

Abstract
Testing the efficacy of cancer drugs requires functional assays that recapitulate the cell populations, anatomy and biological responses of human tumors. Although current animal models and in vitro cell culture platforms are informative, they have significant shortcomings. Mouse models can reproduce tissue-level and systemic responses to tumor growth and treatments observed in humans, but xenografts from patients often do not grow, or require months to develop. On the other hand, current in vitro assays are useful for studying the molecular bases of tumorigenesis or drug activity, but often lack the appropriate in vivo cell heterogeneity and natural microenvironment. Therefore, there is a need for novel tools that allow rapid analysis of patient-derived tumors in a robust and representative microenvironment. We have developed methodology for maintaining harvested tumor tissue in vitro by placing them in a support bed with self-assembled stroma and vasculature. The harvested biopsy or tumor explant integrates with the stromal bed and vasculature, providing the correct extracellular matrix (collagen I, IV, fibronectin), associated stromal cells, and a lumenized vessel network. Our system provides a new tool that will allow ex vivo drug-screening and can be adapted for the guidance of patient-specific therapeutic strategies.
Keywords: tumor microenvironment, self-assembly, vascular network, extracellular matrix, tumor explant
Graphical Abstract
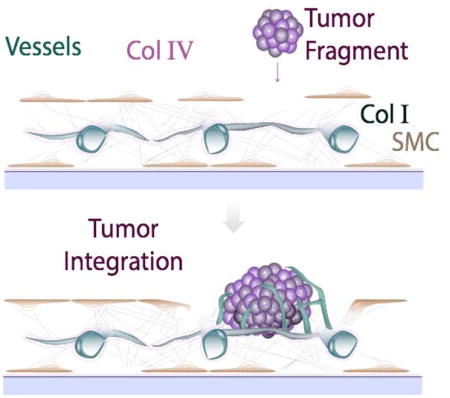
Integration of tumor biopsies into a pre-formed vascularized supporting bed provides a new tool for studying tumor tissue ex vivo.
1. Introduction
The inconsistent ability of cancer assay systems to predict clinical success has generated substantial interest in developing human-based tissue-mimetic constructs for disease modeling and drug testing also referred to as “organs-on-a-chip”1, 2. These organs-on-a-chip are conducive to repetitive, systematic and quantitative investigation of cell and tissue physiology for the purpose of drug discovery and development3–5. Clinical oncology is in great need of such tools to improve the reliability of drug screening and applicability of personalized medicine.
Many approaches have been developed to deconstruct tumors or synthesize them in culture for the purpose of analyzing cancer biology and identifying promising drugs. Cultures of cancer cell lines or circulating cancer cells (CTCs) in 2-D or as spheroids are valuable tools to identify target cancer cell pathways, but are suboptimal to predict clinical response, as they lack important interactions between tumor components. Xenografting human tumors in immunodeficient mice – whether from stable cell lines or newly established patient-derived tumors – provides a better model to dissect tumor biology, tumor-stroma interactions and treatment response. A recent thrust to grow patient-derived xenograft (PDX) tumors in mice for the purpose of drug testing is promising, but this approach necessitates that the original human tumor be distorted, anatomically and potentially biologically, as it is passaged through immunodeficient mice, often in ectopic (subcutaneous) locations. In addition, it can take many months to form a PDX from a patient tumor6, 7. Treatment decisions need to be made much more rapidly than this.
Any effort to synthesize tumors in vitro will be faced with a crucial challenge of reproducing the correct composition and architecture of a tumor. Table 1 summarizes the advantages and challenges of current cell culture systems used to study solid tumors in vitro. Tumor growth and response to treatment are shaped by the interactions with host tissue-derived stroma. In order to re-create the correct anatomy – and thus, a truly biomimetic tumor – the cancer and the host tissue have to co-evolve naturally. Only in this way will the correct mixture and organization of host macrophages, fibroblasts, stromal matrix, blood vessels, immune cells and cancer cells be produced. For these reasons, we explored the possibility of using ex vivo tumor tissue directly rather than using PDX-derived cells or synthesizing tumors ab initio. The challenge was to maintain the excised tumor integrity and biology so that it could be used for drug testing.
Table 1.
Biomimetic approaches to engineering tumors
| Advantages | Disadvantages | ||
|---|---|---|---|
| Single and multicellular tumor spheroids |
Inch et al. (1970)8 Sutherland (1988)9 Helmlinger et al. (1997)10 Hirschhaeuser et al. (2010)11 Kondo et al. (2011)12 La Barbera et al. (2012)13 Seano et al. (2013)14 |
|
|
| Tumor organoids | Cheung et al. (2013)15 |
|
|
| Huang et al. (2015)16 |
|
|
|
| Li et al. (2014)17 |
|
|
|
| Ex vivo tumor slice cultures |
Merz et al. (2013)18 Gerlach et al. (2014)19 Chadwick et al. (2015)20 |
|
|
| Cell sheet technology | Kushida et al. (1999)21 |
|
|
| Microfluidic models | Ehsan et al. (2014)22 |
|
|
| Bioreactors | Ferranini et al (2013)23 |
|
|
| Domansky et al. (2010)24 |
|
|
|
| Self-assembly | Bazou et al. |
|
|
To create vascularized tissues however in vitro, we first need to reproduce the conditions for the assembly of vascular structures. Proper vascular development requires cooperation between endothelial cells (ECs) and the perivascular cells that participate in vessel formation and stabilization. Endothelial cell - smooth muscle cell (SMC) communication begins early in embryogenesis as blood vessels begin to form and continues during vessel maturation. Vascular development is regulated by extrinsic factors, such as growth factor gradients and tissue hypoxia, but the signaling between ECs and SMCs at these earlier stages is essential for vessel formation and ultimately proper function25.
