

Abstract
The self-assembly of higher-order anion helicates in solution remains an elusive goal. Here we present the first triple helicate to encapsulate iodide in organic and aqueous media as well as the solid state. The triple helicate self-assembles from three tricationic arylethynyl strands and resembles a tubular anion channel lined with nine halogen bond donors. Eight strong iodine⋯iodide halogen bonds and numerous buried π-surfaces endow the triplex with remarkable stability even at elevated temperatures. We suggest that the natural rise of a single-strand helix renders its linear halogen bond donors non-convergent. Thus, the stringent linearity of halogen bonding is a powerful tool for the synthesis of multi-strand anion helicates.
Keywords: halogen bonding, self-assembly, supramolecular chemistry, helical structures, host-guest systems
Graphical abstract
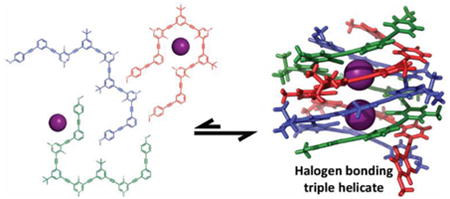
Three tricationic arylethynyl strands self-assemble to form a tubular anion channel lined with nine halogen bond donors in solution and the solid state. Eight strong iodine⋯iodide halogen bonds and numerous buried π-surfaces endow the triplex with remarkable stability even at elevated temperatures. We hypothesize that the stringent linearity of halogen bonding is a powerful tool for the synthesis of multi-strand anion helicates.
The helical folding of a molecule confers extraordinary higher-order structure and function. Examples are rife in nature, ranging from the structural role of collagen to the safeguarding of genetic information in polynucleotides. By implementing this privileged pattern, cation[1] and neutral guest[2] induced helices and foldamers[1e,3] have led to myriad applications, including biomolecular and chiral recognition, supramolecular catalysis, and materials. In contrast, the progression of anion helicates, especially those involving multiple strands, has lagged. This delay is understandable given the complexities of guest induced helical folding, which are magnified by the high solvation energies and variable coordination geometries of anions. To date, a small but growing number of single-strand anion helicates[4] have been synthesized and investigated. However, only four hydrogen bonding (HBing) solid-state[5] and three solution-phase[6] duplexes have been developed. Li et al. have produced the only triplex,[7] which enfolded two trianionic phosphates with HBing bis(biurea) ligands. Only one example of a solid-state halogen bonding (XBing) double helicate exists.[8] Here, we describe an alternative approach to assemble higher-order anion helicates. Exploiting the stringent linearity of XBing, the first triple helicate to bind iodide in solution and the solid state is presented. This cylindrical structure self-assembles from three arylethynyl strands that encircle two iodide anions with XBs. The helix demonstrates remarkable stability at high temperatures and in aqueous and organic solvents. The linearity of XBing facilitates multi-strand complexation and offers a tractable approach to self-assemble large tubular containers with high affinity for complementary anions.
During the last two decades, XBing molecular hosts[9] have evolved with increasing sophistication, while crystallographic,[10] gas-phase,[11] and biomolecular[11a,b,12] investigations have continued to refine our understanding of this emerging noncovalent bond. XBing is an attractive interaction between an electrophilic region of a halogen atom and a nucleophilic region of an atomic or molecular entity.[10g,13] Although analogous to HBing with regard to strength, the XB is far more directional (the angle R–X⋯Y tends to be close to 180°, where X is a halogen, R a covalently bound group, and Y the XB acceptor).
Recently, we synthesized a bidentate XBing receptor (1) that demonstrated notable affinity for perrhenate in solution and the solid state (Scheme 1).[14] Receptor 1 employed two convergent 3-iodopyridinium XB donors extending from a 1,3-diethynylbenzene core. Expanding on this design, we envisioned an oligomer with three 4-iodopyridinium XB donors, spaced by two 1-tert-butyl-3,5-diethynylbenzenes, and capped with two 4-methoxytolans. Design principles were drawn from Moore's seminal work with meta-phenylene ethynylene foldamers[2e,f,3k,l,15] and Flood's elegant chloride encapsulating double helicate[6a] to encourage the favorable π-stacking of alternating electron-deficient and -rich aromatic rings. Our departure from previous work is the strategic placement of inwardly directed XB donors.
