

Abstract
We synthesized a series of benzoic acids and phenylphosphonic acids and investigated their effects on the growth of Staphylococcus aureus and Bacillus subtilis. One of the most active compounds (7, 5-fluoro-2-(3-(octyloxy)benzamido)benzoic acid, ED50 ~ 0.15 μg/mL) acted synergistically with seven antibiotics known to target bacterial cell wall biosynthesis (a fractional inhibitory concentration index, FICI~0.35, on average) but had indifferent effects in combinations with six non cell-wall biosynthesis inhibitors (FICI~1.45, on average). The most active compounds were found to inhibit two enzymes involved in isoprenoid/bacterial cell wall biosynthesis: undecaprenyl diphosphate synthase (UPPS) and undecaprenyl diphophate phosphatase (UPPP), but not farnesyl diphosphate synthase, and there were good correlations between bacterial cell growth inhibition, UPPS inhibition and UPPP inhibition.
Keywords: benzoic acids, cell wall biosynthesis, drug discovery, membrane proteins, Staphylococcus aureus
Graphical abstract
We synthesized 30 benzoic and phenylphosphonic acids and discovered several that inhibited Staphylococcus aureus and Bacillus subtilis growth. They acted synergistically with antibiotics that target cell wall biosynthesis but not with other antibiotics. The most potent compounds targeted undecaprenyl diphosphate phosphatase and undecaprenyl diphosphate synthase and there were good correlations between enzyme and cell growth inhibition.
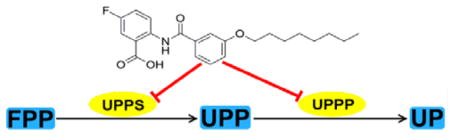
Introduction
There is without doubt a pressing need for the development of novel antibiotics having new structures and new targets, to help combat the development of drug resistance.[1] Over the past ~70 years, many antibiotics that target bacterial cell wall biosynthesis, compounds such as penicillin, methicillin and bacitracin, have been discovered and developed and so the enzymes that are used in cell wall biosynthesis are attractive drug targets. A simplified version of bacterial cell wall biosynthesis is shown in Figure 1. Initial steps involve formation of the (C5) isoprenoids dimethylallyl diphosphate (DMAPP, 1) and isopentenyl diphosphate (IPP, 2), formed in either the mevalonate pathway (in e.g. Staphylococcus aureus) or the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway (in e.g. E. coli), with fosmidomycin inhibiting the MEP pathway.[2] DMAPP then condenses, sequentially, with two molecules of IPP to form the (C15) species, farnesyl diphosphate (FPP, 3) in a reaction catalyzed by farnesyl diphosphate synthase (FPPS). The FPP so produced then condenses with 8 additional IPP molecules to form (C55) undecaprenyl diphosphate (UPP, 4) in a reaction catalyzed by undecaprenyl diphosphate synthase (UPPS). UPPS is an attractive drug target since it is not used by humans and several inhibitors (e.g. tetramic/tetronic acids; diamidines and benzoic acids) have been reported.[3] UPP is then converted by undecaprenyl diphosphate phosphatase (UPPP) to undecaprenyl phosphate (UP, 5) which acts as a membrane “anchor” for formation of glycosylated products (Lipid I, Lipid II) which are then converted to peptidoglycan cell wall products, as outlined in Figure 1. Antibiotics such as bacitracin, vancomycin and methicillin target these later stages in cell wall biosynthesis, again as shown in Figure 1. In this work, we sought to find novel benzoic acid inhibitors of UPPS and potentially UPPP since in earlier work,[3b] we had found that (2-(3-(decyloxy)benzamido)-5-nitrobenzoic acid, 6, Figure 1) inhibited UPPS and we reasoned that similar lipophilic, anionic species might also mimic the UPPP substrate, UPP. We thus synthesized 30 analogs of 6, 7–36, primarily benzoic acids but also, four phosphonic acid analogs, and tested them for activity against Staphylococcus aureus and Bacillus subtilis, as well as against UPPS and UPPP, and in several cases FPPS and a human cell line. In addition, we investigated their possible synergistic activity with a range of known antibiotics that either target, or do not target, bacterial cell wall biosynthesis.
Figure 1.

Schematic outline of cell wall biosynthesis (in most bacteria) delineating the role of isoprenoid biosynthesis in the early stages of peptidoglycan formation, together with the reactions targeted by several compounds discussed in the Text.
Results and Discussion
In previous work we found that the benzoic acid 6 (Figure 1) was a promising inhibitor of both E. coli UPPS (EcUPPS; IC50 = 3.0 μM) and S. aureus UPPS (SaUPPS; IC50 = 0.49 μM). In later work we found that several related lipophilic benzoic acids had activity against gram-positive (but not gram-negative) bacteria with 6 having a 1.3 μM ED50 value against B. subtilis, but no activity against E. coli, leading us to synthesize the 30 compounds whose structures and activities are shown in Table 1. Full synthesis and characterization details are in the Supporting Information.
Table 1.