It has been shown that endothelial cells will create vascular structures in vitro if the growth media26, 27, cell populations28–32 and supporting 3D matrix are appropriate27, 33. However, the creation of vessels in vitro is not robust (i.e. they form inconsistently or regress within days), and we have no mechanistic explanation for why endothelial morphogenesis is facilitated in some protocols but not others. Furthermore, it is not known how faithfully these various protocols mimic the process of vessel formation in vivo. Using mouse models in which blood vessels express GFP (driven by the TIE2 promoter) and aSMA-expressing cells produce DsRed, it is possible to follow the cellular dynamics that enable vascular expansion. Observing tumor vascularization through transparent windows, the extension of the existing vasculature requires: 1) mobilization of aSMA-expressing cells on a laminar structure and 2) the creation and assembly of ECM by the SMCs. This ECM then provides the structure into which the nascent vessels form (Fig. 1a).
Fig. 1.

Vascular network formation in vivo and in vitro. a) Intravital microscopy of vascularization on a silicone elastomer implanted in a transparent window of the mouse. Vascular sprouts (green) enter the system from left to right, preceded by α-SMA positive cells and the extracellular matrix they produce (blue). In this system the α-SMA cells exist in a layer that is distinct from the collagen and vessels. Scale bar, 50 μm. b) Recreating vascularization in vitro. Co-culturing HUVECs (green, CD31-AlexaFluor488 labeled) with SMCs (not shown here for clarity), the system self-assembles into a stable, interconnected network over 21 days (shown are images from days 1, 3, 7, 10, 14 and 21). Scale bar, 200 μm. c) Co-cultures (shown at Day 7 in culture) form a multi-layered tissue where SMCs are seen underneath as well as above the vessel network. HUVECs are shown in green (CD31-AlexaFluor488 labeled) while the F-actin of both cell populations is stained with Phalloidin-568 (red). Nuclear stain is DAPI (blue). Scale bar is 20 μm. d) Side view of a confocal stack showing the resulting multilayered tissue, consisting of cells and matrix that has a thickness of approximately 150 μm. e) 3-D reconstruction of part of an in vitro network. Scale bar, 50 μm. f) The network is complete, and spans across the entire well of the 96-well plate. Scale bar, 800 μm. g) qRT-PCR array for extracellular matrix and angiogenesis genes comparing Day 10 over Day1 co-cultures. Up-regulated genes are shown in red while down-regulated genes are shown in green.
In this study we develop vascularized tissue beds for supporting tumor explants. The stromal matrix and vasculature self-assemble, and the tissue is stable for more than 3 weeks. We then show that tumor tissue harvested from mice and added to the vessel bed integrates into the preformed tissue. The resulting “vascularized tumor explants” (VTEs) provide a new, unique tool with potential for high take rate and rapid turn-around time for drug screening and personalized medicine.
2. Experimental
2.1 Cell Culture
Human umbilical vein endothelial cells (HUVECs) were acquired from the Centre for Excellence in Vascular Biology, Brigham & Women’s Hospital, Harvard Medical School, Boston, MA and maintained at 37°C under an atmosphere of 95 % air and 5 % CO2 in EGM medium (Lonza). HUVECs (passage number 1–5) were used non-labeled or were stably transfected with green fluorescent protein (GFP) as previously described34. Pulmonary artery smooth muscle cells (SMCs) were purchased from Lonza and were maintained at 37°C/5% CO2 in SmGM-2 medium (Lonza). The growth media was changed every third day for both cell lines.
2.2 Co-culturing Endothelial cells (ECs) and SMCs
ECs and SMCs were seeded together, mixed briefly to achieve an even cell distribution and cultured in EGM-2 (Lonza). The cell mixture was cultured in 96-well plates at concentrations of 104 (ECs) and 5×104 (SMCs). Co-cultures were monitored for up to 14–21 days; during observation, the culture media was changed every third day.
2.3 Tumor explant preparation and vascularized tumor explant (VTE) culture
The Massachusetts General Hospital Subcommittee on Research Animal Care approved all mouse experiments. Human melanoma (Mu89) tumors were grown orthotopically in 8-week-old severe combined immunodeficient (SCID) females while human breast (BT474) tumors were grown in the mammary fat pad of female nude mice. Tumors were grown in the mice until they reached 4–5 mm diameter; tumors were then resected and cut into explants for addition into the co-culture wells. Incorporation of the tumor explants took place at Day 1 of vasculature network formation (one explant per well), unless otherwise stated. Explant size varied between 0.2 – 0.5 mm. VTEs (i.e. tumor explants with the vascular network) were monitored for up to 10 days with the culture media being changed every third day. All experiments were performed in triplicate with at least three individual explants generated from each biological replicate.
