiolipin as a retrograde response to activate mitochondrial proliferation. The lipid-binding switch mechanism of prohibitin with phosphatidylinositol-3,4,5-triphosphate (PIP3) and cardiolipin may suggest the role of prohibitin effects on energy metabolism and age-related diseases. The current study examined the region-specific expressions of prohibitin with respect to the retina and retinal pigment epithelium (RPE) in age-related macular degeneration (AMD).
A detailed understanding of prohibitin binding with lipids, nucleotides, and proteins shown in the current study may suggest how molecular interactions control apoptosis and how we can intervene against the apoptotic pathway in AMD. Our data imply that decreased prohibitin in the peripheral RPE is a significant step leading to mitochondrial dysfunction that may promote AMD progression.
Keywords: prohibitin, retinal pigment epithelium, oxidative stress, macular degeneration, mitochondria
1. INTRODUCTION
Oxidative stress from mitochondria and uncontrolled activation of the alternative complement pathways at the membrane surface of RPE cells are thought to be independently associated with age-related macular degeneration (AMD). Emerging evidence suggests that development and progression of AMD involves significant alteration in cell signaling proteins, especially apoptosis and immune signaling. However, the mechanism of apoptosis initiation and the downstream interactions of AMD pathophysiology, remain unknown.
Our proteomics and metabolomics studies demonstrated potential new biomarkers of apoptosis in the RPE [1-9], including erythropoietin, RPE65, and prohibitin [3,5,7]. Prohibitin was originally identified as an anti-proliferative protein that is highly expressed in cancer cells in human; however, it exists in bacteria and plants, suggesting an essential cellular survival role, but the molecular mechanisms underlying prohibitin’s effect on aging and lipid metabolism in the retina are not clearly understood [10-11]. Prohibitin may regulate a cell cycle [10,12] and a transcription [13,14]. Further, prohibitin was reported as an inflammatory switch [15,16], an immune cell receptor [17], a mitochondrial protector [18-20], and a phospholipid binding shuttle [7,21]. Prohibitin stabilizes the respiratory enzymes in mitochondria [22-24] but its functions in the retina are not known. Prohibitin is localized in cell surface (β-cells), mitochondria (epithelial cells), or the nucleus (cancer cells) in different cells [14,25].
Mitochondrial dysfunction leads to oxidative damage and altered energy levels which may accelerate aging and age-related diseases, including AMD. Early stage of AMD shows the extracellular lipoprotein fragments, called drusen, on the surface of Bruch’s membrane. Later, AMD progresses into two distinct advanced forms as nonneovascular (atrophic, dry) and neovascular (exudative, wet) phenotypes [34,35].
Knockdown experiments using siRNA demonstrated that prohibitin depletion initiates apoptosis through p53 and BCLxl pathways.
The current study used the lipid, nucleotide and protein binding assays to provide the underlying mechanisms involved in prohibitin signaling in the RPE.
2. METHODS
2.1. Treatment of Animals
We followed the NIH Guide and the Association for Research in Vision and Ophthalmology (ARVO) statement for in vivo experiments. The animal protocol was approved by the Institutional Animal Care and Use Committee.
Sprague-Dawley (SD) rats (male, 250-300 g) and mice (C57/BL6 genetic background) were purchased from Charles River Laboratories (Wilmington, MA). As a positive control of oxidative stress in the retina, diabetic condition was induced by intravenous injection with streptozotocin (65 mg/kg in 0.1M sodium citrate, pH 4.5, Sigma-Aldrich, St. Louis, MO). Negative control animals received injections of vehicle alone. Rats and mice (n=3 for each group, triplicate) were considered to be diabetic at blood sugar levels >350 mg/dL. Animals were euthanized 2 or 4 weeks following onset of diabetes by an overdose of anesthetic. Comparative analysis was conducted with an identical animal strain (SD). Control SD rats and mice were sacrificed at 12 weeks of age and aged SD rats and mice were sacrificed at 52 and 54 weeks of age. Retinas from rats and mice were removed and frozen in liquid nitrogen. Retinas from different subgroups were collected and prepared for biochemical analysis.
All experiments were repeated (n=2 or n=3 biological samples) with technical triplicate (n=3). Stat View software was used for statistical analysis. Statistical significance was analyzed using unpaired Student’s t test or variance (ANOVA) when appropriate.
2.2. Donor Eye Tissue
Human postmortem donor eye tissues were used following the tenets of the Declaration of Helsinki. Diabetic retinopathy (DR) human retinal tissues (n=3, aged and control) were obtained from the Georgia Eye Bank (Atlanta, GA,). Human age-related macular degeneration (AMD) retina (8 mm macular and peripheral punches), RPE (8 mm central and peripheral punches), and age-matching control eyes (n=2) were provided by the Lions Eye Bank (Moran Eye Center, University of Utah).
2.3. ARPE-19 and HRP Cells
For in vitro experiments, retinal pigment epithelial cells (ARPE-19) were purchased from ATCC (Manassas, VA) and retinal progenitor cells (HRP) were kindly donated (Dr. Harold J. Sheeldo at the University of North Texas Health Science Center). ARPE-19 and HRP cells were cultured in a 5% CO2 incubator at 37 °C in 56 cm2 dishes (Nalge Nunc International, Naperville, IL) using Dulbecco’s modified Eagle’s medium (DMEM) with fetal bovine serum (10%) and penicillin/streptomycin (1%). Confluent cells were retrypsinized (5-7 minutes at 37 °C) using a trypsin-EDTA buffer (0.1%, Sigma-Aldrich, St. Louis, MO), followed by centrifugation (125 × g, 7 min). Cells in fresh culture dishes were grown to confluence for 2–4 days and were treated for oxidative stress (eight to nine passage cells).
2.4. Prohibitin-Lipid Interaction
Subcellular fractionation of bovine retinal/RPE tissue and APRE19 cells, based on differential centrifugation in density gradient buffer to separate mitochondrial, nuclear, cytoplasmic and microsomal fractions. Prohibitin was purified by immunoprecipation. The purity of each fraction by Western blotting using subcellular specific markers, inlcuding RNA polymerase 2 large subunit (nucleus), cytochrome C (mitochondria) and transketolase (cytoplasmic).
The lipid strips were prepared using nitrocellulose membrane. Lipids (1-2 μL, 100 pmol to 10 nmol) were spotted on the membrane dissolved in ethanol. All lipids were commercially available (Sigma-Aldrich, St Louis MO). The protein-lipid complex is incubated overnight at 4 °C along using prohibin antibody. As a negative control, lipids without protein lysate were spotted.
2.5. Oxidative Stress and Melatonin Treatment
To induce oxidative stress in vitro, confluent HRP and ARPE-19 cells were treated using tert-butyl hydroperoxide (200 μM, Sigma-Aldrich, St. Louis, MO) in serum-free medium for 1 or 2 hours. Cells were rinsed (phosphate-buffered saline) and lysed using a mild detergent-based lysis buffer, followed by coimmunoprecipitation using an antiprohibitin antibody.
For melatonin treatment, ARPE-19 cells (70% confluence) were treated for 30 minutes using 100μM melatonin dissolved in dimethyl sulfoxide (DMSO) and incubated for 12 hours in untreated medium. Cells were probed with antibodies against prohibitin and prepared for immunocytochemistry using AlexaFluor488 conjugated secondary antibody. Mitochondria and the nucleus were fluorescently labeled with MitoTracker Orange CMTMRos (Molecular Probes, Carlsbad, CA) and DAPI (VECTASHIELD), respectively. Cell morphology was monitored at various time points (0, 0.5, 8, 12, and 24 hours). Prohibitin from whole cell lysates was analyzed using Western blotting and densitometry.
2.6. Prohibitin-DNA Binding Assay
Mitochondrial and nuclear fractionation was followed as previously reported using bovine retina [7]. Isolated bovine retina cells were washed in PBS buffer three times and homogenized in a prechilled Dounce homogenizer using protease inhibitors and centrifuged (800 × g, 15 minutes). The pellet was saved for the nuclear fraction and the supernatant was centrifuged (6,000 × g, 15 minutes) and the pellet from the supernatant was used for the mitochondrial fraction. Mitochondrial and nuclear fractions were applied to Zymo DNA isolation Kit (Quick-gDNA™ MiniPrep) to purify mitochondrial and nuclear DNA. DNA samples were loaded on 0.5% agarose gel. Formation of DNA-prohibitin complexes was followed as reported [26]. Ten μg of total mitochondrial proteins and nuclear proteins were incubated (room temperature, 40 minutes) using 5 μg of mitochondrial DNA or nuclear DNA. Protein-DNA mixtures were centrifuged (16,000 × g, 30 minutes) and supernatant/pellet were separated using SDS-PAGE (8 16%). Prohibitin and DNA were analyzed using Western blotting and SYBR® Safe DNA dye (Invitrogen).
2.7. Co-Immunoprecipitation (Co-IP)
HRP and ARPE-19 cells were rinsed (Modified Dulbecco’s PBS) and lysed using IP lysis buffer containing Tris (25 mM), NaCl (150 mM), EDTA (1 mM), NP-40 (1%), glycerol (5%), protease inhibitor cocktail at pH 7.4 by incubating on ice for 5 minutes with periodic sonication (3 × 5 min), followed by centrifugation (13,000 × g, 10 minutes). Proteins (1 mg/ml, 200-400 μL) were loaded for immunoprecipitation and nonspecific bindings were avoided using control agarose resin cross-linked by 4% bead agarose. Amino-linked protein-A beads were used to immorbilize antiprohibitin antibody with a coupling buffer (1 mM sodium phosphate, 150 mM NaCl, pH 7.2), followed by incubation (room temperature, 2 hours) with sodium cyanoborohydride (3 μL, 5 M). Columns were washed using a washing buffer (1 M NaCl), and protein lysate was incubated in the protein A-antibody column with gentle rocking overnight at 4 °C. The unbound proteins were spun down as flowthrough, and the columns were washed three times using washing buffer to remove nonspecific binding proteins. The interacting proteins were eluted by incubating with elution buffer for 5 minutes at RT. The eluted proteins were equilibrated with Laemmli sample buffer (5X, 5% β-mercaptoethanol). Eluted proteins were separated using SDS–PAGE and stained using Coomassie blue (Pierce, IL) or silver staining kit (Bio-Rad, Hercules, CA).