ED50 values of benzoic acid and phosphonic acid inhibitors against B. subtilis and S. aureus cell growth (in μg/mL) and IC50 values against SaUPPS and EcUPPP enzymes (in μM). Inhibitors discussed in the Text are shown in blue. logD values were estimated using the chemicalize.org server (http://www.chemaxon.com). Standard errors are shown in Table S1.
| Compound | logD | B. subtilis | S. aureus | SaUPPS | EcUPPP |
|---|---|---|---|---|---|
|
|
3.5 | 0.14 | 0.16 | 0.32 | 2.7 |
|
8 |
3.2 | 0.21 | 0.24 | 0.33 | 1.3 |
|
9 |
4.2 | 0.42 | 0.79 | 0.54 | 4.2 |
|
10 |
4.0 | 0.56 | 0.23 | 0.60 | 3.0 |
|
|
4.7 | 0.21 | 0.082 | 0.78 | 0.83 |
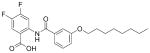 12 |
3.7 | 0.53 | 0.18 | 0.96 | 3.4 |
|
|
3.0 | 1.4 | 0.42 | 1.3 | 11 |
|
14 |
3.1 | 1.2 | 0.64 | 1.3 | >200 |
|
15 |
3.3 | 0.85 | 1.2 | 1.6 | 10 |
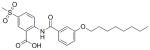 16 |
2.3 | 2.5 | 1.4 | 2.0 | 9.8 |
|
17 |
2.7 | 4.1 | 0.82 | 2.4 | 45 |
|
|
−1.6 | 23 | 11 | 2.5 | 6.7 |
|
19 |
3.6 | >100 | >100 | 3.0 | 4.2 |
|
20 |
3.2 | 2.7 | 3.1 | 3.2 | 8.5 |
|
21 |
0.80 | 2.3 | 3.9 | 3.4 | >200 |
|
22 |
2.6 | >100 | >100 | 4.5 | >200 |
|
23 |
2.8 | 3.6 | 1.3 | 4.9 | 20 |
|
24 |
3.1 | >100 | >100 | 4.9 | 31 |
|
25 |
3.1 | 3.0 | 1.6 | 6.0 | >200 |
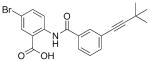 26 |
−0.28 | 3.4 | 0.6 | 6.9 | >200 |
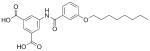 27 |
2.6 | >100 | >100 | 7.0 | >200 |
|
28 |
0.46 | 20 | 18 | 8.0 | >200 |
|
29 |
3.1 | >100 | 3.0 | 8.5 | >200 |
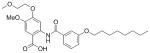 30 |
2.5 | 2.7 | 2.2 | 10 | >200 |
|
31 |
2.1 | >100 | 41 | 39 | 6.5 |
|
32 |
5.6 | >100 | >100 | 60 | >200 |
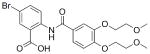 33 |
0.79 | >100 | 33 | 130 | >200 |
|
34 |
2.8 | 20 | 42 | >200 | 73 |
|
35 |
2.9 | >100 | >100 | >200 | >200 |
|
36 |
1.5 | >100 | >100 | >200 | 7.1 |
| Bacitracin | −2.6 | 84 | 145 | N.D. | 32 |
The rationale for the synthesis of these compounds was first, to investigate a series of compounds (7–16, 19, 21) in which we varied the substituent meta to the carboxyl group, covering a wide range of Hammett σm values (0 for −H, 0.71 for −NO2) which we reasoned would affect the acidity of the carboxyl group, the more electron-withdrawing groups yielding, perhaps, a better analog (carboxylate) of the diphosphate group (in FPP and UPP). We also investigated whether phosphonic acids (18, 31, 36) might be better inhibitors than benzoic acids, plus, we investigated a broad range of other benzoic acid ring-substitution patterns, as well as different “side-chains” (corresponding to the octyloxy group in e.g. 7), in order to probe both enzyme as well as cell activity. We then investigated the cell growth inhibition activity of these compounds against S. aureus and B. subtilis and ED50 values (in μg/mL) are shown in Table 1 (rank ordered by SaUPPS inhibition, as discussed below). We also screened for activity against the yeast Saccharomyces cerevisiae and the gram-negative, E. coli, but there was no activity with any compound (ED50 values > 200 μM). Typical dose-response curves for 3 compounds: benzoic acids with electron withdrawing (7) or electron donating (13) ring substituents, plus a phosphonic acid analog (18), are shown in Figure 2. Dose-response curves for all compounds tested are shown in Supporting Information Figure S1. There were several active compounds amongst the 30 investigated, with the most active being the m-trifluoromethoxy analog 11, with an ED50 value of 0.082 μg/mL against S. aureus. The most active bacterial cell growth inhibitors were all lipophilic benzoic acids with highly electron-withdrawing (−NO2, −OCF3) ring substituents, Table 1. We screened 11 and three other compounds (7, 14, 18) against a human cell line (NCI-H460) to assess toxicity finding low activity (a selectivity index of ~1000 for 11, Figure S2). The presence of electron-donating groups (13, 14) decreased anti-bacterial activity, as did the presence of substituents that might hydrogen-bond to water (19, 22, 27, 29). The phosphonic acids (19, 31, 36) had little or no antibacterial activity although the diethyl phosphonate 21 was active, consistent with the observation that all compounds with additional ionizable groups on the benzoic acid have poor activity.