2.4 Immunofluorescence staining of ECs-SMCs co-cultures
SMC-EC co-cultures at Day 1, 3, 7, 10 and 14 in culture, were initially washed with Phosphate Buffered Saline (PBS) to remove any cell media, fixed with 4% formaldehyde for 20 min, washed three times with PBS and subsequently permeabilized with 0.5% Triton in PBS for 10 min at 4°C. Co-cultures were then rinsed three time with PBS-Glycine followed by serum blockade (5% donkey serum/0.1% Triton in PBS for 60 min). Co-cultures were labeled with CD31 (Ready to use, DAKO), α-SMA (1:500, Sigma), Collagen I (1:500, AbCAM), Collagen IV (1:10, Millipore), Fibronectin (1:500, AbCAM) overnight at 4°C and Phalloidin-AlexaFluor 568 (Thermofisher) for 30 min at room temperature. Appropriate fluorescently labeled secondary antibodies were applied for 60 min and washed three times with saline. Cell nuclei were stained with DAPI nuclear stain (Invitrogen, 1:200) and washed again three times with saline prior to confocal microscopy. Cultures containing VTEs were analyzed after 10 days in culture. They were fixed, permeabilized and serum-blocked as described above for the EC-SMC co-cultures. They were then labeled with CD31 (1:100, Millipore), Tenascin-C (1:250, R&D systems) and α-SMA (1:500, Sigma) Appropriate fluorescently labeled secondary antibodies were applied for 60 min and washed three times with saline. Cell nuclei were stained with DAPI nuclear stain (Invitrogen, 1:200) and washed three more times with saline prior to confocal microscopy.
2.5 ELISAs for growth factors
Growth factor levels were quantified using ELISA kits (Qiantikine®, R & D Systems). Culture media was collected from Day 1, 3, 7, 10 and 14 cultures and assayed for Vascular Endothelial Growth Factor (VEGF), soluble Vascular Endothelial Growth Factor Receptor 1 (s-FLT1) and Placental Growth Factor (PLGF). Growth factor levels in the Mu89 VTE cultures were quantified using the same ELISA kits. Culture media was collected from days 3 and 10 in culture and also assayed for VEGF, s-FLT1 and PLGF. All experiments were performed in triplicate.
2.6 Image acquisition
Phase contrast and corresponding fluorescence images were acquired with an epi-fluorescence microscope (Olympus IX70, 4× and 10× air lens, PRIOR automated stage, OpenLab software). Confocal fluorescence images were acquired with an Olympus IX81 microscope (20× air lens and 60× oil lens) equipped with the Fluoview software. Slice thickness varied between 1 and 5 μm. Projections of confocal images were produced using Image J (NIH, Bethesda, MA). Multiple-field mosaic images were acquired using a confocal Olympus IX81 microscope with a slice thickness of 2 μm.
2.7 Fabrication of PDMS wells for high-resolution imaging of co-cultures
To image using a 60× oil emersion lens we fabricated customized wells on a 150 mm tissue culture dish using poly-(dimethylsiloxane) PDMS (Sylgard 184, Dow Corning). PDMS was mixed with the curing agent in a 12:1 ratio respectively, degased and poured into a 100 mm tissue culture dish that was then placed at a 60°C oven for 3 h. After curing, the PDMS slab was peeled and multiple wells, 7 mm in diameter each, were created using a biopsy punch. The slab was then cleaned and washed using Milli-Q water and dried for 1 h in the oven. The dry PDMS slab was then placed on a 150 mm tissue culture dish and sterilized using a Mini 209 UV light sterilizer for 15–20min. ECs and SMCs were seeded in the wells defined by the PDMS at the same ratio and cell density as our co-cultures in 96-well plates. Samples were fixed at Day 7 in culture and stained as described above. Post staining, the PDMS slab was removed leaving the co-culture tissue on the surface. One drop of ProLong® Gold Antifade Mountant with Dapi was place on each tissue before it was covered with thin coverslip and sealed using nail polish. The walls of the tissue culture dish were removed and the dish was placed upside down on the confocal microscope for imaging.
2.8 Image quantification
Anisotropy measurements
Anisotropy measurements were conducted using the Image J plug-in FibrilTool, as previously described by Boudaoud et al.35. Triplicate co-cultures were analyzed (3 images were analyzed per repeat, i.e. 9 images in total per group –control and treated).
Vesiculo-vacuolar organelle (VVO) quantification
Vesiculo-vacuolar organelles were identified as intracellular spherical structures that lacked GFP but were surrounded by the cytoplasmic GFP signal; they were observed either completely within the cytoplasm or associated with the border of a neighboring cell. The number of VVOs in each image was normalized to the cell area of the corresponding image as calculated by image analysis in ImageJ. Three different fields (3 images were analyzed per repeat, i.e. 9 images in total) were analyzed for each time point.