2.8. Mass Spectrometry Analysis
Protein bands were excised (1×1×1 mm). The Coomassie-stained or silver-stained gel pieces were incubated using a Coomassie destaining buffer (200 μL of 50% MeCN in 25 mM NH4HCO3, pH 8.0, room temperature, 20 minutes) or silver destaining buffer (50% of 30 mM potassium ferricyanide, 50% of 100 mM sodium thiosulfate, 5-10 minutes). The gel pieces were dehydrated (200 μL, MeCN) and vacuum-dried (Speed Vac, Savant, Holbrook, NY). Proteins were reduced (10 mM DTT, 100 mM NH4HCO3, 30 minutes, 56 °C) and were alkylated (55 mM iodoacetamide, 100 mM NH4HCO3, 20 minutes, room temperature in dark). Proteins were digested using trypsin (13 ng/μL sequencing-grade from Promega, 37 °C, overnight) in NH4HCO3 (10 mM) containing MeCN (10%). The peptides were enriched using a buffer (50 μL of 50% MeCN in NH4HCO3, 5% formic acid, 20 minutes, 37 °C). Dried peptides were dissolved in a mass spectrometry sample buffer (5–10 μL, 75% MeCN in NH4HCO3, 1% trifluoroacetic acid). Alpha-cyano-4-hydroxycinnamic acid (5 mg/mL, MW 189.04, Sigma-Aldrich, St. Louis, MO) was freshly dissolved in a matrix buffer (50% MeCN, 50% NH4HCO3, 1% triflouroacetic acid) and centrifuged (13,000 × g, 5 minutes). The peptide-matrix mixtures (0.5 μL) were spotted onto the MALDI Plate (Ground steel, Bruker Daltonics, Germany). Mass spectrometer and all spectra were calibrated using a known peptide, including trypsin (842.5099 Da, 2211.105 Da). The mass spectrum was recorded in 800–3000 Da range using Flex MALDI-TOF mass spectrometer (Bruker Daltonics, Germany, 70–75% laser intensity, 100–300 shots). Mass spectrometry data were analyzed using Flex analysis software (Bruker Daltonics, Germany). Peptides were analyzed using the Mascot software (Matrix Science) and NCBI/SwissProt database (zero mismatch cleavage, carbamidomethyl cysteine, methionine oxidation, 50-300 ppm mass tolerance of 50–300 ppm). Peptide identification was evaluated based on Mascot MOWSE score, number of matched peptides, and protein sequence coverage. MOWSE score is expressed as -10logP as a probability value to compute the composite probability P.
2.9. Small Interfering RNA
ARPE-19 cells (6 × 104 cells/well, 0.1 mL DMEM, 10% FBS, 1% penicillin/streptomycin) were transfected with siRNA against prohibitin (175 ng, 23 nM final concentration, AAAGCCAGCTTCCTCGCATCT, FlexiTube siRNA) and random sequence negative control siRNA in serum-free culture medium (100 μL) using HiPerFect reagent (Qiagen, Valencia, CA). Cells were harvested (36–72- hour intervals) and prohibitin was analyzed using Western blotting and immunocytochemistry.
2.10. Immunocytochemistry
Cells were incubated on sterile glass cover slips using DMEM/F12 medium with 10% FBS and 1% penicillin/streptomycin (Hyclone) in 5% CO2 incubator at 37 °C. Cells were washed with PBS and incubated with MitoTracker Orange CMTMRos (100 nM, Molecular Probes, Carlsbad, CA) in serum-free culture medium (30 minutes, 37 °C), followed by washing (PBS) and fixing (10% formaldehyde, 30 minutes, room temperature). Cells were treated using Triton X-100 (0.2%, Sigma-Aldrich, St. Louis, MO) in PBS (30 minutes) for permeabilization and blocked using complete medium (10% FBS, 0.05% Tween-20, 1 hour). Cells were incubated using anti-prohibitin antibody (1:1000, Genemed Synthesis Inc., San Antonio, TX, overnight, 4 °C) and washed with PBS, followed by incubation with Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (1:700; Molecular Probes, Carlsbad, CA, 1 hour, room temperature). Cells were mounted with VECTASHIELD medium with DAPI (4,6-diamidino-2-phenylindole, the nucleus) and visualized using a Zeiss AxioVert fluorescent microscope (200 M Apo Tome, 63× magnification). Data were analyzed using ImageJ software (NIH).
2.11. Prohibitin Analysis
Proteins were analyzed quantitatively using a BSA standard curve with a BCA protein assay kit (Pierce). Proteins were solubilized in Laemmli buffer (5X, 240 mM Tris, pH 6.8, 8% SDS, 40% glycerol, 0.4% bromophenol blue, 10% β- mercaptoethanol) and denatured at 90 °C for 10 minutes. Proteins were separated using a gradient polyacrylamide precast gel (8-16%, Bio-Rad, Hercules, CA), followed by semidry electrotransfer (15 V, 30 minutes) onto the methanol-activated PVDF membrane (Bio-Rad). Nonspecific proteins were blocked (5% nonfat dry milk, 2 hours, 4 °C) in Tween-20 (0.1%). Membranes were incubated using a primary antibody (rabbit polyclonal, 1:1000, Genemed Synthesis, San Antonio, TX, 4 °C, overnight) and an anti-rabbit HRP conjugated secondary antibody (1:10,000, Agrisera, Vännäs, Sweden, 2 hours, room temperature). Protein bands were visualized using West Pico Chemiluminescent reagent (Pierce, 1–2 minutes) and a LAS 4000 mini luminescent image analyzer (GE, Piscataway, NJ). Membranes were incubated again using an anti-β-actin antibody (mouse monoclonal, 1:5000, Sigma-Aldrich, St. Louis, MO) followed by a secondary antibody (1:7,000, anti-mouse, horse radish peroxidase conjugated, Santa Cruz Biotechnology, Santa Cruz, CA) as a loading/positive control. Proteins were analyzed quantitatively based on pixels (area and intensity) using Quantity One software (Bio-Rad) and GraphPad Prism software.
2.12. Multiple Sequence Alignment and 3D Modeling
A 3D model of the protein was built using the 3D structure 2DYB chain ‘A’ as a template. 2DYB chain A is the Protein Data Bank structure for p47 Phox. This template shares 17% identity with the query sequence using the ALIGN program. The conserved residues were M1, L27, R35, F39, F42, R43, P59, P64, D68, R72, R97, R105, L119, K219 and L228. The modeling was performed using EsyPred3D, an automated homology modeling program protocol used for predicting 3D structures of proteins and viewed using PyMol, an open-source molecular visualization system (DeLano Scientific LLC). Based on p47 PX domain structure, the conserved basic amino acids of prohibitin were considered to serve as a good interaction site with phospholipids. P47 has PX domain residues between residues 4–125. SNX6 has PX residues between residues 26–173, and the SGK3 PX domain is located at 12–124. Conserved amino acids of prohibitin aligned within the PX domain of p47 were detected.
3. RESULTS
3.1. Lipid Binding Sequences in Prohibitin
Controlled translocation of prohibitin to specific subcellular organelles under stress conditions is a critical event for apoptosis, proliferation, lipid metabolism, and aging. We speculate that prohibitin translocalization and trafficking might be influenced by specific lipid binding sequences and temporal lipid concentrations. We hypothesized that prohibitin may have a putative lipid binding domain that includes conserved basic amino acids. To test our hypothesis, we examined whether prohibitin has any homologous lipid binding domain or conserved basic amino acids using the PFAM database (http://www.sanger.ac.uk/software/pfam), the Ensembl human genome browser (http://www.ensembl.org/homo_sapiens), and support vector machine-based functional prediction (http://jing.cz3.nus.edu.sg/cgi-bin/svmprot.cgi). Lipid binding modules, including C1, C2, epsin amino-terminal homology (ENTH), phox homology (PX), pleckstrin homology (PH), fab1-Yop-Vac1-Eea1 (FVVE), four-point-one-Ezrin-Radixin-Moesin (FERM) domains, ANTH, BAR, PDZ, tubby, START, and Src homology 3 (SH3) domain, were investigated.
First, we compared the potential PX domain of prohibitin with the known p47phox structure (Figure 1). Multiple sequence alignment demonstrated that two of the most well-conserved basic amino acids in prohibitin are R41 and R72/74 that are homologous to R43 and R90 in the PX domain of p47phox (Figure 1). PIP3 interacts with PX domain containing proteins that include p40phox, p47phox, phospholipase D1 (PLD1), SNX6, and cytokine-independent serine/threonine kinase (CISK or SGK3). The conserved basic amino acids, including R41 and R72 in prohibitin, are homologous to the cationic residues in the putative PIP3 binding pocket in p47phox. In addition, R35, R43, R72 in prohibitin are conserved and homologous to K75, R90, and R135 of SGK3, which represent the phosphoinositide binding pocket. Other conserved basic residues are K11, K83, R97, R105, R143, R157, K207, and K208 in prohibitin are homologous to basic amino acids in SNX6 and SGK3 proteins, both known as phosphoinositide binding proteins. Next, based on sequence alignment, a 3D model of prohibitin was built using the Protein Databank structure of 2DYB chain A as template (p47Phox). 2DYB chain A of p47Phox showed the putative lipid binding pocket residues (Figure 2A). Alignment with amino acids in the prohibitin sequence suggests a potential phosphoinositide binding pocket (Figure 2B). The p47Phox template shares 17% identities with the query sequence using the ALIGN program.
Figure 1. Multiple Sequence Alignment of Prohibitin and p47.

(A) Sequence alignment of prohibitin and PX domain containing p47 Phox (A). The conserved basic amino acid residues (R 41 and R72) in prohibitin are homologous to the cationic residues in the putative PIP3 binding pocket (R43, R90) of p47phox. (B) The sequence alignment SH3 domain containing PI3K and prohibitin is also shown to compare the presence of PX and SH3 domains.
Figure 2. Predicted Lipid Binding Pocket of Prohibitin.