Figure 2.

Representative dose-response curves for three inhibitors (7, 13 and 18) against B. subtilis and S. aureus. Data points are reported as mean±SD for duplicate experiments.
The question then arises: what might the targets of these inhibitors be? The compounds were designed based on the UPPS inhibition precedent with 6, but FPPS and UPPP inhibition also seemed possible (with some carboxylic acids being known, potent FPPS inhibitors,[4] binding to an allosteric site). To try and narrow down the possibilities as well as open up potential routes to synergistic combinations, we next investigated whether or not one of the most active compounds, the fluoro-benzoic acid 7, exhibited synergistic, additive, indifferent or antagonistic activity with a broad range of antibiotics that act either by inhibiting bacterial cell wall biosynthesis, or by other mechanisms. The antibiotics that act by targeting cell wall biosynthesis were ampicillin, bacitracin, fosmidomycin, carbenicillin, vancomycin, fosfomycin, and cefotaxime. The compounds that do not target bacterial cell wall biosynthesis were kanamycin, tetracycline, sulfamethoxazole, trimethoprim, spectinomycin, and chloramphenicol We determined the fractional inhibitory concentration index (FICI) values for each combination using the FICI formula:[5]
where FICA, FICB are the fractional inhibitory concentrations of drugs A and B, MIC(A) and MIC(B) are the MIC values of drugs A and B acting alone, and MIC(AB) and MIC(BA) are the MIC values of the most effective combination of drug A or B in the presence of drug B or A. Using this method, FICI values of <0.5 indicate synergy, >0.5 and <1.0 indicate additivity, >1 and <2 indicate an indifferent effect, and ≥2 indicates antagonism.[6] In addition, we evaluated isobolograms using the method of Berenbaum.[7] FICI values are shown in Table 2 and representative isobolograms are in Figure 3. All isobolograms are given in SI Figure S3.
Table 2.
Combinations of 7 and antibiotics against B. subtilis and S. aureus. FIC values are reported as mean±SD for duplicate experiments.
|
B. subtilis
| ||||||
|---|---|---|---|---|---|---|
| Antibiotic | MIC antibiotic (μg/mL) | FIC antibiotic | MIC 7 (μg/mL) | FIC 7 | FIC Index | |
| Cell Wall Biosynthesis Inhibitors | Fosmidomycin | 5 | 0.039 ± 0.0053 | 1 | 0.13 ± 0.056 | 0.17 ± 0.056 |
| Carbenicillin | 5 | 0.22 ± 0.073 | 1 | 0.15 ± 0.071 | 0.37 ± 0.10 | |
| Cefotaxime | 2.5 | 0.17 ± 0.026 | 1 | 0.14 ± 0.081 | 0.31 ± 0.085 | |
| Vancomycin | 0.5 | 0.27 ± 0.24 | 1 | 0.17 ± 0.063 | 0.44 ± 0.25 | |
| Fosfomycin | 200 | 0.16 ± 0.15 | 1 | 0.063 ± 0.022 | 0.22 ± 0.15 | |
| Ampicillin | 0.5 | 0.30 ± 0.072 | 1 | 0.23 ± 0.084 | 0.53 ± 0.11 | |
| Bacitracin | 200 | 0.18 ± 0.089 | 1 | 0.36 ± 0.14 | 0.53 ± 0.17 | |
|
|
||||||
| Protein Biosynthesis Inhibitors | Chloramphenicol | 0.5 | 0.75 ± 0.18 | 1 | 0.97 ± 0.42 | 1.72 ± 0.46 |
| Kanamycin | 1.5 | 0.64 ± 0.14 | 1 | 0.71 ± 0.30 | 1.36 ± 0.33 | |
| Tetracycline | 5 | 0.48 ± 0.047 | 1 | 0.57 ± 0.50 | 1.04 ± 0.50 | |
|
|
||||||
| Nucleic Acid Inhibitors | Sulfamethoxazole | 200 | 0.78 ± 0.21 | 1 | 0.94 ± 0.64 | 1.72 ± 0.67 |
| Trimethoprim | 0.5 | 0.66 ± 0.13 | 1 | 0.85 ± 0.29 | 1.51 ± 0.32 | |
| S. aureus | ||||||
|
| ||||||
| Antibiotic | MIC antibiotic (μg/mL) | FIC antibiotic | MIC 7 (μg/mL) | FIC 7 | FIC Index | |
|
| ||||||
| Cell Wall Biosynthesis Inhibitors | Carbenicillin | 15 | 0.23 ± 0.048 | 1 | 0.077 ± 0.026 | 0.30 ± 0.055 |
| Cefotaxime | 2 | 0.18 ± 0.055 | 1 | 0.084 ± 0.031 | 0.27 ± 0.063 | |
| Vancomycin | 1.5 | 0.14 ± 0.034 | 1 | 0.15 ± 0.062 | 0.29 ± 0.071 | |
| Fosfomycin | 200 | 0.20 ± 0.11 | 1 | 0.091 ± 0.034 | 0.29 ± 0.36 | |