ECM quantification
ECM deposition in the co-cultures was quantified by manually thresholding the corresponding images to include the Collagen I, IV and fibronectin-positive signals. Images (triplicate co-cultures with 2 images randomly selected from each repeat were analyzed, i.e. 6 images in total) were then converted to binary and the % area fraction was quantified using the ImageJ software.
VTE area measurements
VTE area measurements were extracted by manually defining the tumor boundary in the brightfield image (using ImageJ). Nine and five VTEs from three different mice were analyzed for the Mu89 and BT474 tumors respectively.
2.9 Statistical analysis
The data are shown as mean ± SEM. Analysis of means was performed with a two-tailed two sample t-test or one-way analysis of variance (ANOVA) (GraphPad Prism software, San Diego, CA, USA). Differences were considered significant at P values less than 0.05. ****: p < 0.00005; ***: p < 0.0005; **: p < 0.005; *: p < 0.05.
3. Results and discussion
3.1 Forming the vascular bed
Our strategy was to create a vascularized tissue bed for the tumor explant. This tissue should supply angiocrine factors that are important in the growth and progression of solid cancers36, 37, thus, creating a more native environment. To create the tissue, we drew upon observations made in animal models of neovascularization. Using mice in which blood vessels express GFP (driven by the TIE2 promoter) and αSMA-expressing cells produce DsRed, it is possible to follow the cellular dynamics that enable vascular expansion using intravital microscopy. In previous studies, we found that the extension of the existing vasculature requires mobilization of αSMA-expressing cells and the assembly of extracellular matrix by the SMCs. This ECM then provides the structure into which the nascent vessels form (Fig. 1a)38.
Inspired by these observations in vivo, we cultured ECs together with SMCs in an attempt to biomimetically reproduce this process in vitro. By culturing these cell populations in 96-well plates in the correct ratio (1:5, ECs: SMCs), we were able to reproduce lumenized vessel networks similar to those created in the mouse models of angiogenesis. The ECs undergo morphogenesis under these conditions, and produce a basement membrane. In this system, there is no exogenous matrix added – the ECM is created by the cells.
Within the first 24 h (Day 1) in culture, individual ECs migrate, elongate and begin connecting with neighboring ECs (Fig. 1b, shown in green). Vessels grow in a matrix produced by –and structurally contiguous with– the SMCs, giving rise to a multilayered tissue where SMCs can be seen underneath as well as above the vessel network (Fig. 1c and ESI, SVideo 1). The resulting multilayered tissue, consisting of cells and matrix, has a thickness of approximately 150 μm (Fig. 1d).
A complete lumenized network is formed within 14-days (Fig. 1e and ESI, SVideo 2). The networks are stable for at least 21 days and span the well of the 96-well plate (Fig. 1f). The formation of a stable network is accompanied by the up-regulation of several genes including growth factors (Angiopoietin 1) matrix components (Collagens 8, 14, 16, 18), and adhesion molecules (ICAM1, VCAM1 and Integrins α4, 6, 7, V and β2, 3, 4) (Fig. 1g).
The formation of the vascular network is greatly affected by the ratio of ECs to SMCs. To determine the optimum ratio, we maintained the overall number of cells/well the same (i.e. 6×104) while changing the ratio of ECs to SMCs (Fig. 2a). Increasing the number of ECs and decreasing the number of SMCs resulted in patches of ECs (arrows), which did not participate in the vessel network. As expected, both ECs and SMCs form monolayers when cultured alone (6:0 and 0:6, respectively). In addition, seeding ECs (104) 24 h before SMCs (5×104) did not significantly affect vascular network formation (Fig. 2b), while seeding SMCs 24 hours prior to ECs resulted in a significant reduction in the vascularization (Fig. 2b). In the latter case the tissue also contracted. Taken together, this data confirm that co-seeding ECs and SMCs presents the optimum topology/configuration for vascular network formation.
Fig. 2.

Development of vascular networks. a) Micrographs illustrating the resultant vascular network when the ratio of ECs to SMCs changes. Vessels are shown in green. Scale bar is 50 μm. b) Live imaging of vascular network formation at Day 3 in culture following different seeding conditions. ECs are shown in green, while for reasons of clarity SMCs are not labelled. Scale bar, 1 mm. c) Vessels that form in the EC-SMC co-culture contain contiguous lumens (arrows) that extend throughout the network (three different examples are shown). Scale bar, 30 μm. d) Lumen formation by vesicle fusion. At Day 1 VVO structures (arrows) develop within individual endothelial cells. Significantly more VVOs were observed on Day 3; by Day 7, extensive fusion of these structures creates the lumens. Three different image fields are shown for each time point. e) VVO quantification shows that the number of individual vesicles was highest on day 3; after this, fusion reduced the number on distinct structures. Scale bar, 30 μm.
The self-assembled vessel networks contain lumen structures (Fig. 2c, arrows) created by fusion of intracellular vesicles. Structures resembling vesiculo-vacuolar organelles (VVOs)39 were observed at Day 1 (Fig. 2d-arrows, e) and became more abundant at Day 3 (Fig. 2d-arrows, e). By Day 7 (Fig. 2d-arrows, e) most had merged together – both within individual cells, and between adjacent cells– to form the lumens.