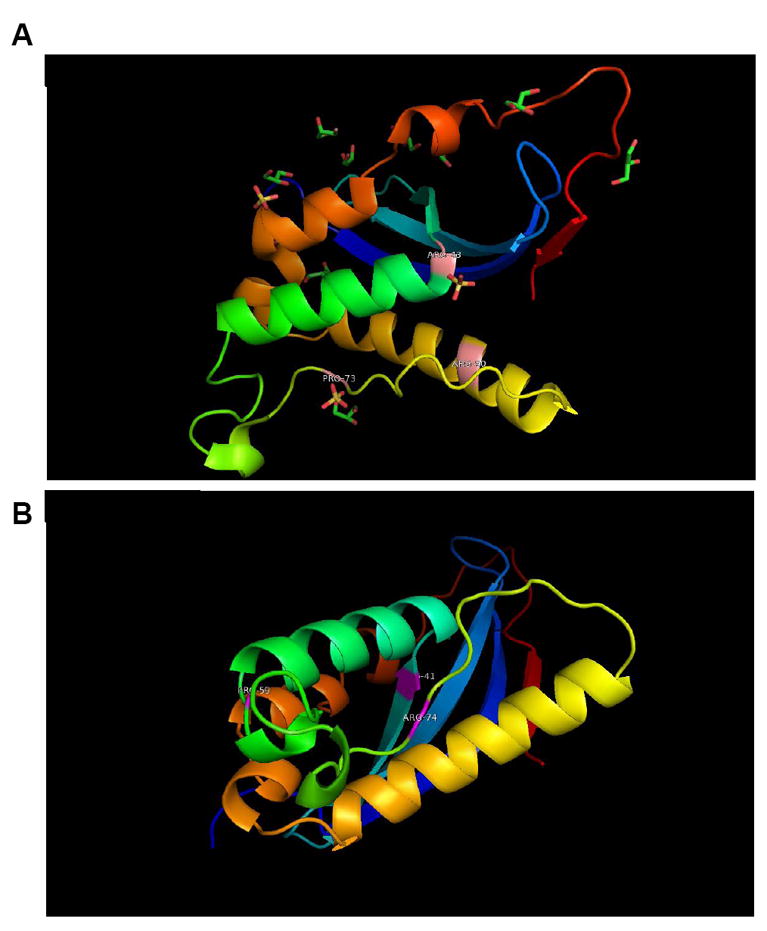
Based on sequence alignment, a 3D model of prohibitin was built using the 3D structure 2DYB chain ‘A’ as template. (A) 2DYB chain A is the Protein Databank structure for p47 Phox and it is based on the putative binding pocket residues that alignment with amino acids in the prohibitin sequence that can serve as a binding pocket in prohibitin-PIP3 binding (B). The p47 Phox template shares 17% identities with the query sequence using the ALIGN program.
Phosphate groups in PIP3 may interact with two arginine residues, R41/43 and R72/74 of prohibitin. A conserved F42 (F44 in p47phox) seems essential for the inositide ring interaction to make its U shaped PIP3 binding pocket. Two conserved proline residues, P64 and P98, suggest a β-turn structure, mimicking the second anion-binding site of p47phox (P73, P76). Other conserved basic residues include R143, R157, K207, and K208, implying a putative second anion-binding site (e.g. cardiolipin) at the C terminus.
3.2. Prohibitin-Lipid Interactions
To understand oxidative damage and mitochondrial dysfunction in aging by prohibitin, we determined the molecular mechanism through prohibitin-ligand interactions. Previously, prohibitin-lipid binding assays demonstrated that prohibitin binds with cardiolipin, a major mitochondrial phospholipid, under oxidative stress [7]. In the current study, we asked whether prohibitin in various organelles or cells may have different cardiolipin affinity to determine subcellular organelle-specific prohibitin-lipid interactions. The current lipid interaction assay demonstrates that microsomal and cytosolic prohibitin showed stronger interaction with cardiolipin compared to mitochondrial and nuclear prohibitin (Figure 3A). In addition, prohibitin from bovine RPE or ARPE19 cells did not show any interaction with cardiolipin, whereas prohibitin from bovine retina or HRPC showed cardiolipin binding. Microsomal prohibitin showed a strong interaction with cholesterol and a weaker interaction with phosphatidylserine (Figure 3A).
Figure 3. Lipid binding and lipid-dependent prohibitin regulation.
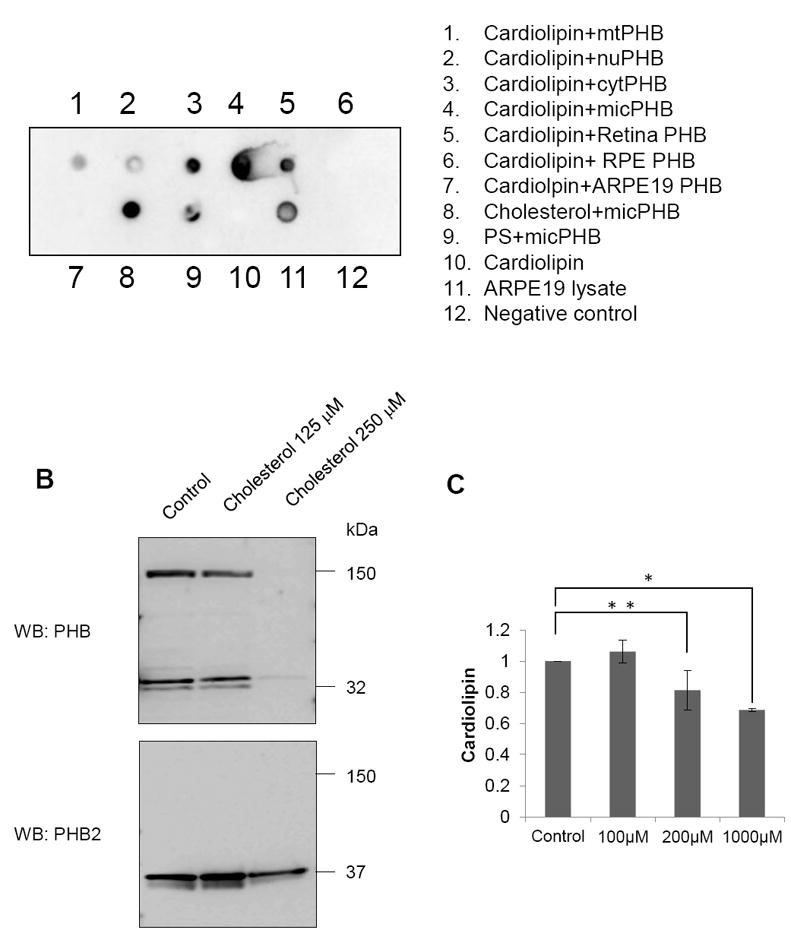
A. Western blotting using anti-prohibitin primary antibody demonstrates that prohibitin has different interactions with phospholipids, including cardiolipin and phosphatidylserine. Prohibitin in the retina has high affinity to cardiolipin, while prohibitin in the RPE shows weaker affinity. Microsomal prohibitin in the retina shows high affinity to phosphotidylserine, cardiolipin, and cholesterol. Microsomal fraction applied on the PVDF membrane is considered as a positive control.
B. Cholesterol-dependent prohibitin expression in the RPE. ARPE-19 cells were treated with cholesterol (16 hours). Increased cholesterol levels significantly downregulate PHB1 expression whereas PHB2 levels show less sensitivity toward the elevated cholesterol levels. C. Graphical representation shows that cardiolipin levels decreased in human RPE under elevated oxidative stress.
To test the hypothesis of the inverse correlation between prohibitin expression and cholesterol based on the cholesterol-binding promoter region, we added cholesterol (125-250 μM) to APRE19 cells during cell culture and observed downregulation of prohibitin and prohibitin2 expression (Figure 3B) under higher cholesterol concentration. To determine cardiolipin and cholesterol changes under oxidative stress, relative lipid concentrations were analyzed by MALDI-TOF mass spectrometry. Cardiolipin decreased 20% under oxidative stress (200 μM H2O2) in a dose and time-dependent manner (Figure 3C). Our data suggest that prohibitin from different subcellular organelle or different cells may have a different affinity toward phospholipids that include cardiolipin. Further, our experiments show that prohibitin expression is inversely correlated with cholesterol concentration in RPE cells.
3.3. Prohibitin-Nucleotide Interactions
Mitochondrial deficits contribute to cellular and tissue aging through the mechanisms of proton leakage, decreased membrane fluidity, decreased unsaturated lipids such as cardiolipin, and oxidative damage to mitochondrial DNA. Nuclear DNA (nuDNA) is more resistant than mitochondrial DNA (mtDNA) toward oxidative stress due to oxidative metabolism in mitochondria [18]. Previously, we demonstrated that leaking of cytochrome C and prohibitin may indicate mitochondrial membrane depolarization induced by oxidative stress. The current study examined prohibitin-nucleotide interaction to determine the protection mechanism of mitochondrial localization of prohibitin. Our experiments were designed to determine whether prohibitin binds to mitochondrial or nuclear DNA to answer a molecular chaperone function of prohibitin. We hypothesized that mitochondrial prohibitin may show different affinity toward mtDNA compared to nuDNA. Prohibitin from mitochondria and the nucleus was incubated with mtDNA and nuDNA, and separated by native gel assay to determine higher molecular weight complex. It was found that mitochondrial prohibitin forms protein-mtDNA complex at 720 kDa as shown in Figure 3A (lane 2 and 3), whereas it forms a 150 kDa complex with nuclear DNA. Nuclear prohibitin showed a weaker interaction with mitochondrial and nuclear DNA, forming mixtures of 720 and 150 kDa complexes (Figure 4A, lane 5–8). Higher concentration of mtDNA and nuDNA with nuclear prohibitin breaks molecular interactions shown as monomeric prohibitin at 32 kDa (Figure 4A, lane 6 and 8). Further, to confirm prohibitin-mtDNA interaction, we used the sedimentation assay using mitochondrial prohibitin with or without mtDNA. Prohibitin from the mitochondrial fraction formed a high-density complex with mtDNA as shown in Figure 3B. The prohibitin-DNA binding assay suggests a mtDNA binding chaperone function of prohibitin in mitochondria.
Figure 4. Prohibitin-DNA binding analysis.