| Ampicillin | 0.5 | 0.41 ± 0.17 | 1 | 0.15 ± 0.071 | 0.56 ± 0.18 | |
| Bacitracin | 200 | 0.16 ± 0.11 | 1 | 0.044 ± 0.019 | 0.20 ± 0.11 | |
|
|
||||||
| Protein Biosynthesis Inhibitors | Spectinomycin | 40 | 0.75 ± 0.43 | 1 | 0.81 ± 0.32 | 1.56 ± 0.54 |
| Chloramphenicol | 5 | 0.88 ± 0.47 | 1 | 0.81 ± 0.31 | 1.69 ± 0.56 | |
| Kanamycin | 1.5 | 0.89 ± 0.68 | 1 | 0.83 ± 0.29 | 1.72 ± 0.74 | |
| Tetracycline | 0.5 | 0.24 ± 0.11 | 1 | 0.51 ± 0.24 | 0.75 ± 0.26 | |
|
|
||||||
| Nucleic Acid Inhibitors | Sulfamethoxazole | 200 | 0.54 ± 0.13 | 1 | 0.98 ± 0.37 | 1.53 ± 0.39 |
| Trimethoprim | 15 | 0.69 ± 0.32 | 1 | 0.57 ± 0.25 | 1.26 ± 0.41 | |
|
| ||||||
| Mean FICIs: | B. subtilis | inhibitors targeting cell wall biosynthesis | 0.37 ± 0.14 | |||
| inhibitors targeting nucleic acids and protein biosynthesis | 1.47 ± 0.28 | |||||
| S. aureus | inhibitors targeting cell wall biosynthesis | 0.32 ± 0.12 | ||||
| inhibitors targeting nucleic acids and protein biosynthesis | 1.42 ± 0.37 | |||||
Figure 3.

Representative isobolograms for 7 with antibiotics having known mechanisms of action. a) 7+fosmidomycin in B. subtilis showing synergy (FICI=0.17) of 7 with a cell wall biosynthesis inhibitor (that targets DXR, 1-deoxy-D-xylulose 5-phosphate reductoisomerase, in the non-mevalonate pathway); b) 7+sulfamethoxazole in B. subtilis showing an indifferent effect (FICI=1.72) of 7 with a nucleic acid biosynthesis inhibitor (that targets dihydropteroate synthase); c) 7+bacitracin in S. aureus showing synergy (FICI=0.20) of 7 with a cell wall biosynthesis inhibitor (that targets UPPP); d) 7+ kanamycin in S. aureus showing an indifferent effect (FICI=1.72) of 7 with a protein biosynthesis inhibitor (that targets ribosome function).
As can be seen in Table 2, there are very distinct differences between the FICI values obtained with 7 and the cell-wall biosynthesis inhibitors, and those obtained with 7 and the non cell-wall biosynthesis inhibitors. For S. aureus, the mean FICI with a cell wall biosynthesis inhibitor is 0.32, for B. subtilis, 0.37. These values represent synergism (i.e. the FICI is <0.5) and suggest that 7 acts in the cell wall biosynthesis pathway. With the non cell-wall biosynthesis inhibitors, the mean FICI for S. aureus in 1.42, and for B. subtilis, 1.47, meaning indifferent effects, as expected. Clearly then, in both S. aureus as well as in B. subtilis, 7 is targeting primarily one or more enzymes involved in cell wall biosynthesis and likely candidates that utilize long, lipophilic anionic substrates are UPPS and UPPP, and perhaps, FPPS. We thus next tested all compounds against UPPS (from S. aureus), since in earlier work we had found 6 was a UPPS inhibitor, plus, we tested all compounds against a UPP-phosphatase. UPPP is a membrane protein and we used the fusion hybrid of E. coli UPPP with Haloarcula marismortui bacteriorhodopsin,[8] which is active in detergent-based assays and is inhibited by bacitracin. We also tested the most active compounds against an EcFPPS, but there was no inhibition (up to 200 μM). Typical dose-response curves for SaUPPS and EcUPPP are shown in Figure 4 and include for UPPP, results for the known inhibitor, bacitracin, which has an IC50 = 32 μM, as also reported by Chang et al.[9] Full dose response curves for all compounds are in SI Figure S4 and all numerical results are shown in Table 1.
Figure 4.

Dose-response curves of various benzoic acid and phenyl phosphonic acid derivatives against SaUPPS and EcUPPP. The benzoic acids are up to ~40× more potent UPPP inhibitors than bacitracin, a known UPPP inhibitor used as a topical antibiotic. Data points are reported as mean±SD, for duplicate experiments.