Because vasculature is usually supported by matrix structures, we next investigated whether EC-SMC co-cultures produce their own basement membrane and extracellular matrix. Collagen IV staining co-localized with the vessel network by Day 3 (Fig. 3a, top row and ESI, SFig. 1), becoming more pronounced on Day 10. Some collagen IV structures did not co-localize with ECs (Fig. 3a, red arrows), suggesting that vessel structures occasionally recede from the basement membrane sleeves. Collagen I was also produced with a similar peri-endothelial localization pattern (Fig. 3a, middle row and ESI, SFig. 2). Fibronectin was also abundantly produced (Fig. 3a, bottom row and ESI, SFig. 3). Expression and deposition of collagen I and fibronectin (Fig. 3b) increased significantly between Day 1 and 3 in culture, consistent with the active vasculogenesis in this time period.
Fig. 3.

Co-cultures define their own microenvironment. a) Top row: Confocal immunofluorescence analysis revealed significant collagen IV (grey) staining around the vessel network (green) at Day 3. By Day 10 collagen IV had completely ensheathed the vessel network. Middle row: Collagen I (grey) was also produced and showed a similar peri-endothelial localization pattern in co-culture. Bottom row: Fibronectin (grey) was also abundantly produced by the co-cultures. Scale bar, 50 μm. b) Collagen I, collagen IV and fibronectin levels increased over time in EC-SMC co-cultures (assessed by the fractional area of staining in the IHC images). Error bars are standard errors from n = 3. c) ELISA results show a significant increase in VEGF levels at Day 3. s-FLT1 and PLGF were more constant. d) Top row: collective alignment of the SMCs was associated with vessel network formation and is apparent in the bright field image, At right: brightfield only; at left: brightfield with overlay of HUVECs (green). Bottom row: In fibronectin-treated wells (FN), SMCs form a non-aligned monolayer, and vessel network formation is minimal (bottom row, superimposed HUVECs-GFP). The SMC alignment is highlighted with red arrows in the last column. Scale bar, 150 μm. e) Measurements of the anisotropy index showed the loss of directionality of the SMCs. Control co-cultures at Day 3 have an anisotropy index of 0.32 ± 0.01, while fibronectin-treated co-cultures have a significantly lower anisotropy index (0.17 ± 0.01).
Finally, the vascular networks also produce essential growth factors required to drive vasculogenesis. ELISA assays revealed that at Day 3 there was a significant accumulation of VEGF (Fig. 3c), coinciding with the time of active network formation. s-FLT1 and PLGF on the other hand, remained at constant levels throughout the 14 days in culture (Fig. 3c).
3.2 Collective alignment of the SMCs is essential for vessel network formation
In cultures that form connected, stable vessel networks, the SMCs collectively align in patterns (Fig. 3d) usually observed above the vessel network (Fig. 1c). We hypothesized that preventing this collective behavior would stop vasculogenesis. Treating the co-cultures with fibronectin sufficiently altered the matrix dynamics so that aligned patterns no longer formed, and indeed, this prevented network formation (Fig. 3d, bottom row, brightfield image). Thus, supplying exogenous fibronectin interferes with the auto-assembly of the structures, even though the cells produce large quantities of the same component on their own (Fig. 3a, bottom row). The change in collective alignment (highlighted with the overlaying red arrows, Fig. 3d) with FN treatment can be quantified using an anisotropy index (Fig. 3e).
3.3 Addition of Exogenous Matrix components or substitution of fibroblasts for SMCs interferes with network formation
Because the formation of vascular structures in our EC-SMC co-cultures appeared to require the production of matrix components, we next tested whether exogenous addition of these components would facilitate the process. Mixing ECs and SMCs with collagen I (3 mg/ml) (Fig. 4a–c) or fibrin (2.5 mg/ml) (Fig. 4d–f) did not result in vessel formation. Collagen I cultures contained very few vessel segments at Day 3 and there was significant contraction of the supplied matrix (Collagen I-cell mass outlined in red- Fig. 4c). Similarly, ECs and SMCs mixed in fibrin formed even fewer vascular structures; fibrin was also significantly contracted by Day 3, although it remained anchored to the surface at some locations (Fig. 4f, arrows). These results show that the cells interact differently with exogenously-added matrix than with endogenously-produced matrix. Furthermore, it appears that the correct topology is necessary for evolution of the system: the cells must attach to the surface, providing structural support for the 3D matrix so that the ECs can then undergo morphogenesis within this matrix layer. Adding a 3D ECM at the start of the culture prevents the evolution of this topology. Further supporting this concept are experiments performed with mesenchymal 10T1/2 fibroblasts rather than the vascular SMCs. We previously used these fibroblasts to support creation of vascular networks in collagen gels implanted into mice. In that context, vessels form and connect to the host vasculature34. However, when the same 10T1/2 fibroblasts are used in our in vitro co-culture system, the correct topology did not occur, and networks did not form. Instead of forming a multi-layered, structurally connected tissue, the cells formed a mosaic monolayer of fibroblasts and endothelial cells (Fig. 4g, h).