Scheme 1.

Synthesis of the bromo- and iodopyridinium nonamers. Reagents and conditions: a) 2, 1-tert-butyl-3,5-diethynylbenzene, PdCl2(PPh3)2, CuI, Et3N, DMF, RT, 12 h, 21%; b) 4-bromo-3,5-diiodopyridine, PdCl2(PPh3)2, CuI, Et3N, DMF, 50 °C, 12 h, 75%; then TBAF, THF, 0 °C to RT, 10 min, quantitative; c) 5, PdCl2(PPh3)2, CuI, Et3N, DMF, 50 °C, 24 h, 61%; then MeOTf, DCM, RT, 12 h, 93% (6); then NaI, 1:3 v/v DMF-MeCN, RT, 12 h, 90% (7); then AgPF6, 1:1 v/v DMF-EtOAc, 30 min, RT, 80% (8).
Synthesis of the arylethynyl oligomers began with the Sonogashira mono-cross-coupling of known 4-bromo-3,5-diiodopyridine and commercially available (triisopropylsilyl)acetylene to create the monoacetylated halopyridine 2 (Scheme 1). Mono-cross-coupling 2 with known 1-tert-butyl-3,5-diethynylbenzene afforded the arylethynyl dimer 3. Cross-coupling two equivalents of 3 to 4-bromo-3,5-diiodopyridine followed by removing both triisopropylsilyl protecting groups yielded arylethynyl pentamer 4. Synthesis of the 4-methoxytolan cap 5 was conducted by mono-cross-coupling commercially available 4-ethynylanisole and 1,3-diiodobenzene. Cross-coupling two equivalents of 5 to 4 and subsequent methylation of the bromopyridines with methyl triflate resulted in the tricationic bromopyridinium nonamer 6. Exchange of the halogens (bromine for iodine) and counteranions (triflate for iodide) was achieved by stirring 6 with excess sodium iodide, providing the iodopyridinium target 7 (for further synthetic details, see Sections S2–3 in the Supporting Information, SI).
Yellow plates of 7 suitable for X-ray diffraction were grown by the vapor diffusion of methyl-tert-butyl ether into a 1:2 v/v DMF-MeCN solution of 6 and excess tetra-n-butylammonium (TBA) iodide.[16] Triple helicate 7 crystallized in space group C2/c, adopting both M- and P-helical conformations. Each complex is composed of three intertwined tricationic nonameric strands, offset along a common screw axis as defined by the two intrachannel iodides (Figure 1 a). Each iodide is bound tightly by four strong and linear XBs within the helical channel (average XB I⋯I– distance is 3.4 Å, 83% of ΣvdW radii; average C–I⋯I– angle is 171°). Consequently, pseudo square planar coordination is achieved (Figure 1 c). The XBs are complemented by numerous aromatic and ethynyl π-stacking interactions (44 buried aromatic surfaces, Figure 1 b; average ring-ring distance is ca. 3.7 Å). Additionally, seven iodides held to the helicate's exterior by ion pairing interactions help balance the nine positive charges associated with the cationic strands (see Figure S23 in the SI). Each triplex exhibits an approximate height and width of 13 and 19 Å, respectively, and a pitch of 10 Å. Finally, a 2.7 Å pore adorned with XB donors highlights the unique microenvironment found within the triple helicate (Figures 1 a–b). The only molecular axis of symmetry (C2) for the triplex aligns with the C–I bond of the nonbonding iodopyridinium donor (Figures 1 a and c, yellow sticks; for further crystallographic details, see Section S4 in the SI).
Figure 1.

Solid-state representations of triple helicate 7 and DFT-minimized nonamer. a) Solid-state structure of the triple helicate binding two intrachannel iodides. b) Crystal structure of the triplex looking down its anion channel (iodides removed). c) Pseudo square planar coordination geometry of the helicate's XB donors (scaffolding removed). Black dashes denote XBs. d) DFT-minimized nonamer of 7. Black dashes and iodides were added to emphasize the non-convergence of the XB donors. Figures (a)–(c): external iodide atoms removed for clarity. Figures (a) and (c): yellow C–X stick demarcates the nonbonding XB donor and axis of molecular C2 symmetry of the complex. Not all colors are representative of atom identity.