As can be seen in Table 1, the most active cell growth inhibitors are also some of the most potent UPPS and UPPP inhibitors with IC50 values as low as 320 nM (for UPPS) and 1.3 μM (for UPPP). For the top 8 most potent SaUPPS inhibitors (7, 8, 9, 10, 11, 13, 14, 15) there is a good correlation with reported Hammett σm values (R2=0.79) but this breaks down with bulkier ring substituents. For example, the electron-withdrawing group (EtO)2PO (σm=0.42) has weak activity. We have not been able to obtain X-ray structures of SaUPPS with bound inhibitors, but we have reported the structure of 6 bound to EcUPPS (PDB ID code 4H2O), which suggests a possible explantion for the results reported here. Specifically, 6 has a decyloxy sidechain and the two molecules that bind to UPPS have relatively solvent exposed nitrobenzoate sidechains. This exposure will have an associated energy penalty. The shorter C8 sidechain in 8 could improve enzyme inhibition by reducing this solvent exposure, but the effect is small (0.33 μM vs 0.49 μM). However, with another pair of ligands: the fluorobenzoate 7 with a C8 sidechain and the fluorobenzoate 23 with a C6 sidechain, we find that the shorter chain species has a 4.9 μM IC50 while the longer-chain (C8) species has a much smaller IC50 (IC50=0.32 μM). Taken together, these results indicate that inhibitors with a C8 side-chain and small, electron-withdrawing ring substituents have close to optimum activity. The bulkier methyl sulfone and the diethyl phosphonate groups then—while being very electron withdrawing—likely contribute to a decrease in protein binding affinity because the added ligand volume is more solvent exposed than with the smaller substituents. This binding mode (as seen with 6 in PDB ID code 4H2O) in which the alkyloxy groups are buried in the interior of UPPS while the benzoates are at or close to the the surface could also explain why adding extra carboxylates or phosphonates reduces activity—the added groups can hydrogen bond with the solvent.
Interestingly, as can be seen in Figure 5a, there is a very good correlation between the pED50 (= −log10 ED50 [μM]) values for S. aureus and B. subtilis cell growth inhibition (a Pearson r-value correlation-coefficient, r = 0.90), strongly suggesting that the same targets are involved in both systems. And as shown in Figure 5b, the correlation for S. aureus cell growth versus SaUPPS inhibition is also high with an r = 0.85. Figure 5c shows a Pearson r-value correlation-coefficient matrix[10] for all cell and enzyme activities as well as for logD (estimated using the chemicalize.org web portal, http://www.chemaxon.com), from which it can be seen that both UPPS inhibition as well as UPPP inhibition correlate with cell growth inhibition. There are weaker correlations between cell growth inhibition and logD, while the enzyme inhibition results are essentially uncorrelated with logD so, as expected, permeabilty/transport plays a role in overall activity in cell growth inhibition. The enzyme/cell correlations are better than those we discussed in a previous study[11] where we found for 10 different cell/putative enzyme target inhibition assays (three from our group, seven randomly chosen from the literature) that on average the correlation between the enzyme and cell assay results was r~0.55.
Figure 5.

Correlations between cell growth and enzyme inhibition results. a) Correlation between S. aureus and B. subtilis cell growth inhibition based on pED50 (=−log10ED50 [μM]) results; b) Correlation between S. aureus cell growth inhibition and SaUPPS inhibition; c) Pearson r-value correlation matrix/heat map for S. aureus cell growth inhibition, B. subtilis cell growth inhibition, SaUPPS and EcUPPP enzyme inhibition (all based on pED50 or pIC50 values), and logD. The Pearson r-values are indicated and red/orange=high correlation, green=low correlation.
What is also apparent from the results shown in Table 1 is that, in general, the IC50 values for UPPP inhibition are somewhat larger than the IC50 values for UPPS inhibition, suggesting that in cells, UPPS and not UPPP is the primary target. This is, however, likely to be an over-simplification since the enzyme assays were all detergent based; substrate concentrations in cells are not known; UPPP is a membrane protein and inhibitors may accumulate in membranes; and we used an EcUPPP-bacteriorhodopsin fusion in our assay together with FPP, and not UPP. Nevertheless, as can be seen in Table 1, several of the most potent cell growth inhibitors do target both enzymes, and inhibition of two consecutive enzymes in the bacterial cell wall biosynthesis pathway is expected to result in enhanced activity over single target inhibition. Moreover, we find good enzyme inhibition/cell growth correlations, plus, as shown in Figure 3, addition of any of the known cell wall biosynthesis inhibitors in assays with 7 results in synergistic cell growth inhibition with, on average, FICI values of ~0.35.
Conclusions
The results reported here are of interest since we find that several lipophilic benzoic acids with electron-withdrawing ring substituents have activity against S. aureus and B. subtilis cell growth. One of the most potent compounds exhibited synergistic activity with numerous known cell wall biosynthesis inhibitors (FICIavg~0.35, n=13 different cell/inhibitor combinations), but an indifferent effect (FICIavg~1.45, n=12 cell/inhibitor combinations) with non cell-wall biosynthesis antibiotics. We tested all compounds against UPPS and UPPP and in some cases FPPS finding several promising UPPS as well as UPPP inhibitors. The most potent UPPP inhibitor was the trifluoromethoxy analog 11 which was ~40× more active than the known UPPP inhibitor, the antibiotic bacitracin. It also inhibited UPPS and was active against S. aureus (ED50 ~82 ng/mL). Electron-withdrawing substituents were essential for enzyme/cell inhibition by the benzoic acids, presumably because they make the benzoates much stronger acids (that are fully dissociated), although the presence of multiple acid groups reduced both enzyme and cell activity. The phosphonic acids were worse UPPS/UPPP inhibitors and were mostly inactive in cells. There were good correlations between cell growth inhibition and UPPS inhibition, as well as between cell growth inhibition and UPPP inhibition, but UPPS inhibition was more potent. Overall, the results are of interest since we show for the first time that lipophilic benzoic acids inhibit both UPPP as well as UPPS and are active in cells where they act synergistically with known cell wall biosynthesis inhibitors and as such, they may represent new leads for developing bacterial cell wall biosynthesis inhibitors.