Fig. 4.

Addition of Exogenous Matrix components or substitution of fibroblasts for SMCs interferes with network formation. a, b) Mixing ECs and SMCs in collagen I (3 mg/ml) at Day 1 (a) resulted in isolated vessel segments at Day 3 (b). c) Collagen I matrix contraction was observed within 3 days- matrix-ECs-SMCs mass outlined with the red solid line. d) Mixing ECs and SMCs in fibrin (2.5 mg/ml) at Day 1 did not lead to vessel formation as shown in the fluorescent micrograph (e- Day 3). f) Fibrin also significantly contracted by Day 3, showing some anchor points to the underlying substratum (arrows). g) Co-culturing ECs with 10T1/2 cells did not induce vessel formation. ECs (shown in green) formed patches of monolayers within the 10T1/2 monolayer. h) superimposed brightfield and fluorescence micrographs showing the mosaic monolayer on the surface. Scale bar, 150 μm.
3.4 Creating Vascularized Tissue Explants (VTEs)
With the protocol for creating a vascular bed established we next assessed whether the cultured vessel bed would integrate with tumor explants. We first added the tumor explants to the culture together with the ECs and SMCs. In this case (or in the case when the explant was placed directly on plastic), the results were inconsistent. Explants that contact the plastic tend to disperse, as cells from the tumor migrate away on the plastic. When this happened in the co-cultures, cells from the explant were able to displace the vascular bed (Fig. 5a). Consequently, explants that spread on the plastic create an avascular boundary, as the vessels remain associated with the surrounding SMCs-matrix “tissue” (Fig. 5a, corresponding fluorescence micrograph).
Fig. 5.

VTE cultures. a) Mu89 tumors disrupted, with many cells migrating away from the main tumor explant body when co-implanted with ECs-SMCs at Dy 0, as shown in the brightfield image. Vessels (green), in the mosaic confocal micrograph of the same tumor (purple outline), did not penetrate the explant but formed a ‘ring’ around the tumor explant-cancer cell area. b) Vascularization of Mu89 tumor explants (asterisk) in the co-culture system. Vessels (green) surround and penetrated the tumor explants (due to autofluorescence, also shown in green) over 10 days. Scale bar, 150 μm. c) BT474 tumor explants were also successfully vascularized over 10 days. Scale bar, 150 μm. d) Growth curves for VTEs. e) Mu89 explants retained strong levels of tenascin-C (grey) ex-vivo. Scale bar, 100 μm. f) Mu89 VETs released increased levels of VEGF in relation to EC-SMC cultures alone, while, s-FLT1 and PLGF were at similar levels in comparison to EC-SMC cultures alone.
To prevent the explant from contacting the surface, we pre-formed the vascularized support bed and added Mu89 or BT474 tumor explants after one day of culture. In this case, the explants retained their morphology, and the vasculature migrated to surround the tumors over 10 days in culture (Fig. 5b, c). During this time, Mu89 explants grew approximately 40% of their initial size, while no significant size increase was observed for the BT474 tumor explants (Fig. 5d). In culture, tumor explants retained important stromal and immune components. Tenascin-C, an extracellular matrix molecule highly associated with melanoma, was expressed in Mu89 explants (Fig. 5e), and these explants also produced angiogenic growth factors including VEGF, s-FLT1 and PLGF (Fig. 5f). Thus, the tumor explants appear to maintain important properties associated with primary tumors when cultured in this system, while the underlying vascular bed is essential to the integrity of the tumor explants. Mu89 tumors continuously released increased levels of VEGF, while s-FLT1 and PLGF were at similar levels over the 10 day culture period. This may be due to the increase in size of the Mu89 explants, which would presumably lead to an increase in hypoxia and a corresponding upregulation of VEGF. For the Mu89 VTEs, the levels of VEGF were significantly higher than in cultures containing only ECs and SMCs, further indicating that VTEs are actively releasing VEGF.
One of the important findings from this study is that auto-assembly of tissue is enabled only if the cells can establish the correct spatial relationships (Fig. 2a). While it is possible for this to happen in tissue culture plates, it is also easy for the process to be disrupted because cells are attracted to the highly adhesive plastic surface. Furthermore, providing a pre-defined 3D environment may not always work. Our system for forming the vasculature seemed to require three steps: i) the SMCs and ECs cover the surface; ii) The SMCs produce ECM components that polymerize (collagen I and fibronectin), with the collagen I forming a 3D environment for the ECs; iii) The ECs undergo morphogenesis within this matrix, and create a collagen IV basement membrane for the vasculature. SMC collective alignment is likely important for this step. In time-lapse experiments, it was evident that the resulting “tissue” was structurally connected, as collective migration of SMCs resulted in deformation of the EC network multiple cell diameters away (ESI, SVideo 3). Thus, it is likely that the ECM provides the structural integrity (just as it does in vivo), anchored both at the vessel wall and at the SMCs. It is also possible that tension produced in the ECM by the migrating SMCs somehow initiates the EC morphogenesis.