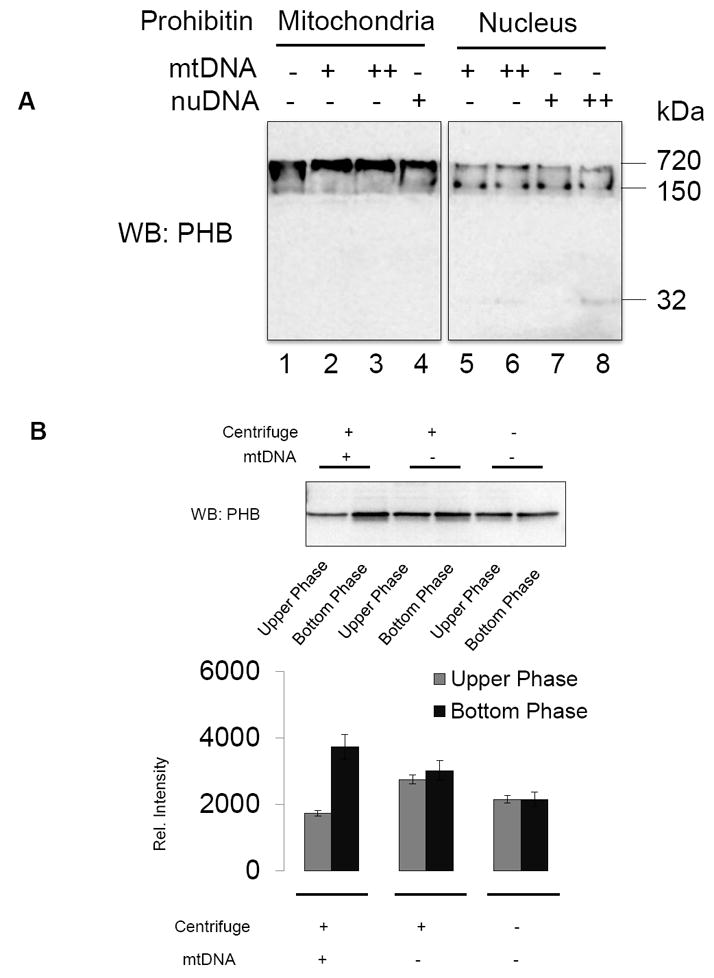
A. Mitochondrial and nuclear PHB fractions from bovine retina were mixed with gel-purified mitochondrial (mt) and nuclear (nu) DNA extracted from bovine retina. DNA-protein complexes were analyzed by native-PAGE and prohibitin was quantitated by Western blotting analysis.
B. Ten μg of total proteins in mitochondria (mt) were mixed with 5μg of mitochondrial DNA or TE buffer (negative control) followed by 40 minutes incubation at RT and centrifugation (16,000 × g, 30 minutes). Protein-DNA mixtures from upper half supernatant vs. bottom portion were subjected to SDS-PAGE (8–16%), and prohibitin expression was visualized by Western blotting analysis. Data showed mtPHB and mtDNA formed complexes and precipitated down to the bottom of the sample. Bands were quantified by QuantityOne software.
3.4. Prohibitin-Binding Proteins
To determine the downstream signaling of prohibitin in mitochondrial response under stress conditions, prohibitin-binding proteins were examined using immunoprecipitation (IP) analysis (Table 1, Supplement Figure 1A–B). ARPE-19 cells were treated with or without 200 μM, t-BuOOH (oxidative stress vs. control) and 49 putative prohibitin binding proteins in elution fractions were analyzed by MALDI-TOF mass spectrometry. IP with pre-immune serum and a blank gel with trypsin digestion were used as negative controls and prohibitin-p53 binding was considered as a positive control. Identified protein name, accession number, MOWSE probability score, sequence coverage, predicted mass, predicted pI, and identified peptide number are presented in Table 1. Several cytoskeletal proteins, including kinesin, actin, and myosin, were found in both control and oxidative stress as prohibitin binding proteins. Actin is involved in cell motility as well as nuclear and mitochondrial structural integrity. Actin has a nucleotide binding domain which is crucial in the proper function of ion channels. Nucleoporin transports macromolecules between the nucleus and cytoplasm. Nucleoporin and laminins may regulate the translocation of prohibitin between mitochondria and the nucleus. PI3K, the p85 binding domain, suggests that prohibitin is involved in the PIP3 pathway to regulate lipid and insulin signaling. Nuclear mitotic apparatus protein 1 isoform (NUMA) belongs to the same coiled-coil family as prohibitin, myosins, laminins and intermediate filaments. NUMA is proposed to play a role in maintaining and establishing nuclear structure, possibly as a structural component of the nuclear matrix.
TABLE 1.
Prohibitin-Binding Proteins. A1-A49 represents prohibitin binding proteins in ARPE19 cells and H1-H48 shows prohibitin binding proteins in human retinal progenitor cells.
| Band | Accession | Protein identified | MOWSE score | %coverage | Mass | PI | Peptide |
|---|---|---|---|---|---|---|---|
| A1 | 83404899 | Kinesin Family member 19 protein | 47325 | 8.2 | 93161 | 6.2 | 7 |
| A1 | 194388398 | Unknown, BAG65583, 110kDa myosin motor domain, ATPase, SH3 | 253758 | 9.0 | 110912 | 8.5 | 7 |
| A6 | 15277503 | ACTB protein partial | 1.59e+7 | 30.0 | 40221 | 5.6 | 9 |
| A13 | 26330300 | Unknown | 633431 | 10.0 | 88790 | 4.7 | 6 |
| A14 | 15277503 | ACTB protein partial | 1.59e+7 | 30.0 | 40221 | 5.6 | 9 |
| A16 | 292352 | Collagen, partial | 189998 | 11.0 | 72107 | 7.3 | 5 |
| A18 | 535012 | NADP+-dependent malic enzyme | 1.06e+6 | 14.9 | 67055 | 7.9 | 6 |
| A18 | 74187742 | Nucleoporin-like protein | 1.95e+6 | 12.0 | 86380 | 6.5 | 6 |
| A22 | 2217933 | PKU-beta (Serine/Threonine kinase) | 2211 | 7.6 | 88883 | 8.9 | 4 |
| A22 | 8392959 | Beta-1,4-galactosyltransferase 2 | 3975 | 15.4 | 41910 | 9.4 | 4 |
| A22 | 113195686 | Lamin-B2 | 3513 | 10.9 | 67319 | 5.4 | 4 |
| A23 | 194388398 | Unknown, BAG65583, 110 kDa myosin motor domain, ATPase, SH3 | 9.80e+7 | 15.1 | 110912 | 8.5 | 12 |
| A25 | 158259341 | PI3Kinase, p85 binding domain protein | 645600 | 11.6 | 124285 | 6.9 | 8 |
| A28 | 40352738 | Ion transport protein | 306309 | 18.5 | 61801 | 5.5 | 5 |
| A32 | 4220898 | Transcriptional co-activator CRSP130 | 441010 | 8.4 | 106209 | 8.5 | 6 |
| A33 | 41945518 | Tiam1 PH domain lipid binding protein | 105719 | 8.6 | 99654 | 8.7 | 7 |
| A34 | 38569540 | 5'-3' exoribonuclease 2 isoform 3 | 140868 | 10.6 | 103802 | 8.2 | 7 |
| A34 | 148705232 | Leucine rich repeats,pyrin death domain | 179814 | 9.4 | 112138 | 8.0 | 8 |
| A35 | 624871 | a1(XIX) collagen chain precursor | 2.94e+7 | 12.9 | 115096 | 8.4 | 8 |
| A41 | 74187742 | Nucleoporin-like protein | 379387 | 10.2 | 86380 | 6.5 | 5 |
| A44 | 62897409 | Beta actin variant | 2.47e+8 | 35.5 | 41723 | 5.3 | 10 |
| A45 | 3603443 | Fibrousheathin I | 192098 | 11.7 | 94617 | 6.0 | 5 |
| A45 | 5834582 | E3 ubiquitin-protein ligase TRIM33 | 66846 | 8.7 | 115982 | 7.1 | 6 |
| A47 | 119582405 | Cadherin tandem repeat,ion binding domain | 4.44e+8 | 18.6 | 88669 | 5.0 | 10 |
| A49 | 13278786 | Nuclear mitotic apparatus protein 1 isoform | 1.85e+7 | 14.2 | 109280 | 8.1 | 11 |
| H1 | 194388398 | Unknown, BAG65583, 110kDa myosin motor domain, ATPase, SH3 | 161675 | 8.6 | 110912 | 8.5 | 7 |
| H3 | 15277503 | ACTB protein, partial | 8.45e+6 | 33.6 | 40221 | 5.6 | 9 |
| H4 | 158256650 | Myosin head motor domain Transmembrane 67, Meckelin | 1.14e+9 | 14.3 | 116422 | 6.2 | 11 |
| H5 | 119603632 | arginine methyltransferase 7, isoform CRA_b | 77142 | 7.3 | 69472 | 5.2 | 4 |
| H5 | 119603470 | Esterase 31, isoform CRA_e | 43420 | 7.7 | 59890 | 5.4 | 4 |
| H6 | 7019942 | 5’-3’ Exonuclease | 6.28e+6 | 17.1 | 63785 | 9.0 | 6 |
| H8 | 56204817 | Actin, alpha 1, skeletal muscle | 1.79e+6 | 30.7 | 32049 | .3 | 8 |
| H11 | 11908000 | BCL-6 corepressor short isoform | 1.78e+7 | 17.8 | 107405 | 8.8 | 9 |
| H11 | 119584312 | Inositol 1,4,5-triphosphate receptor, type 1 | 1.47e+6. | 8.8 | 117553 | 6.2 | 6 |
| H13 | 119612108 | Transmembrane protein 67, isoform CRA_b | 752714 | 10.2 | 109958 | 6.2 | 6 |
| H14 | 5882259 | Genethonin 3 | 9.41e+6 | 15.3 | 89342 | 5.2 | 8 |
| H19 | 14250401 | Actin, beta, partial | 4.89e+6 | 32.3 | 41005 | 5.6 | 9 |
| H21S | 74213524 | ACTB protein, partial | 1.22e+9 | 33.6 | 41751 | 5.3 | 10 |
| H22 | 21758574 | Organic solute transport protein 1 | 63.8 | 20.4 | 38752 | 7.8 | 4 |
| H23 | 14250401 | Actin, beta, partial | 3.70e+7 | 35.9 | 41005 | 5.6 | 10 |
| H23S | 2656123 | p53 binding protein | 2881 | 7.4 | 93501 | 9.6 | 4 |
| H25 | 288541297 | leucine-rich repeat-containing protein 15 isoform b precursor | 1.44e+8 | 27.7 | 64367 | 6.2 | 10 |
| H26 | 2102696 | Karyopherin beta 3 | 6.53e+6 | 13.2 | 123599 | 4.8 | 10 |
| H29 | 115529728 | Guanine nucleotide exchange factor OSTIII | 5.98e+6 | 8.8 | 123524 | 5.7 | 8 |
| H32 | 34189309 | Nuclear Pore Protein 107 | 2.40e+11 | 19.2 | 79971 | 5.2 | 8 |
| H33 | 119597872 | Unknown | 52313 | 8.2 | 112707 | 5.1 | 6 |
| H34 | 272982598 | Anti-HIV-1 gp120 Ig heavy chain | 4.02e+8 | 22.4 | 51810 | 8.1 | 8 |
| H36 | 5305448 | PH domain containing signaling mediator | 175808 | 16.8 | 70047 | 5.7 | 6 |
| H38 | 39644830 | Pleckstrin 2 protein, partial | 152085 | 24.2 | 39711 | 9.5 | 5 |
| H39 | 7661922 | Ras related protein Rab-21 | 38925 | 33.8 | 24348 | 8.1 | 5 |
| H41 | 148692949 | Leucine rich repeat containing 21 | 169639 | 9.6 | 111679 | 6.8 | 5 |
| H44 | 226958677 | Guanine nucleotide exchange factor DSB 3 | 872951 | 7.6 | 123694 | 5.7 | 8 |
| H45 | 148682292 | Retinis pigmentosa 1 homolog (human) | 30379 | 5.1 | 124037 | 9.5 | 6 |
| H48 | 74213524 | Actin like nucleotide binding sugar kinase | 2.37e+7 | 44.3 | 41751 | 5.3 | 12 |
Next, we used human retinal progenitor (HRP) cells to gain better understanding as to whether or not prohibitin in the retina may have similar binding partners. Actin and myosin binding of prohibitin suggest a mitochondrial DNA maintenance function of prohibitin in the retina. Pleckstrin (PH) domain containing protein, PH2, and Rab21 binding implies prohibitin-induced specific inositide and Ras signaling in the retina.