Experimental Section
Chemical Syntheses: General Methods
All chemicals were reagent grade. 1H NMR and 13C NMR spectra were obtained on Varian (Palo Alto, CA) Unity spectrometers at 400 and 500 MHz for 1H. HPLC/MS analyses were performed by using an Agilent LC/MSD Trap XCT Plus system (Agilent Technologies, Santa Clara, CA) with an 1100 series HPLC system including a degasser, an autosampler, a binary pump, and a multiple wavelength detector. Purity was determined by HPLC-MS and structures were characterized by 1H NMR and HRMS. We synthesized 26 benzoic acids and 4 phenylphosphonates using the general methods shown in Scheme 1a, 1b. Full synthesis and characterization detail are in the Supporting Information and details of the synthesis of two representative compounds are shown below.
Scheme 1.

General synthesis methods. a) for benzoic acids; b) for phenylphosphonates.
5-Fluoro-2-(3-(octyloxy)benzamido)benzoic acid (7) The scheme used to synthesize 7 is as shown in Scheme 1a. 1-Bromooctane b (7.7 g, 40 mmol) was added to a solution of methyl 3-hydroxybenzoate a (3.0 g, 20 mmol) in 20 mL DMF followed by addition of potassium carbonate (5.5 g, 20 mmol). The solution was heated to 80°C and stirred for 12 h. After cooling to room temperature, the solution was washed with water (100 mL), then extracted with ethyl acetate (50 mL ×3). The organic layer was dried over Na2SO4 and solvent removed under vacuum. The residue was purified by flash chromatography on silica gel (hexane/EtOAc = 20:1) to give methyl 3-(octyloxy)benzoate c as a colorless oil (4.8 g, 90%). Compound c (4.0 g, 15 mmol) was dissolved in 20 mL of THF, and aqueous LiOH (1.8 g in 10 mL water) added. The reaction mixture was stirred at room temperature for 12 h, then concentrated under vacuum to remove solvent. The aqueous solution was acidified with HCl to pH 1 upon which a white solid precipitated. The suspension was extracted with ethyl acetate (30 mL ×3). The organic layer was dried over Na2SO4 and solvent removed under vacuum. The 3-(octyloxy)benzoic acid d was obtained as a white solid (2.5 g, 95%). Compound d (2.5 g, 10 mmol) was dissolved in 20 mL of CH2Cl2, and 2 mL of oxalyl chloride added. Then, one drop of DMF was added as a catalyst. The reaction mixture was stirred at room temperature for 12 h, then concentrated under vacuum. The residue 3-(octyloxy)benzoyl chloride e was obtained as a yellow liquid (3.4 g, 90%). Compound e (2.7 g, 10 mmol) was dissolved in 20 mL of CH2Cl2, then 2-amino-5-fluorobenzoic acid f (1.6 g, 10 mmol) added. After stirring for 5 min, 2 mL of NEt3 was added. The reaction mixture was stirred at room temperature for 12 h then washed with 2 M HCl (30 mL ×2) and water (30 mL). The residue was concentrated under vacuum to give the final product g (7) as a light yellow solid (3.0 g, 80%).1H NMR (DMSO-d6, 500 MHz) δ (ppm): 8.66 (dd, J =5, 9.5 Hz, 1 H), 7.75 (d, J =9.5 Hz, 1 H), 7.54 (t, J =9.5 Hz, 1 H), 7.48 (m, 3 H), 7.19 (d, J =8.0 Hz, 1 H), 4.03 (t, J =6.5 Hz, 2 H), 1.72 (m, 2 H), 1.41 (m, 2 H), 1.26–1.22 (m, 8 H), 0.84 (t, J =7.0 Hz, 3 H). 13C NMR (125 MHz): d 168.8, 164.3, 158.9, 156.9 (d, J = 240 Hz), 137.4, 135.7, 130.1, 122.1 (d, J = 7.6 Hz), 121.0 (d, J = 22.0 Hz), 118.9, 118.7 (d, J = 6.5 Hz), 118.3, 117.0 (d, J = 23.9 Hz), 112.1, 67.6, 31.2, 28.7, 28.6, 28.5, 25.4, 22.0, 13.9. ESI HRMS: m/z [M+H]+ calculated for C22H27FNO4+: 388.1924, found: 388.1924. Purity of the product determined by HPLC (Phenomenex C6-Phenyl 110A. 100×2 mm, 3 μm, 250 nm, retention time = 8.1 min): 99.7%