4. Conclusions
We have shown that co-culturing ECs with SMCs leads to the formation of a stable, lumenized vessel network embedded in basement membrane and extracellular matrix, and that the culture produces growth factors required to drive vasculogenesis. The vessel network remains stable for at least 21 days, and can potentially be interfaced with microfluidics technology to perfuse the lumenized vasculature and control nutrient delivery to the tumor40, 41. An application of this methodology is to culture and maintain patient tumors ex vivo for biological studies, drug screening, or to guide personalized treatment regimens42. Even without perfusion, the inclusion of the vasculature in the VTE culture supplies angiocrine factors that are important in tumor development and response to treatment36, 37. This provides a more relevant system for testing not only anti-cancer drugs, but also anti-angiogenic drugs and combination therapies. In this way, the technology will become a valuable tool to improve in vitro assays and complement existing animal studies.
Supplementary Material
Insight, innovation, integration.
This work presents a novel strategy for providing excised tissues with a supporting tissue bed that includes stroma and a microvascular network with morphological and biochemical markers of blood vessels found in vivo, including patent lumens, representative extracellular matrix and growth factor microenvironments. We demonstrate feasibility by vascularizing tumor explants derived from mouse samples. The technology is straightforward, relying on self-assembly of the tissue, and provides a novel tool for analyzing tumor biology and treatment response in an environment more representative than tissue culture assays. The technology can be adapted to high-throughput screening of drugs in a physiologically-relevant context– a capability not previously available.
Acknowledgments
The authors would like to acknowledge funding from the National Institutes of Health (R01HL106584). We thank Vera Verbruggen for generating the vascular bed mosaic image and Julia Kahn for her help with tumor implantations.
References
- 1.Neuzi P, Giselbrecht S, Lange K, Huang TJ, Manz A. Nature reviews Drug discovery. 2012;11:620–632. doi: 10.1038/nrd3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Meer AD, van den Berg A. Integrative biology: quantitative biosciences from nano to macro. 2012;4:461–470. doi: 10.1039/c2ib00176d. [DOI] [PubMed] [Google Scholar]
- 3.Bersini S, Jeon JS, Dubini G, Arrigoni C, Chung S, Charest JL, Moretti M, Kamm RD. Biomaterials. 2014;35:2454–2461. doi: 10.1016/j.biomaterials.2013.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elliott NT, Yuan F. Journal of pharmaceutical sciences. 2011;100:59–74. doi: 10.1002/jps.22257. [DOI] [PubMed] [Google Scholar]
- 5.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:214–219. doi: 10.1073/pnas.1417115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siolas D, Hannon GJ. Cancer research. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Nature reviews Clinical oncology. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inch WR, McCredie JA, Sutherland RM. Growth. 1970;34:271–282. [PubMed] [Google Scholar]
- 9.Sutherland RM. Science. 1988;240:177–184. doi: 10.1126/science.2451290. [DOI] [PubMed] [Google Scholar]
- 10.Helmlinger G, Netti PA, Lichtenbeld HC, Melder RJ, Jain RK. Nature biotechnology. 1997;15:778–783. doi: 10.1038/nbt0897-778. [DOI] [PubMed] [Google Scholar]
- 11.Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA. Journal of biotechnology. 2010;148:3–15. doi: 10.1016/j.jbiotec.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Kondo J, Endo H, Okuyama H, Ishikawa O, Iishi H, Tsujii M, Ohue M, Inoue M. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6235–6240. doi: 10.1073/pnas.1015938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaBarbera DV, Reid BG, Yoo BH. Expert opinion on drug discovery. 2012;7:819–830. doi: 10.1517/17460441.2012.708334. [DOI] [PubMed] [Google Scholar]
- 14.Seano G, Chiaverina G, Gagliardi PA, di Blasio L, Sessa R, Bussolino F, Primo L. Blood. 2013;121:e129–137. doi: 10.1182/blood-2012-08-452292. [DOI] [PubMed] [Google Scholar]