3.5. Prohibitin Depletion in the RPE by siRNA
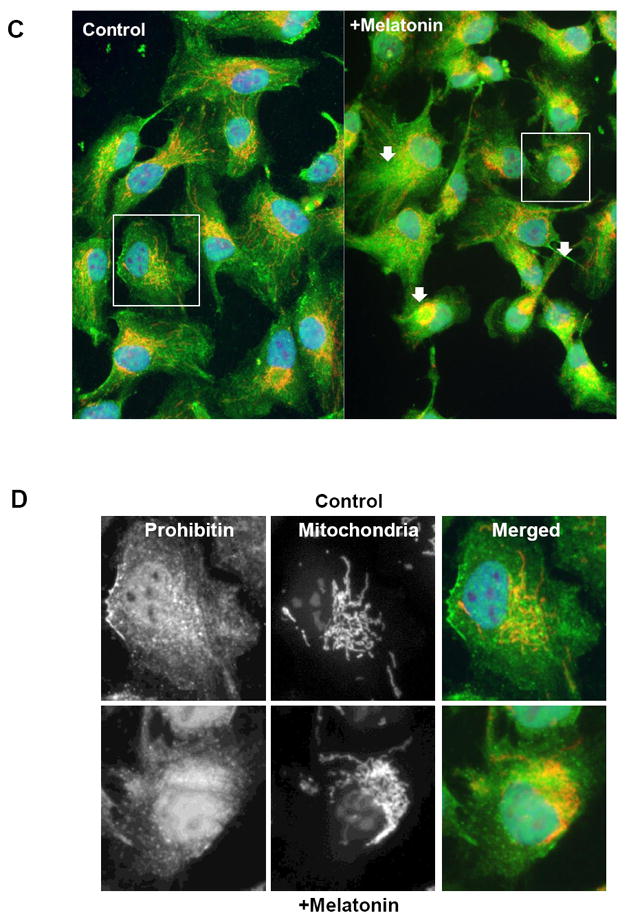
To examine prohibitin depletion phenotype as shown in the RPE of AMD eyes, we conducted a functional study using siRNA targeting prohibitin. Prohibitin knockdown experiments demonstrate that a decreased prohibitin (Figure 5A) breaks the regular lamellar cristae, whereas random sequence control and t=0 control do not affect the structure of mitochondria. Mitochondrial morphology, visualized by MitoTracker, suggested that prohibitin depletion may lead to mitochondrial fragmentation as a dramatic change from the tubular shape as shown in control cells (Figure 5B). MitoTracker Orange (mitochondria), DAPI (nucleus), and Alexa Fluor 488 (prohibitin) were used to label subcellular organelles and prohibitin, respectively. Next, we tested the hypothesis that melatonin may control prohibitin concentration, considering mitochondrial protection mechanism by melatonin.6 Previously, our data implied that melatonin may change the RPE proteome to protect the RPE from oxidative stress [6].
Figure 5.
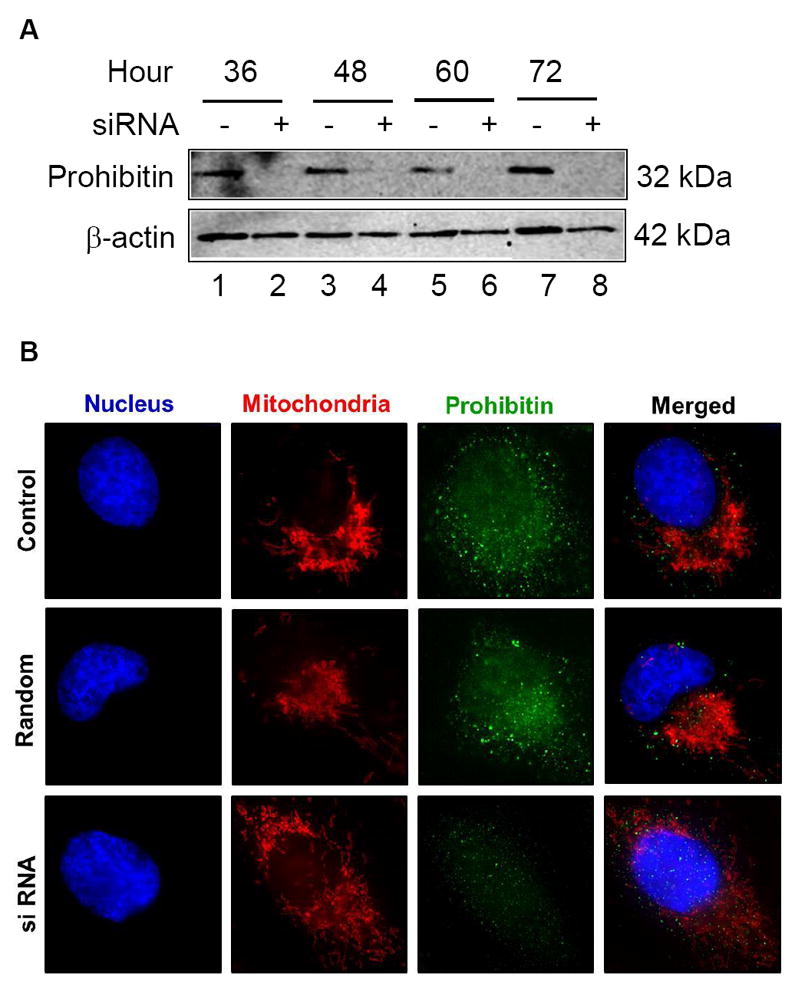
A. Downregulated prohibitin is shown by siRNA knockdown.
ARPE-19 cells were transfected by prohibitin specific siRNA in serum free medium in a time-dependent manner (36 to 72 h). Prohibitin expression and depleted levels were analyzed by Western blotting analysis from cell extracts. Prohibitin levels were diminished during siRNA knockdown. β-actin was used as a loading control.
B. Mitochondrial morphological changes during prohibitin siRNA knockdown analysis. ARPE-19 cells were incubated using prohibitin specific siRNA (175 ng for 48 h) and random sequence control. Prohibitin and organelles were visualized by immunocytochemical analysis using DAPI (blue, nucleus), MitoTracker Orange (red, mitochondria), and Alexa-Fluor 488 (green, prohibitin). Disrupted mitochondrial morphological changes were observed under prohibitin depleted levels. The scale bar represents 5 μm.
C, D. ARPE19 cells were incubated using 100μM DMSO-dissolved melatonin for 30 mintures, followed by incubation in untreated medium (12 hours). Cells were visualized using prohibitin antibody and Alexa-Fluor-488 secondary antibody. Mitochondria and nucleus were labeled using MitoTracker Orange and DAPI respectively. Cell morphology was tracked at initial, half-hour, and 12-hour time points. Cells incubated for 12 hours are shown.
Immunocytochemistry of melatonin-treated cells illustrates an increase and dissimilar distribution of prohibitin compared to non-melatonin-treated cells (Figure 5C). Mitochondrial arrangement is also changed by melatonin treatment (Figure 5D). The initial cell shape of ARPE-19 is epithelioid, but after 0.5-hour treatment with melatonin cells undergo a morphological change to a fusiform shape with spindle outgrowths. Cells return to epithelioid shape after 12 hours in untreated medium, confirming melatonin effect on prohibitin expression.
3.6. Prohibitin Expression Increased in the Retina but Decreased in the RPE of AMD
Since the susceptibility towards oxidative stress significantly varies from the retina and RPE cells, we hypothesized that prohibitin expressions in the macular and peripheral retinal and RPE region could be affected differentially by aging. Our Western blot analyses demonstrated that prohibitin is up-regulated significantly in both macular (8 mm) and peripheral retinal regions from postmortem AMD donor eyes (Figure 6A). On the other hand, prohibitin is down-regulated dramatically in both macular RPE region and peripheral RPE in AMD eyes compared to age-matching controls (Figure 6B). Differences in region-specific protein content were reported in central and peripheral retina in the progression of AMD [43].
Figure 6.
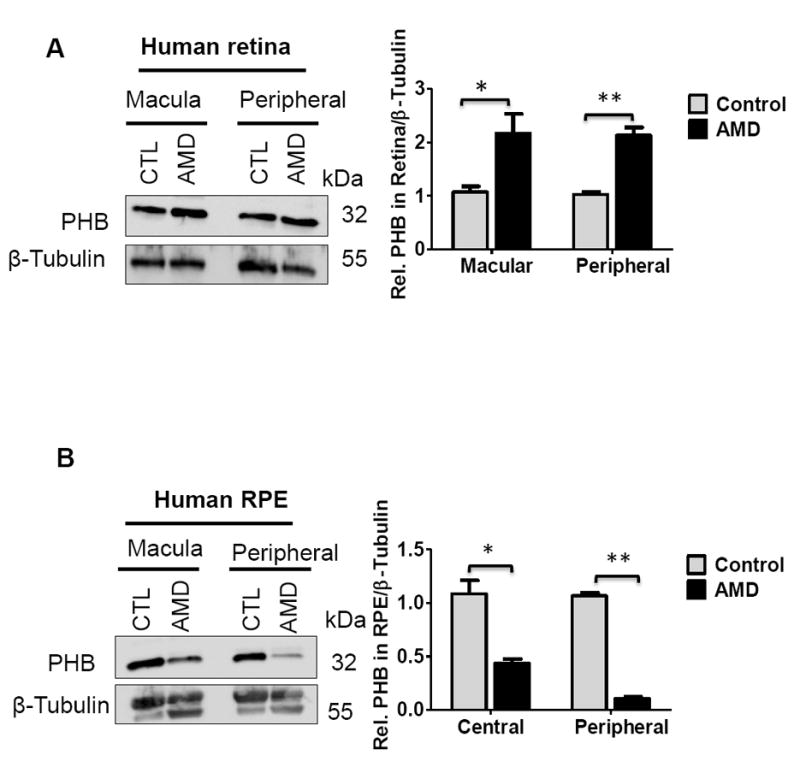
Prohibitin expressions in human AMD retina (8mm macular, peripheral retina) and RPE (8mm central and peripheral region, n=6 for two biological sample for triplicate experiments)) were analyzed by Western blotting. Retinal tissue and RPE cells were homogenized in RIPA buffer followed by sonication. As a loading control, β-tubulin was used. Prohibitin was analyzed quantitatively based on pixel size and intensity. A. Prohibitin in the macular and peripheral region from AMD retina. B. Prohibitin in the central and peripheral region from AMD RPE. Statistical comparisons between means were performed by 2-tailed t test. A p value of ≤0.05 was considered as statistically significant (P > 0.05 not significant; P ≤ 0.05 *; P ≤ 0.01 **; P 0.001 ***).