2-(N-Methyl-3-(octyloxy)benzamido)phenylphosphonic acid (31) The scheme used to synthesize 31 is as shown in Scheme 1b. Compound e (540 mg, 2.0 mmol) was dissolved in 20 mL of CH2Cl2 and then diethyl(2-aminophenyl)phosphonate h (460 mg, 2.0 mmol) was added. After stirring for 5 min, 2 mL of NEt3 was added. The reaction mixture was stirred at room temperature for 12 h then washed with 2 M HCl (30 mL ×2) and water (30 mL). The residue was concentrated under vacuum to give compound i as a white solid (780 mg, 85%). Compound i (460 mg, 1 mmol) was dissolved in dry CH2Cl2 (15 mL), cooled to 0°C and Me3SiBr (1.2 mL, 9 mmol) added drop-wise over 30 min. The mixture was then stirred for 2 d at room temperature. The solvent was evaporated and the residue dried in vacuum for approximately 1 h. Then, 20 mL dry methanol was added and the mixture stirred for 20 min at room temperature. The solvent was removed and the residue dried overnight to give the final product j (31) as a white solid (390 mg, 95%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 12.14 (s, 1 H), 8.64 (dd, J =4.5, 8.5 Hz, 1 H), 7.65 (dd, J =7.5, 14.5 Hz, 1 H), 7.59 (d, J =7.5 Hz, 1 H), 7.54 (m, 2 H), 7.44 (t, J =8.0 Hz, 1 H), 7.16 (m, 2 H), 4.03 (t, J =6.5 Hz, 2 H), 1.73 (m, 2 H), 1.42 (m, 2 H), 1.26–1.22 (m, 12 H), 0.85 (t, J =7.0 Hz, 3 H). ESI HRMS: m/z [M-H]− calculated for C21H27NO5P−: 404.1627, found: 404.1622. Purity of the product determined by HPLC (Phenomenex C6-Phenyl 110A. 100×2 mm, 3 μm, 250 nm, retention time = 7.3 min): 93.1%.
B. subtilis growth inhibition assay
ED50 values for B. subtilis cell growth inhibition were determined using a microdilution method. A stationary overnight starter culture of B. subtilis (Bacillus subtilis subsp. subtilis (Ehrenberg) Cohn ATCC 6051) was diluted 1000-fold and grown to an OD600 of ~0.3. This log-phase culture was again diluted 500-fold into fresh LB broth to generate the “working solution”. 200 μL of working solution was transferred into each well of a 96-well culture plate (Corning 3370). Inhibitors were then added at 1 mM and sequentially diluted 3× to 46 nM, keeping volume and culture broth composition constant. Plates were incubated for 12 hours at 37°C, shaking at 200 RPM, then absorbance at 600 nm was measured to assess bacterial cell growth. ED50 values were determined using nonlinear regression whereas minimum inhibitory concentration (MIC) values for each antibiotic and 7 in the synergy assays were calculated by using a Gompertz function in Prism 5 (GraphPad Software, Inc, La Jolla, CA).
S. aureus growth inhibition assay
As with the B. subtilis growth inhibition assay, an overnight starter culture of S. aureus (Newman strain) was diluted 1000-fold to create a “working solution”. Working solutions were transferred into flat-bottom 96-well plates and inhibitors added at 1 mM and sequentially diluted 3× to 46 nM. Plates were incubated at 37°C, shaking at 200 RPM for 24 hours. The OD600 was then measured to determine bacterial growth inhibition.
H460 cell toxicity
A broth microdilution method was used to determine the growth inhibition ED50 values. Briefly, ~ 104 NCI-H460 cells suspended in 100 μL of DMEM supplemented with 10% fetal bovine serum (FBS), 4.5 g/L glucose and L-glutamine and preserved with 1% penicillin–streptomycin were seeded in 96-well plates (Corning Inc., Corning, NY) and incubated at 37 °C in a 5% CO2 atmosphere. The cells were cultured for 24 h then incubated with different concentrations of compounds for another 24 h. Then, an MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cell proliferation assay (ATCC, Manassas, VA) was performed. Data from 4 experiments for each inhibitor were pooled and then fitted to single dose-response curves. The reported values are thus +/− SEM (n=4).
Synergy/Antagonism Assays
In order to investigate possible synergistic interactions between compound 7 and a range of antibiotics, we carried out two-drug combination assays. Bacterial cells were incubated with a 3× gradient of antibiotic typically ranging from 40 μg/mL to 18 ng/mL (200 μg/mL to 90 ng/mL for bacitracin, fosfomycin, and sulfamethoxazole) in the presence half-MIC concentrations of 7, in addition to a 3× gradient of 7 ranging from 40 μg/mL to 18 ng/mL in the presence of half-MIC concentrations of each antibiotic. New MIC values were calculated by using a Gompertz function in Prism 5 (GraphPad Software, Inc, La Jolla, CA).