To explore the implications of helical rise and XB linearity, we calculated the conformation of a single strand of 7 using Density Functional Theory (DFT). The added black dashes and iodides in Figure 1 d emphasize the poor preorganization of a single strand. Iodide was placed in this non-convergent binding pocket, and the energies of both tridentate and bidentate XBing were calculated. Regardless of guest placement, nonbonding or repulsive interactions were inevitable (for computational details, see Section S5 in the SI). These calculations suggest that the strict linearity of XBing disfavors 1:1 binding.
The elucidation of triple helicate 7 in solution began with 1H NMR spectroscopic titrations. Compared to the relatively simple 1H NMR spectrum of 6 in [D7]DMF, the spectrum of triplex 7 suggested a thermodynamically stable aggregate (Figure 2 a). Even an excess of TBA bromide failed to convolute the spectrum of 6 (see Figure S30 in the SI). Given the superior XBing ability of iodines,[9f,10f,11a] these data provided qualitative evidence that 7 persisted as a XB induced aggregate. Furthermore, titrating silver hexafluorophosphate (AgPF6)—which precipitated silver iodide leaving non-coordinating PF6– anions—to a solution of 7 resulted in complete spectral deconvolution (see Figure S31 in the SI). The isolation of PF6– salt 8 (Scheme 1) permitted the reverse titration, holding the concentration of 8 constant while titrating TBA iodide. Surprisingly, even 0.2 equivalents of guest induced significant complex formation that displayed slow exchange with single strands of 7 on the NMR timescale (Figure 2 b). The aggregate's pyridinium and anisole signals were markedly shifted upfield (up to -0.79 and -0.54 ppm, respectively; for proton assignments, see Figure S32 in the SI), suggesting significant π-stacking in solution.[2a,3k,l,4a,4c,7] With three equivalents of TBA iodide, the resulting 1H NMR spectrum was analogous to that of 7, indicating strong XBing in solution (Figure 2 b).
Figure 2.

1H NMR variable temperature and titration experiments. a) 1H NMR spectra of triple helicate 7 subjected to variable temperature (500 MHz, 1:4 v/v [D7]DMF/CD3CN). b) 1H NMR titration involving 8 with additions of TBA iodide (600 MHz, 1:3 v/v [D7]DMF/CD3CN, 298 K).
The 2D NOESY spectrum of triplex 7 provided further evidence of higher-order helication in solution. Strong in-phase cross peaks corresponding to pyridinium methyl and tert-butyl signals were consistent with the solid-state structure but impossible for a single strand (over 7 Å apart; see Figure S33 in the SI). Likewise, medium in-phase cross peaks between tert-butyl and pyridinium protons as well as tert-butylbenzene and pyridinium methyl protons agreed with the X-ray crystal structure but could not originate from a single strand (over 5 and 6 Å apart, respectively). In stark contrast, the 2D NOESY spectrum of 8 manifested none of these features. Instead, only opposite-phase cross peaks between aromatic protons and same-ring substituents were evident, consistent with conformational heterogeneity in solution (see Figure S34 in the SI).
Further comparisons between the 1H NMR spectrum of 7 and its solid-state structure confirmed triple helicate fidelity in solution. The numbers and intensities of 1H NMR signals corresponding to the solid-state triplex were readily predictable due to its molecular C2 symmetry (see Figure S35 in the SI). The spectrum of 7 should exhibit three tert-butyl and three methoxy methyl signals of equal intensities, four equal intensity pyridinium methyl signals and one of half intensity, and nine equal intensity pyridinium signals. The 1H NMR spectrum of 7 is in full agreement with these predictions (see Figure S36 in the SI), indicating solution and solid-state structural congruence. Higher- or lower-order helicates would produce more or fewer 1H NMR signals, and variations in molecular symmetry would result in altered ratios between peak counts and relative intensities (for further solution and crystallographic structure analysis, see Section S9 in the SI).