3.7. Prohibitin Expression is Downregulated in the Retina of the Diabetic Retinopathy Model
Prohibitin regulates lipid metabolism by binding specific phospholipids, including cardiolipin and PIP3 [7,21]. Increased prohibitin levels negatively affect insulin signaling through tyrosine phosphorylation as phosphatidylinositol 3 kinase (PI3K) downstream [21]. The next experiments tested the hypothesis that prohibitin signaling could be altered in both diabetic and aging models. We first examined whether mitochondrial prohibitin was altered during hyperglycemic conditions using the streptozotocin (STZ)-treated murine model and diabetic human tissue. Our Western blotting analysis showed that the prohibitin levels decreased significantly in the diabetic retinopathy retina from the STZ-induced mouse (Figure 7A) and rat (Figure 7B).
Figure 7.
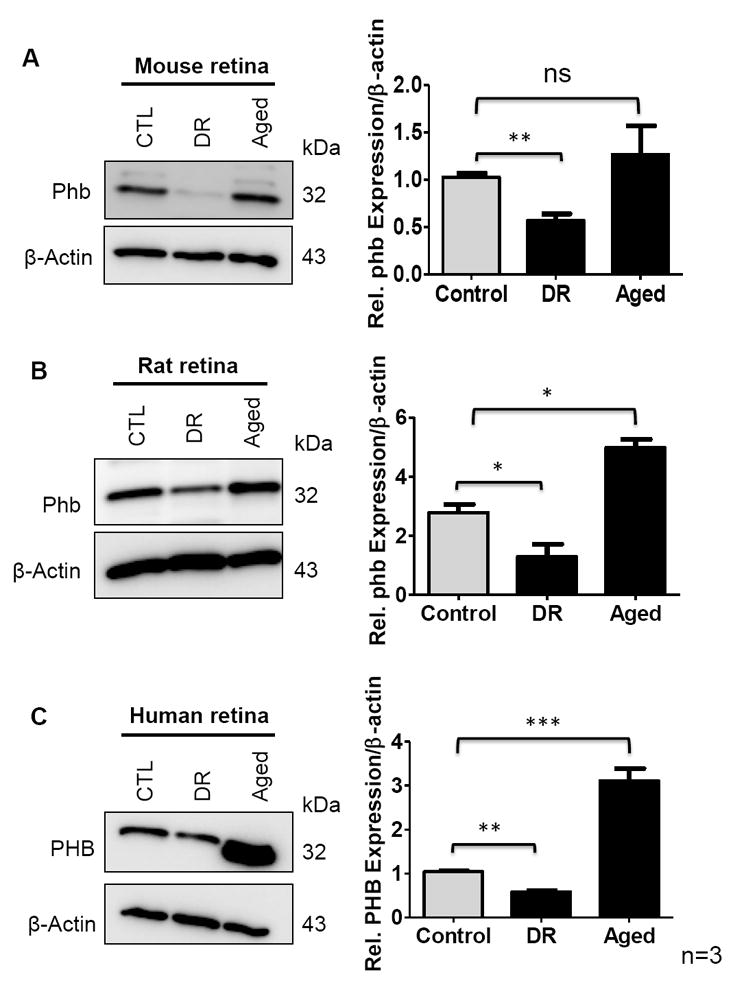
Prohibitin in control, diabetic retinopathy (DR) and aged retina (n=9 for biological triplicate and technical triplicate) were analyzed. Retinal tissue was homogenized in RIPA buffer followed by sonication. β-actin blot was used as a loading control. A. Prohibitin from diabetic retinopathy (DR) and aged mouse retina was compared to control. B. Prohibitin from diabetic retinopathy (DR) and aged rat retina was compared to control. C. Prohibitin from diabetic retinopathy (DR) and aged human retina was compared to control. Statistical comparisons between means were performed by 2-tailed t test. A p value of ≤ 0.05 was considered as statistically significant (P > 0.05 ns (not significant); P ≤ 0.05 *; P ≤ 0.01 **; P ≤ 0.001 ***).
Next, considering that reactive oxygen species (ROS) accumulation leads to the disruption of the mitochondrial network during aging, we tested our hypothesis that prohibitin expression could be changed in aged retinas from mouse, rat (12 months), and human (71 years) compared to controls. In contrast to the diabetic retinopathy condition, we observed that the level of prohibitin is significantly increased in rats and humans, whereas there were minimal changes in the mouse retina (Figure 7A–C).
4. DISCUSSION
Prohibitin protects the retina and the RPE in light-induced stress, aging, and diabetes [2]. Previsouly, we demonstrated that: (1) stress-dependent translocalization of prohibitin between mitochondria and the nucleus as a retrograde signal, (2) positive correlation between PIP3 and cardiolipin binding with prohibitin under stress, (3) prohibitin interaction with the tumor suppressor p53 and phosphorylation in the nucleus, and (4) accelerated apoptosis by prohibitin depletion. The oxidative stress-induced changes in location and the expression of prohibitin are crucial events for mitochondrial integrity. As the susceptibilities of the RPE and retina to oxidative stress are different, altered expression patterns of prohibitin in the RPE and the retina are anticipated in the diabetic and aging models.
The current study aimed to show that AMD leads to prohibitin downregulation in the RPE. Decreased prohibitin may modulate lipid metabolism, transcriptional activation, and RPE apoptosis to accelerate the progression of AMD. We tested our working hypothesis of inverse correlation of prohibitin levels and AMD using in vivo human models. Prohibitin knockdown using siRNA and molecular interaction assays demonstrate that prohibitin is a phospholipid and mitochondrial DNA binding molecule to maintain mitochondrial integrity. As a positive control of oxidative stress, we introduced a diabetic eye model to compare prohibitin expressions in aged and normal conditions. Our in vivo data from human donors demonstrate that prohibitin is depleted in the RPE during AMD pathogenesis.
Conventional proteomic profiling studies reported human RPE proteome [33], drusen composition [34], lipofuscin components [35,36] and proteins in native differentiated RPE cells and cultured dedifferentiated RPE [37,38]. Proteomic changes in RPE from AMD [39-41] and diabetic eyes were known [42]. Proteins in the vitreous humor from glaucoma model and diabetic retinopathy were reported [42-44]. The current study identified the potential binding partners of prohibitin and their putative functional roles in the retina and RPE. Our biochemical and proteomic analyses imply that prohibitin is a specific lipid binding modulator in diabetes-induced retinopathy and AMD.
4.1. Prohibitin Interacts with Cardiolipin and PIP3
Previously, we demonstrated that prohibitin is a lipid metabolism switch that binds to PIP3 and cardiolipin in a stress-dependent manner [7]. We speculated that prohibitin may contain a lipid binding domain, including conserved basic amino acids. It is reported that prohibitin-PIP3 interaction may regulate the insulin signaling [21].
Our multiple sequence alignment suggests that prohibitin may have a putative phospholipid binding sequence such as a PX domain that may influence PIP3 and cardiolipin interaction. The PX domain binds to phosphoinositide that includes PIP3. Conserved basic residues that include R43, R72, K83, R97, and R105 are aligned with the PX domain in p47phox, SNX6, and SGK3. PX domain residues are not highly conserved, as shown by 25–50% similarity compared to other PX domain proteins; however, essential basic amino acids with hydrophobic (F, I, L, A) and structural (P, G) amino acids seem enough to make a phospholipid binding pocket as shown in p47phox. A putative secondary pocket also suggests that allosteric or post-translational modification-dependent regulatory mechanisms on lipid binding may exist in prohibitin-phospholipid binding. We speculate that prohibitin may accelerate or inhibit aging signaling by altered lipid bindings, including cardiolipin and PIP3, under stress conditions. Prohibitin showed stronger interaction toward PIP3 compared to PI(4,5)P2 by a fivefold ratio [7]. In normal conditions, PI(4,5)P2 has higher concentration than PIP3 in the cell, thus strong PIP3 binding will facilitate the forward reactions catalyzed by PI3K. Prohibitin-PIP3 interaction may shift toward prohibitin-cardiolipin binding under oxidative stress, suggesting a switch-off function of phosphoinositide reaction under stress.
RPE phagocytosis is the major mechanism by which apoptotic cells and photoreceptor discs are eliminated in a circadian-dependent manner. PI(4,5)P2 interventions, both degradation and synthesis, compromise the phagocytic pathway. During phagocytosis, PIP3 and PI3K are regulated for clustering receptors and actin polymerization. Considering weak binding of PI(4,5)P2 and strong binding with PIP3, prohibitin may act as a cell fate determinant by altering lipid interactions under oxidative stress, including aging and diabetes. Prohibitin has an anti-apoptotic function in mitochondria as the default function showing high affinity toward PIP3 under normal conditions as a positive regulator of the PI3K pathway. Under oxidative stress, prohibitin may become a transcriptional regulator in the nucleus with weaker interaction with PIP3. Considering PIP3 is an important regulator of cell proliferation, a tight binding of PIP3-prohibitin may explain the anti-proliferative function of prohibitin. The lipid binding switch may regulate uncontrolled proliferation of RPE cells under oxidative stress that includes aging and diabetes. Depletion of prohibitin may control lipid metabolism in an aging- and oxidative stress-dependent manner.