Enzyme Inhibition Assays
SaUPPS and EcUPPP were expressed and purified as described previously.[3b, 9] UPPS assays were carried out using a phosphate release assay.[3b] Benzoic acid and phosphonic acid derivatives were prepared as 10 mM stock solutions in DMSO, and were then serially diluted from 1 mM to 1 nM. Inhibitors were incubated with 25 ng of SaUPPS at room temperature for 10 minutes in a pH 7.5 buffer (50 mM HEPES, 150 mM NaCl, 10 mM MgCl2, and 0.02% n-dodecyl-β-D-maltopyranoside) before adding “reaction mixture” containing 5 μM FPP, 50 μM IPP, 3 U/mL purine nucleoside phosphorylase, 1 U/mL inorganic phosphatase, and ~600 μM 7-methyl-6-thioguanosine (MESG), again in the same buffer. Reactions were monitored for 15 minutes with the rate of increase in absorbance at 360 nm taken as the rate of UPP synthesis. IC50 values were calculated by using Prism 5 (GraphPad Software, Inc, La Jolla, CA). The UPPP inhibition assay was carried out using a malachite-green reagent as described previously.[12] The same 10 mM inhibitor stock solutions and assay buffer as for the SaUPPS assays were used to test for UPPP inhibition. Inhibitors were incubated with 20 nM EcUPPP at room temperature for 15 minutes before adding FPP to 35 μM. Reaction mixtures were incubated at 37°C for 20 minutes, then quenched by adding 30 μL of malachite-green reagent. In this assay, the phosphate released from FPP reacts with ammonium molybdate to form phosphomolybdate (yellow) which then forms a complex (λmax ~ 620 nm) with malachite-green, used to assess phosphatase activity. Phosphate release was measured at 620 nm and quantified based on a phosphate standard curve, and the OD620 values used to construct dose-response curves.
Supplementary Material
Acknowledgments
This work was supported by the United States Public Health Service (NIH grants CA158191 and GM065307), a Harriet A. Harlin Professorship, the University of Illinois Foundation/Oldfield Research Fund, and the National Natural Science Foundation of China (grants 31200053, 31300615, 31400678 and 31470240).
Footnotes
Supporting information for this article is available on the WWW under XXXX; enzyme and cell growth inhibition Tables and graphs; isobolograms; synthesis and characterization; HPLC purity results; 1H NMR spectra of inhibitor compounds.
References
- 1.Antibiotic resistance threats in the United States. Executive Summary. 2013 http://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf.
- 2.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 3.a) Peukert S, Sun Y, Zhang R, Hurley B, Sabio M, Shen X, Gray C, Dzink-Fox J, Tao J, Cebula R, Wattanasin SM. Bioorg Med Chem Lett. 2008;18:1840–1844. doi: 10.1016/j.bmcl.2008.02.009. [DOI] [PubMed] [Google Scholar]; b) Zhu W, Zhang Y, Sinko W, Hensler ME, Olson J, Molohon KJ, Lindert S, Cao R, Li K, Wang KM. Proc Natl Acad Sci USA. 2013;110:123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Czarny TL, Brown ED. ACS Infectious Diseases. 2016 doi: 10.1021/acsinfecdis.6b00044. [DOI] [PubMed] [Google Scholar]
- 4.Jahnke W, Rondeau JM, Cotesta S, Marzinzik A, Pelle X, Geiser M, Strauss A, Gotte M, Bitsch F, Hemmig R, Henry C, Lehmann S, Glickman JF, Roddy TP, Stout SJ, Grenn JR. Nat Chem Biol. 2010;6:660–666. doi: 10.1038/nchembio.421. [DOI] [PubMed] [Google Scholar]
- 5.a) Eliopoulos GM, Moellering RC. In: Antibiotics in Laboratory Medicine. Lorian V, editor. Williams & Wilkins Publishing Co; 1998. pp. 330–396. [Google Scholar]; b) Singh PK, Tack BF, McCray PB, Welsh MJM. Am J Physiol Lung Cell Mol Physiol. 2000;279:L799–805. doi: 10.1152/ajplung.2000.279.5.L799. [DOI] [PubMed] [Google Scholar]
- 6.European Committee for Antimicrobial Susceptibility Testing (EUCAST) Clin Microbiol Infect. 2000;6:503–508. doi: 10.1111/j.1469-0691.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 7.Berenbaum MC. Pharmacol Rev. 1989;41:93–141. [PubMed] [Google Scholar]
- 8.Hsu MF, Yu TF, Chou CC, Fu HY, Yang CS, Wang AH. PLoS One. 2013;8:e56363. doi: 10.1371/journal.pone.0056363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang HY, Chou CC, Hsu MF, Wang AH. J Biol Chem. 2014;289:18719–18735. doi: 10.1074/jbc.M114.575076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng X, Zhu W, Shurig-Briccio LA, Lindert S, Shoen C, Hitchings R, Li J, Wang Y, Baig N, Zhou T, Kim BK, Cric DC, Cynamon M, McCammon JA, Gennis RB, Oldfield E. Proc Natl Acad Sci USA. 2015;112:E7H073–E7082. doi: 10.1073/pnas.1521988112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukkamala D, No JH, Cass LM, Chang TK, Oldfield E. J Med Chem. 2008;51:7827–7833. doi: 10.1021/jm8009074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baykov AA, Evtushenko OA, Avaeva SM. Anal Biochem. 1988;171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.