2D DOSY data were collected to further characterize triple helicate 7 in solution. The 1H NMR resonances of 7 and 8 correlated with discrete diffusion coefficient lines, verifying that both species were distinct and monodispersed (see Figures S40–41 in the SI). Additionally, the hydrodynamic radii (rH) of 7, 8, and an internal standard (dichloromethane) were compared. Unsurprisingly, the rH of 8 was 1.3× larger than that of the triple helicate. Given the dynamics of 8 in solution, a rH inclusive of uncoiled conformations is sensible. In contrast, the π-stacked and coiled conformation of 7 would likely result in a smaller rH. The triple helicate's estimated rH of 8 Å agrees with the crystallographic radii of the complex (roughly 6.4 Å heightwise and 9.5 Å widthwise; for details pertaining to DOSY refinement and analysis, see Section S10 in the SI).
Given that most anion multiplexes require either highly charged anions or low temperatures to form in solution, it was remarkable that the helicate proved stable up to 68 °C (Figure 2 a). Surmising that XBs are critical for triple helicate stability, we probed them directly with UV-Vis titrations. The UV-Vis spectra of 8 suggested significant conformational changes upon titrating TBA iodide (see Figure S42 in the SI). Gradual depression of the 312 nm π-π* band was observed, consistent with the hypochromic effect of π-stacking phenylene ethynylene oligomers.[3k,l] Overall, the absorbance decreased by 22% after titrating two equivalents of guest. Surprisingly, adding as little as 0.01 equivalents of TBA iodide induced half of this total hypochromicity. At higher concentrations of 8, titrating TBA iodide produced a dark yellow solution associated with the appearance and growth of an absorption band at 400 nm (see Figure S43 in the SI). The absorption band is consistent with XB charge-transfer in solution.[10f,17] Alongside the demonstrated anion induced folding and denaturing of the triplex, these data implicate XBing as a vital component of helicate formation.
To ascertain triple helicate stability in aqueous phase, 7 was subjected to 1H NMR and 2D NOESY spectroscopy in a 1:1 v/v D2O-[D7]DMF solution (the limit of solubility). Aside from differences in chemical shifts, the spectroscopic features of 7 were fully consistent with those identified in organic solvents (see Figures S44–45 in the SI). Remarkably, after 20 days the 1H NMR spectrum of 7 exhibited minimal decomposition, notwithstanding the chemical instability of 4-iodopyridiniums (see Figures S46 in the SI). In contrast, residual water hydrolyzed 8 in a matter of hours. The compact and helical conformation of 7 protects the otherwise chemically sensitive 4-iodopyridinium XB donors. This helix conferred chemical stability is not without precedent.[1c,1e]
In conclusion, we have described the first XB induced triple helicate to bind iodide in solution and the solid state. The helicate is stabilized by multiple strong and linear XBs and π-stacking interactions. Furthermore, we have demonstrated that the complex is shape-persistent at high temperatures and in aqueous phase. Given the competing speciation and myriad noncovalent interactions in solution, the thorough characterization of a self-assembled triple anion helicate is an important step towards the rational design of large tubular containers with high affinity for complementary guests. We hypothesize that the combination of helical rise and XB linearity influences higher-order helication by destabilizing 1:1 complexes. Hence, the expedient self-assembly of a convergent, multidentate XBing microenvironment may be realized. These results have exciting implications in anion sensing, nanomaterials, and synthetic ion channeling.