- 15.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Cell. 2013;155:1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC, Arrowsmith C, Kalloger SE, Renouf DJ, Connor AA, Cleary S, Schaeffer DF, Roehrl M, Tsao MS, Gallinger S, Keller G, Muthuswamy SK. Nature medicine. 2015;21:1364–1371. doi: 10.1038/nm.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O, Cantrell MA, Rack PG, Neal JT, Chan CW, Yeung T, Gong X, Yuan J, Wilhelmy J, Robine S, Attardi LD, Plevritis SK, Hung KE, Chen CZ, Ji HP, Kuo CJ. Nature medicine. 2014;20:769–777. doi: 10.1038/nm.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merz F, Gaunitz F, Dehghani F, Renner C, Meixensberger J, Gutenberg A, Giese A, Schopow K, Hellwig C, Schafer M, Bauer M, Stocker H, Taucher-Scholz G, Durante M, Bechmann I. Neuro-oncology. 2013;15:670–681. doi: 10.1093/neuonc/not003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerlach MM, Merz F, Wichmann G, Kubick C, Wittekind C, Lordick F, Dietz A, Bechmann I. British journal of cancer. 2014;110:479–488. doi: 10.1038/bjc.2013.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chadwick EJ, Yang DP, Filbin MG, Mazzola E, Sun Y, Behar O, Pazyra-Murphy MF, Goumnerova L, Ligon KL, Stiles CD, Segal RA. Journal of visualized experiments: JoVE. 2015 doi: 10.3791/53304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kushida A, Yamato M, Konno C, Kikuchi A, Sakurai Y, Okano T. Journal of biomedical materials research. 1999;45:355–362. doi: 10.1002/(sici)1097-4636(19990615)45:4<355::aid-jbm10>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 22.Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CC, George SC. Integrative biology: quantitative biosciences from nano to macro. 2014;6:603–610. doi: 10.1039/c3ib40170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrarini M, Steimberg N, Ponzoni M, Belloni D, Berenzi A, Girlanda S, Caligaris-Cappio F, Mazzoleni G, Ferrero E. PloS one. 2013;8:e71613. doi: 10.1371/journal.pone.0071613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Lab on a chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armulik A, Abramsson A, Betsholtz C. Circulation research. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 26.Berthod F, Germain L, Tremblay N, Auger FA. Journal of cellular physiology. 2006;207:491–498. doi: 10.1002/jcp.20584. [DOI] [PubMed] [Google Scholar]
- 27.Newman AC, Nakatsu MN, Chou W, Gershon PD, Hughes CC. Molecular biology of the cell. 2011;22:3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fillinger MF, Sampson LN, Cronenwett JL, Powell RJ, Wagner RJ. The Journal of surgical research. 1997;67:169–178. doi: 10.1006/jsre.1996.4978. [DOI] [PubMed] [Google Scholar]
- 29.Griffith CK, Miller C, Sainson RC, Calvert JW, Jeon NL, Hughes CC, George SC. Tissue engineering. 2005;11:257–266. doi: 10.1089/ten.2005.11.257. [DOI] [PubMed] [Google Scholar]
- 30.Kim S, Lee H, Chung M, Jeon NL. Lab on a chip. 2013;13:1489–1500. doi: 10.1039/c3lc41320a. [DOI] [PubMed] [Google Scholar]
- 31.Moya ML, Hsu YH, Lee AP, Hughes CC, George SC. Tissue engineering Part C, Methods. 2013;19:730–737. doi: 10.1089/ten.tec.2012.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao RR, Peterson AW, Ceccarelli J, Putnam AJ, Stegemann JP. Angiogenesis. 2012;15:253–264. doi: 10.1007/s10456-012-9257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghajar CM, Chen X, Harris JW, Suresh V, Hughes CC, Jeon NL, Putnam AJ, George SC. Biophysical journal. 2008;94:1930–1941. doi: 10.1529/biophysj.107.120774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng G, Liao S, Kit Wong H, Lacorre DA, di Tomaso E, Au P, Fukumura D, Jain RK, Munn LL. Blood. 2011;118:4740–4749. doi: 10.1182/blood-2011-02-338426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boudaoud A, Burian A, Borowska-Wykret D, Uyttewaal M, Wrzalik R, Kwiatkowska D, Hamant O. Nature protocols. 2014;9:457–463. doi: 10.1038/nprot.2014.024. [DOI] [PubMed] [Google Scholar]
- 36.Brantley-Sieders DM, Dunaway CM, Rao M, Short S, Hwang Y, Gao Y, Li D, Jiang A, Shyr Y, Wu JY, Chen J. Cancer research. 2011;71:976–987. doi: 10.1158/0008-5472.CAN-10-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, Simons M, Tam W, Felsher DW, Shido K, Rafii A, Scandura JM, Rafii S. Cancer cell. 2014;25:350–365. doi: 10.1016/j.ccr.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruionu G, Bazou D, Maimon N, Onita-Lenco M, Gruionu LG, Huang P, Munn LL. Lab on a chip. 2016;16:1840–1851. doi: 10.1039/c6lc00237d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dvorak AM, Feng D. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2001;49:419–432. doi: 10.1177/002215540104900401. [DOI] [PubMed] [Google Scholar]
- 40.Song JW, Bazou D, Munn LL. Integrative biology: quantitative biosciences from nano to macro. 2012;4:857–862. doi: 10.1039/c2ib20061a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song JW, Munn LL. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15342–15347. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boj SF, Hwang CI, Baker LA, Chio, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, Wilson JP, Feigin ME, Ohlund D, Handly-Santana A, Ardito-Abraham CM, Ludwig M, Elyada E, Alagesan B, Biffi G, Yordanov GN, Delcuze B, Creighton B, Wright K, Park Y, Morsink FH, Molenaar IQ, Borel Rinkes IH, Cuppen E, Hao Y, Jin Y, Nijman IJ, Iacobuzio-Donahue C, Leach SD, Pappin DJ, Hammell M, Klimstra DS, Basturk O, Hruban RH, Offerhaus GJ, Vries RG, Clevers H, Tuveson DA. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.