Prohibitin mediates epithelial migration, reversal of obesity, drug sensitivity, longevity, estrogen receptor-dependent nuclear translocation, transcription repression, and histone deacetylase-related activities. Prohibitin is involved in cell cycle regulation and apoptosis6 by interacting with the retinoblastoma tumor suppressor protein Rb [7]. Prohibitin plays a major role in C-Raf activation for epithelial cell adhesion and migration [8]. Oxidative stress-induced implications during mitochondrial biogenesis, disruption of mitochondria-nuclear communication, and damage to the mitochondrial DNA (mtDNA) lead to the development of diabetic retinopathy (DR) [10,11,12]. Mitochondrial dysfunction, including mtDNA damage in the retina and RPE, leads to various age-related diseases including AMD [13] As mitochondrial genes are more susceptible than nuclear genes to oxidative stress, mitochondrial heat shock proteins (mtHsp70) and ATP synthase were downregulated in the RPE under oxidative damage [14]. Frequent retinal irradiation, imbalance in oxygen consumption, and polyunsaturated fatty acids (PUFA) oxidation could be the main causes for the progression of AMD [15].
4.2. Prohibitin-Protein Interaction
Prohibitin depletion leads to the mitochondrial decay and fragments accumulation [26,27]. Decreased prohibitin also reduces mtDNA and destabilizes mitochondrial-encoded subunits of the respiratory chain [28,29]. Our prohibitin-protein binding experiments suggest that prohibitin may change cytoskeletal rearrangement by binding with actin, myosin, and kinesin. Kinesin-like protein is a microtubule-dependent molecular motor that transports organelles and chromosomes during cell division. In addition, prohibitin interacts with myosin head motor domain. β actin is observed to be present in both control and oxidative stress, suggesting a general role of prohibitin-actin interaction on mitochondrial structure maintenance. β actin has a nucleotide binding domain that controls the flow of ions across epithelial cells and regulates cell volume. Prohibitin binds to a hypothetical protein that contains cadherin tandem repeat domains. The cadherin domain participates in cell fate, proliferation, differentiation, and migration. Nucleoporin (86 kDa) has a role of transporting macromolecules between the nucleus and cytoplasm. Prohibitin binds to inositol-1,4,5-triphosphate receptor (IP3R), which may regulate Ca2+ concentration to activate neurotransmitter-gated receptors.
CONCLUSION
Prohibitin showed strong affinity with PIP3 compared to PI(4,5)P2. Prohibitin-PIP3 interaction may facilitate the forward reaction of the PI3K pathway in normal conditions. Under stress conditions, prohibitin interacts with cardiolipin as a retrograde response to activate mitochondrial proliferation. The lipid-binding switch mechanism of prohibitin with PIP3 and cardiolipin may suggest the role of prohibitin effects on energy metabolism and age-related diseases. Differential expression of prohibitin was observed in the postmortem retinas of diabetic retinopathy (DR) and aged human eyes as well as in the retinas of mice and rats in vivo. Region-specific expressions of prohibitin with respect to the retina (macular and peripheral) and RPE (central and peripheral) were observed in the postmortem tissues of AMD.
A detailed understanding of prohibitin interaction with lipids, nucleotides, and proteins shown in the current study may suggest how molecular interactions control apoptosis and how we can intervene against the apoptotic pathway in AMD. Our data suggest that prohibitin depletion in the peripheral RPE is a significant step leading to mitochondrial dysfunction that may promote AMD progression.
Supplementary Material
A. Immunoprecipitation of PHB interacting proteins. Coomassie blue stained gel shows immunoprecipitated proteins from control ARPE-19 cells (lysate) by anti-PHB antibody binding. Cells were harvested, lysed and proteins were analyzed by immunoprecipitation and SDS-PAGE. Proteins in W1 to W3 represents wash fractions; E1 to E3 represents elution fractions; Eb represents the protein elution by immunoprecipitated boiling protein-A beads with SDS-sample buffer. Elution fractions showed the potential binding partners of prohibitin and were analyzed further using mass spectrometry. Coomassie blue stained gel shows immunoprecipitated proteins from oxidative stress (200 μM H2O2 or tBUO2H) treated ARPE-19 cells (lysate) with anti-prohibitin antibody. W1 to W3 represents wash fractions; E1 to E3 represents elution fractions; Eb represents the protein elution by immunoprecipitated boiling protein-A beads with sample buffer (Eb). Protein standard marker represents the size between 10 to 120 kDa. Elution fractions showed the potential binding partners of prohibitin and were further processed for protein identification. B. Prohibitin interacting proteins in the retina. Coomassie blue stained gel shows immunoprecipitated proteins from control human retinal progenitor (HRP) cells using anti-prohibitin antibody. Cells were harvested and lysed, followed by protein analysis using SDS-PAGE and mass spectrometry. Proteins in W1-W3 lanes represents washing fractions; E1 to E3 represents elution fractions; Eb represents the protein elution by protein-A beads boiling with sample buffer. Elution fractions showed the potential binding partners of prohibitin and were further analyzed using mass spectrometry.
Figure 8. Up- and downstream regulation of prohibitin network.
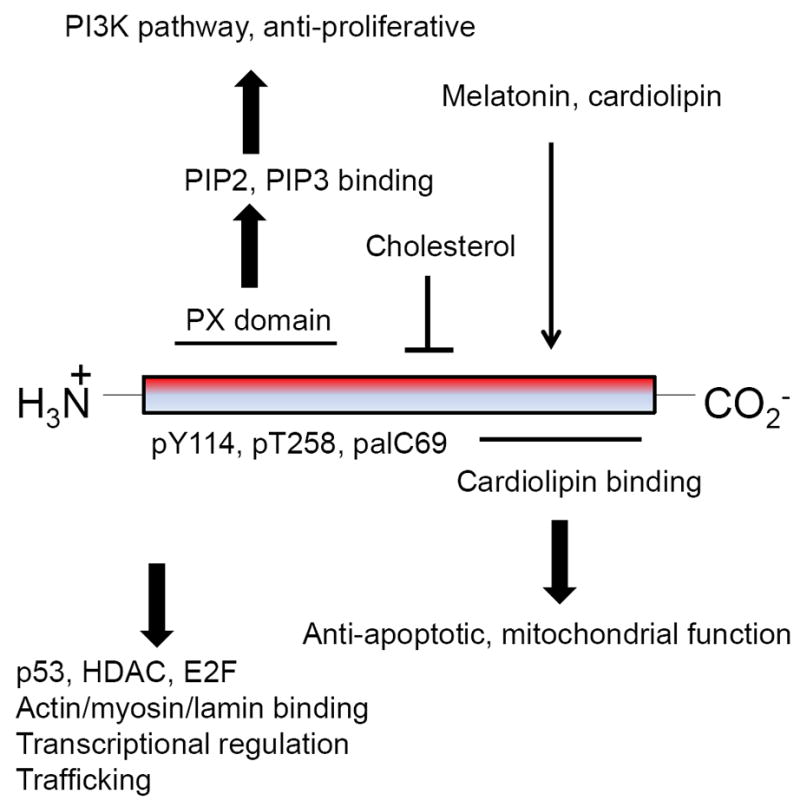
Prohibitin bindings and up/down stream regulations are shown. Basic amino acids of N-terminus may catalyze PI3K pathway by PIP3 binging, whereas C-terminus cardiolipin binding may have a role as an anti-apoptotic signaling by mtDNA binding.
Acknowledgments
The authors thank Jeremy Goldman and Mike Gibson for insightful discussions and sharing equipment. Matthew Durocher and Dr. Tristan Purvis are acknowledged for their suggestions and critical reading. This study was supported by the Century II Equipment fund and the Research Excellence Fund from Michigan Technological University, Research Assistantship and Teaching Assistantship from American University of Nigeria.
LIST OF ABBREVIATIONS
- DAPI
4’,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle Medium
- IP
immunoprecipitation
- ROS
reactive oxygen species
- RPE
retinal pigment epithelium
- siRNA
small interfering RNA
Footnotes
AUTHORS’ CONTRIBUTIONS
SRS and WJJ designed the hypothesis, aim, and experiments. SRS, ODS, FL, WH, TM, JYU conducted the experiments. WR, PSB, MB, WJJ provided materials, equipment, and participated in discussion. All authors wrote the paper.