Supplementary Material
Acknowledgments
This work was funded by the Center for Biomolecular Structure and Dynamics CoBRE (NIH NIGMS grant P20GM103546), National Science Foundation (NSF) CAREER CHE-1555324, Montana University System MREDI 51030-MUSRI2015-02, and the University of Montana (UM). The X-ray crystallographic data were collected using a Bruker D8 Venture, principally supported by NSF MRI CHE-1337908. We thank Allen Oliver and Brian Patrick for valuable crystallographic discussions. Funding for shared instrumentation in the CAMCOR NMR Facility at the University of Oregon was provided by NSF Award CHE-0923589 (MRI/ARRA), CHE-1427987 (MRI) with additional support from the Oregon Nanoscience and Microtechnologies Institute (ONAMI), OregonBEST, and the UO Office for Research and Innovation. We thank Klara Briknarova for help with 2D NMR interpretation, Earle Adams for general NMR guidance, Michael Strain for assistance with DOSY NMR collection and refinement, and Valeriy Smirnov and Ian Chrisman for the use of their glovebox.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Makiguchi W, Kobayashi S, Furusho Y, Yashima E. Angew Chem Int Ed. 2013;52:5275. doi: 10.1002/anie.201301005. [DOI] [PubMed] [Google Scholar]; b) Piguet C, Borkovec M, Hamacek J, Zeckert K. Coord Chem Rev. 2005;249:705. [Google Scholar]; c) Hannon MJ, Childs LJ. Supramol Chem. 2004;16:7. [Google Scholar]; d) Albrecht M. Chem Rev. 2001;101:3457. doi: 10.1021/cr0103672. [DOI] [PubMed] [Google Scholar]; e) Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. Chem Rev. 2001;101:3893. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]; f) Piguet C, Bernardinelli G, Hopfgartner G. Chem Rev. 1997;97:2005. doi: 10.1021/cr960053s. [DOI] [PubMed] [Google Scholar]
- 2.a) Jeon HG, Jung JY, Kang P, Choi MG, Jeong KS. J Am Chem Soc. 2016;138:92. doi: 10.1021/jacs.5b11808. [DOI] [PubMed] [Google Scholar]; b) Chandramouli N, Ferrand Y, Lautrette G, Kauffmann B, Mackereth CD, Laguerre M, Dubreuil D, Huc I. Nature Chem. 2015;7:334. doi: 10.1038/nchem.2195. [DOI] [PubMed] [Google Scholar]; c) Gan Q, Ferrand Y, Bao C, Kauffmann B, Grélard A, Jiang H, Huc I. Science. 2011;331:1172. doi: 10.1126/science.1200143. [DOI] [PubMed] [Google Scholar]; d) Juwarker H, Suk Jm, Jeong KS. Chem Soc Rev. 2009;38:3316. doi: 10.1039/b909034g. [DOI] [PubMed] [Google Scholar]; e) Tanatani A, Mio MJ, Moore JS. J Am Chem Soc. 2001;123:1792. doi: 10.1021/ja003678n. [DOI] [PubMed] [Google Scholar]; f) Prince RB, Barnes SA, Moore JS. J Am Chem Soc. 2000;122:2758. [Google Scholar]
- 3.a) Hartley CS. Acc Chem Res. 2016;49:646. doi: 10.1021/acs.accounts.6b00038. [DOI] [PubMed] [Google Scholar]; b) Zhang DW, Zhao X, Li ZT. Acc Chem Res. 2014;47:1961. doi: 10.1021/ar5000242. [DOI] [PubMed] [Google Scholar]; c) Zhao Y, Cho H, Widanapathirana L, Zhang S. Acc Chem Res. 2013;46:2763. doi: 10.1021/ar300337f. [DOI] [PubMed] [Google Scholar]; d) Zhang DW, Zhao X, Hou JL, Li ZT. Chem Rev. 2012;112:5271. doi: 10.1021/cr300116k. [DOI] [PubMed] [Google Scholar]; e) Guichard G, Huc I. Chem Commun. 2011;47:5933. doi: 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]; f) Roy A, Prabhakaran P, Baruah PK, Sanjayan GJ. Chem Commun. 2011;47:11593. doi: 10.1039/c1cc13313f. [DOI] [PubMed] [Google Scholar]; g) Haldar D, Schmuck C. Chem Soc Rev. 2009;38:363. doi: 10.1039/b803553a. [DOI] [PubMed] [Google Scholar]; h) Saraogi I, Hamilton AD. Chem Soc Rev. 2009;38:1726. doi: 10.1039/b819597h. [DOI] [PubMed] [Google Scholar]; i) Gong B. Acc Chem Res. 2008;41:1376. doi: 10.1021/ar700266f. [DOI] [PubMed] [Google Scholar]; j) Huc I. Eur J Org Chem. 2004;2004:17. [Google Scholar]; k) Prince RB, Saven JG, Wolynes PG, Moore JS. J Am Chem Soc. 1999;121:3114. [Google Scholar]; l) Nelson JC, Saven JG, Moore JS, Wolynes PG. Science. 1997;277:1793. doi: 10.1126/science.277.5333.1793. [DOI] [PubMed] [Google Scholar]
- 4.a) Juwarker H, Jeong KS. Chem Soc Rev. 2010;39:3664. doi: 10.1039/b926162c. [DOI] [PubMed] [Google Scholar]; b) Johnson CA, Berryman OB, Sather AC, Zakharov LN, Haley MM, Johnson DW. Cryst Growth Des. 2009;9:4247. doi: 10.1021/cg5018856. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Suk Jm, Jeong KS. J Am Chem Soc. 2008;130:11868. doi: 10.1021/ja804845m. [DOI] [PubMed] [Google Scholar]
- 5.a) Selvakumar PM, Jebaraj PY, Sahoo J, Suresh E, Prathap KJ, Kureshy RI, Subramanian PS. RSC Adv. 2012;2:7689. [Google Scholar]; b) Haketa Y, Maeda H. Chem Eur J. 2011;17:1485. doi: 10.1002/chem.201002748. [DOI] [PubMed] [Google Scholar]; c) Coles SJ, Frey JG, Gale PA, Hursthouse MB, Light ME, Navakhun K, Thomas GL. Chem Commun. 2003:568. doi: 10.1039/b210847j. [DOI] [PubMed] [Google Scholar]; d) Keegan J, Kruger PE, Nieuwenhuyzen M, O'Brien J, Martin N. Chem Commun. 2001:2192. doi: 10.1039/b106981k. [DOI] [PubMed] [Google Scholar]
- 6.a) Hua Y, Liu Y, Chen CH, Flood AH. J Am Chem Soc. 2013;135:14401. doi: 10.1021/ja4074744. [DOI] [PubMed] [Google Scholar]; b) Maeda H, Kitaguchi K, Haketa Y. Chem Commun. 2011;47:9342. doi: 10.1039/c1cc12822a. [DOI] [PubMed] [Google Scholar]; c) Sánchez-Quesada J, Seel C, Prados P, de Mendoza J, Dalcol I, Giralt E. J Am Chem Soc. 1996;118:277. [Google Scholar]
- 7.Li S, Jia C, Wu B, Luo Q, Huang X, Yang Z, Li QS, Yang XJ. Angew Chem Int Ed. 2011;50:5721. doi: 10.1002/anie.201180593. [DOI] [PubMed] [Google Scholar]
- 8.Casnati A, Liantonio R, Metrangolo P, Resnati G, Ungaro R, Ugozzoli F. Angew Chem Int Ed. 2006;45:1915. doi: 10.1002/anie.200504459. [DOI] [PubMed] [Google Scholar]
- 9.a) Brown A, Beer PD. Chem Commun. 2016 [Google Scholar]; b) Wageling NB, Neuhaus GF, Rose AM, Decato DA, Berryman OB. Supramol Chem. 2016;28:665. doi: 10.1080/10610278.2015.1118101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Beyeh NK, Pan F, Rissanen K. Angew Chem Int Ed. 2015;54:7303. doi: 10.1002/anie.201501855. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:7411. [Google Scholar]; d) Dumele O, Trapp N, Diederich F. Angew Chem Int Ed. 2015;54:12339. doi: 10.1002/anie.201502960. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:12516. [Google Scholar]; e) Gilday LC, Robinson SW, Barendt TA, Langton MJ, Mullaney BR, Beer PD. Chem Rev. 2015;115:7118. doi: 10.1021/cr500674c. [DOI] [PubMed] [Google Scholar]; f) Vargas Jentzsch A. Pure Appl Chem. 2015;87:15. [Google Scholar]; g) Beale TM, Chudzinski MG, Sarwar MG, Taylor MS. Chem Soc Rev. 2013;42:1667. doi: 10.1039/c2cs35213c. [DOI] [PubMed] [Google Scholar]; h) Erdelyi M. Chem Soc Rev. 2012;41:3547. doi: 10.1039/c2cs15292d. [DOI] [PubMed] [Google Scholar]; i) Walter SM, Kniep F, Herdtweck E, Huber SM. Angew Chem Int Ed. 2011;50:7187. doi: 10.1002/anie.201101672. [DOI] [PubMed] [Google Scholar]; j) Sarwar MG, Dragisic B, Sagoo S, Taylor MS. Angew Chem Int Ed. 2010;49:1674. doi: 10.1002/anie.200906488. [DOI] [PubMed] [Google Scholar]