COMPETING INTERESTS
None
References
- 1.Chung H, Lee H, Lamoke F, Hrushesky WJM, Wood PA, Jahng WJ. Neuroprotective role of erythropoietin by antiapoptosis in the retina. J Neurosci Res. 2009;87:2365–2374. doi: 10.1002/jnr.22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee H, Chung H, Arnouk H, Lamoke F, Hunt RC, Hrushesky WJM, Wood PA, Lee SH, Jahng WJ. Cleavage of the retinal pigment epithelium-specific protein RPE65 under oxidative stress. Int J Biol Macromol. 2010;47:104–108. doi: 10.1016/j.ijbiomac.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee H, Arnouk H, Sripathi S, Chen P, Zhang R, Bartoli M, Hunt RC, Hrushesky WJM, Chung H, Lee SH, Jahng WJ. Prohibitin as an oxidative stress biomarker in the eye. Int J Biol Macromol. 2010;47:685–690. doi: 10.1016/j.ijbiomac.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee H, Chung H, Lee SH, Jahng WJ. Light-induced phosphorylation of crystallins in the retinal pigment epithelium. Int J Biol Macromol. 2010;48:194–201. doi: 10.1016/j.ijbiomac.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnouk H, Lee H, Zhang R, Chung H, Hunt RC, Jahng WJ. Early biosignature of oxidative stress in the retinal pigment epithelium. J Proteomcs. 2011;74:254–261. doi: 10.1016/j.jprot.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang R, Hrushesky WJM, Wood PA, Lee SH, Hunt RC, Jahng WJ. Melatonin reprogrammes proteomic profile in light-exposed retina in vivo. Int J Biol Macromol. 2010;47:255–260. doi: 10.1016/j.ijbiomac.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sripathi SR, He W, Atkinson CL, Smith JJ, Liu Z, Elledge BM, Jahng WJ. Mitochondrial-nuclear communication by prohibitin shuttling under oxidative stress. Biochemistry. 2011;50:8342–8351. doi: 10.1021/bi2008933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sripathi SR, He W, Um J, Moser T, Dehnbostel S, Kindt K, Goldman J, Frost M, Jahng WJ. Nitric oxide leads to cytoskeletal reorganization in the retinal pigment epithelium under oxidative stress. Adv Biosci and Biotech. 2012;3:1167–1178. doi: 10.4236/abb.2012.38143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jahng WJ. New Biomarkers in the Retina and RPE Under Oxidative Stress. In: Adio Adedayo., editor. Ocular Diseases. InTech; 2012. pp. 121–156. [DOI] [Google Scholar]
- 10.McClung JK, Jupe ER, Liu XT, Dell’Orco RT. Prohibitin: potential role in senescence, development, and tumor suppression. Exp Gerontol. 1995;30:99–124. doi: 10.1016/0531-5565(94)00069-7. [DOI] [PubMed] [Google Scholar]
- 11.Nuell MJ, Stewart DA, Walker L, Friedman V, Wood CM, Owens GA, Smith JR, Schneider EL, Dell’ Orco R, Lumpkin CK. Prohibitin, an evolutionarily conserved intracellular protein that blocks DNA synthesis in normal fibroblasts and HeLa cells. Mol Cell Biol. 1991;11:1372–1381. doi: 10.1128/mcb.11.3.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He F, Zeng Y, Wu X, Ji Y, He X, Andrus T, Zhu T, Wang T. Endogenous HIV-1 Vpr-mediated apoptosis and proteome alteration of human T-cell leukemia virus-1 transformed C8166 cells. Apoptosis. 2009;14:1212–1226. doi: 10.1007/s10495-009-0380-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem. 2003;278:47853–47861. doi: 10.1074/jbc.M305171200. [DOI] [PubMed] [Google Scholar]
- 14.Wang S, Fusaro G, Padmanabhan J, Chellappan SP. Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene. 2002;21:8388–8396. doi: 10.1038/sj.onc.1205944. [DOI] [PubMed] [Google Scholar]
- 15.Theiss AL, Idell RD, Srinivasan S, Klapproth JM, Jones DP, Merlin D, Sitaraman SV. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB. 2007;21:197–206. doi: 10.1096/fj.06-6801com. [DOI] [PubMed] [Google Scholar]
- 16.Sharma A, Qadri A. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc Natl Acad Sci USA. 2004;101:17492–17497. doi: 10.1073/pnas.0407536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolonin MG, Saha PK, Chan L, Pasqualini R, Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nature Med. 2004;10:625–632. doi: 10.1038/nm1048. [DOI] [PubMed] [Google Scholar]
- 18.Nijtmans LG, De Jong L, Artal Sanz M, Coates PJ, Berden JA, Back JW, Muijsers AO, Van der Spek H, Grivell LA. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Artal-Sanz M, Tsang WY, Willems EM, Grivell LA, Lemire BD, Van der Spek H, Nijtmans LGJ, Sanz MA. The mitochondrial prohibitin complex is essential for embryonic viability and germline function in Caenorhabditis elegans. J Biol Chem. 2003;278:32091–32099. doi: 10.1074/jbc.M304877200. [DOI] [PubMed] [Google Scholar]
- 20.Steglich G, Neupert W, Langer T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol Cell Biol. 1999;19:3435–3442. doi: 10.1128/mcb.19.5.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ande SR, Mishra S. Prohibitin interacts with phosphatidylinositol 3,4,5-triphosphate (PIP3) and modulates insulin signaling. Biochem and Biophys Res Commun. 2009;390:1023–1028. doi: 10.1016/j.bbrc.2009.10.101. [DOI] [PubMed] [Google Scholar]
- 22.Bourges I, Ramus C, Mousson de Camaret B, Beugnot R, Remacle C, Cardol P, Hofhaus G, Issartel JP. Structural organization of mitochondrial human complex I: role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem J. 2004;383:491–499. doi: 10.1042/BJ20040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Back JW, Sanz MA, De Jong L, De Koning LJ, Nijtmans LGJ, De Koster CG, Grivell LA, Van Der Spek H, Muijsers AO. A structure for the yeast prohibitin complex: Structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci. 2002;11:2471–2478. doi: 10.1110/ps.0212602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nijtmans LGJ, Sanz MA, Grivell LA, Coates PJ. The mitochondrial PHB complex : roles in mitochondrial respiratory complex assembly. Cell Mol Life Sci. 2002;59:143–155. doi: 10.1007/s00018-002-8411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivera-Milla E, Stuermer CAO, Málaga-Trillo E. Ancient origin of reggie (flotillin), reggie-like, and other lipid-raft proteins: convergent evolution of the SPFH domain. Cell Mol Life Sci. 2006;63:343–357. doi: 10.1007/s00018-005-5434-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kutsyi MP, Gouliaeva NA, Kuznetsova EA, Gaziev AI. DNA-binding proteins of mammalian mitochondria. Mitochondrion. 2005;5:35–44. doi: 10.1016/j.mito.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Rajalingam K, Wunder C, Brinkmann V, Churin Y, Hekman M, Sievers C, Rapp U, Rudel T. Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nature Cell Biol. 2005;7:837–843. doi: 10.1038/ncb1283. [DOI] [PubMed] [Google Scholar]
- 28.Artal-Sanz M, Tavernarakis N. Prohibitin couples diapause signaling to mitochondrial metabolism during ageing in C. elegans. Nature. 2009;461:793–797. doi: 10.1038/nature08466. [DOI] [PubMed] [Google Scholar]
- 29.Gregory-Bass RC, Olatinwo M, Xu W, Matthews R, Stiles JK, Thomas K, Liu D, Tsang B, Thompson WE. Prohibitin silencing reverses stabilization of mitochondrial integrity and chemoresistance in ovarian cancer cells by increasing their sensitivity to apoptosis. Int J Cancer. 2008;122:1923–1930. doi: 10.1002/ijc.23351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu D, Lin Y, Kang T, Huang B, Xu W, Garcia-Barrio M, Olatinwo M, Matthews R, Chen YE, Thompson WE. Mitochondrial dysfunction and adipogenic reduction by prohibitin silencing in 3T3-L1 cells. PloS One. 2012;7:e34315. doi: 10.1371/journal.pone.0034315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasashima K, Sumitani M, Satoh M, Endo H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp Cell Res. 2008;314:988–996. doi: 10.1016/j.yexcr.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 32.He J, Cooper HM, Reyes A, Di Re M, Sembongi H, Litwin TR, Gao J, Neuman KC, Fearnley IM, Spinazzola A, Walker JE, Holt IJ. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012;40:6109–6121. doi: 10.1093/nar/gks266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonilha VL, Bhattacharya SK, West KA, Sun J, Crabb JW, Rayborn ME, Hollyfield JG. Proteomic characterization of isolated retinal pigment epithelium microvilli. Mol Cell Proteomics. 2004;3:1119–1127. doi: 10.1074/mcp.M400106-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suter M, Remé C, Grimm C, Wenzel A, Jäättela M, Esser P, Kociok N, Leist M, Richter C. Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J Biol Chem. 2000;275:39625–39630. doi: 10.1074/jbc.M007049200. [DOI] [PubMed] [Google Scholar]
- 36.Ng KP, Gugiu B, Renganathan K, Davies MW, Gu X, Crabb JS, Kim SR, Rózanowska MB, Bonilha VL, Rayborn ME, Salomon RG, Sparrow JR, Boulton ME, Hollyfield JG, Crabb JW. Retinal pigment epithelium lipofuscin proteomics. Mol Cell Proteomics. 2008;7:1397–1405. doi: 10.1074/mcp.M700525-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alge CS, Suppmann S, Priglinger SG, Neubauer AS, May CA, Hauck S, Welge-Lussen U, Ueffing M, Kampik A. Comparative Proteome Analysis of Native Differentiated and Cultured Dedifferentiated Human RPE Cells. Invest Ophthalmol Visual Sci. 2003;44:3629–3641. doi: 10.1167/iovs.02-1225. [DOI] [PubMed] [Google Scholar]
- 38.Alge CS, Hauck SM, Priglinger SG, Kampik A, Ueffing M. Differential Protein Profiling of Primary versus Immortalized Human RPE Cells Identifies Expression Patterns Associated with Cytoskeletal Remodeling and Cell Survival. J Proteome Res. 2006;5:862–878. doi: 10.1021/pr050420t. [DOI] [PubMed] [Google Scholar]
- 39.Nordgaard CL, Berg KM, Kapphahn RJ, Reilly C, Feng X, Olsen TW, Ferrington DA. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Visual Sci. 2006;47:815–822. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- 40.Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Visual Sci. 2008;49:2848–55. doi: 10.1167/iovs.07-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cottingham K. Age-related macular degeneration and the RPE secretome. J Proteome Res. 2006;5:2501. doi: 10.1021/pr062762k. [DOI] [PubMed] [Google Scholar]
- 42.Decanini A, Karunadharma PR, Nordgaard CL, Feng X, Olsen TW, Ferrington DA. Human retinal pigment epithelium proteome changes in early diabetes. Diabetologia. 2008;51:1051–1061. doi: 10.1007/s00125-008-0991-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zamora DO, Riviere M, Choi D, Pan Y, Planck SR, Rosenbaum JT, David LL, Smith JR. Proteomic profiling of human retinal and choroidal endothelial cells reveals molecular heterogeneity related to tissue of origin. Mol Vis. 2007;13:2058–2065. [PubMed] [Google Scholar]
- 44.Tezel G, Yang X, Cai J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Invest Ophthalmol Visual Sci. 2005;46:3177–3187. doi: 10.1167/iovs.05-0208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Immunoprecipitation of PHB interacting proteins. Coomassie blue stained gel shows immunoprecipitated proteins from control ARPE-19 cells (lysate) by anti-PHB antibody binding. Cells were harvested, lysed and proteins were analyzed by immunoprecipitation and SDS-PAGE. Proteins in W1 to W3 represents wash fractions; E1 to E3 represents elution fractions; Eb represents the protein elution by immunoprecipitated boiling protein-A beads with SDS-sample buffer. Elution fractions showed the potential binding partners of prohibitin and were analyzed further using mass spectrometry. Coomassie blue stained gel shows immunoprecipitated proteins from oxidative stress (200 μM H2O2 or tBUO2H) treated ARPE-19 cells (lysate) with anti-prohibitin antibody. W1 to W3 represents wash fractions; E1 to E3 represents elution fractions; Eb represents the protein elution by immunoprecipitated boiling protein-A beads with sample buffer (Eb). Protein standard marker represents the size between 10 to 120 kDa. Elution fractions showed the potential binding partners of prohibitin and were further processed for protein identification. B. Prohibitin interacting proteins in the retina. Coomassie blue stained gel shows immunoprecipitated proteins from control human retinal progenitor (HRP) cells using anti-prohibitin antibody. Cells were harvested and lysed, followed by protein analysis using SDS-PAGE and mass spectrometry. Proteins in W1-W3 lanes represents washing fractions; E1 to E3 represents elution fractions; Eb represents the protein elution by protein-A beads boiling with sample buffer. Elution fractions showed the potential binding partners of prohibitin and were further analyzed using mass spectrometry.