- 10.a) Mukherjee A, Tothadi S, Desiraju GR. Acc Chem Res. 2014;47:2514. doi: 10.1021/ar5001555. [DOI] [PubMed] [Google Scholar]; b) Troff RW, Mäkelä T, Topić F, Valkonen A, Raatikainen K, Rissanen K. Eur J Org Chem. 2013;2013:1617. [Google Scholar]; c) Cavallo G, Metrangolo P, Pilati T, Resnati G, Sansotera M, Terraneo G. Chem Soc Rev. 2010;39:3772. doi: 10.1039/b926232f. [DOI] [PubMed] [Google Scholar]; d) Aakeröy CB, Schultheiss NC, Rajbanshi A, Desper J, Moore C. Cryst Growth Des. 2009;9:432. doi: 10.1021/cg8006712. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Fourmigué M. Curr Opin Solid State Mater Sci. 2009;13:36. [Google Scholar]; f) Metrangolo P, Meyer F, Pilati T, Resnati G, Terraneo G. Angew Chem Int Ed. 2008;47:6114. doi: 10.1002/anie.200800128. [DOI] [PubMed] [Google Scholar]; g) Metrangolo P, Neukirch H, Pilati T, Resnati G. Acc Chem Res. 2005;38:386. doi: 10.1021/ar0400995. [DOI] [PubMed] [Google Scholar]; h) Walsh RB, Padgett CW, Metrangolo P, Resnati G, Hanks TW, Pennington WT. Cryst Growth Des. 2001;1:165. [Google Scholar]
- 11.a) Scholfield MR, Zanden CMV, Carter M, Ho PS. Protein Sci. 2013;22:139. doi: 10.1002/pro.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. J Med Chem. 2013;56:1363. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]; c) Politzer P, Murray JS, Clark T. PCCP. 2010;12:7748. doi: 10.1039/c004189k. [DOI] [PubMed] [Google Scholar]; d) Riley KE, Murray JS, Politzer P, Concha MC, Hobza P. Journal of Chemical Theory and Computation. 2009;5:155. doi: 10.1021/ct8004134. [DOI] [PubMed] [Google Scholar]; e) Legon AC. In: Halogen Bonding: Fundamentals and Applications. Metrangolo P, Resnati G, editors. Springer Berlin Heidelberg; Berlin, Heidelberg: 2008. p. 17. [Google Scholar]; f) Zou JW, Jiang YJ, Guo M, Hu GX, Zhang B, Liu HC, Yu QS. Chemistry - A European Journal. 2005;11:740. doi: 10.1002/chem.200400504. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X, Jiang H, Zhu W. J Med Chem. 2009;52:2854. doi: 10.1021/jm9000133. [DOI] [PubMed] [Google Scholar]
- 13.Desiraju GR, Ho PS, Kloo L, Legon AC, Marquardt R, Metrangolo P, Politzer P, Resnati G, Rissanen K. Pure Appl Chem. 2013;85:1711. [Google Scholar]
- 14.Massena CJ, Riel AMS, Neuhaus GF, Decato DA, Berryman OB. Chem Commun. 2015;51:1417. doi: 10.1039/c4cc09242b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuda K, Stone MT, Moore JS. J Am Chem Soc. 2002;124:11836. doi: 10.1021/ja027437m. [DOI] [PubMed] [Google Scholar]
- 16.Crystallographic Data for 7 C80H61I6N3O2, M = 1857.71, monoclinic, space group C2/c (no. 15), a = 54.1200(19), b = 36.8537(14), c = 35.419(2), β = 128.1810(10), V = 55530(5), Z = 24, T = 100 K, μ (CuK α) = 16.102 mm-1, Dcalc = 1.333 g ml-1, 2θmax = 101.124, 291827 reflections collected, 29038 unique (Rint = 0.0668, Rsigma = 0.0322), R1 = 0.0837 (I > 2σ(I)), wR2 = 0.2858 (all data). See Section S4 in the SI. CCDC 1476727 contains the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
- 17.Rosokha SV, Neretin IS, Rosokha TY, Hecht J, Kochi JK. Heteroat Chem. 2006;17:449. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.