

Abstract
Live-attenuated bacterial vectors for antigens delivery have aroused growing interest in the field of cancer immunotherapy. Their potency to stimulate innate immunity and to promote intracellular antigen delivery into antigen-presenting cells could be exploited to elicit a strong and specific cellular immune response against tumor cells. We previously described genetically-modified and attenuated Pseudomonas aeruginosa vectors able to deliver in vivo protein antigens into antigen-presenting cells, through Type 3 secretion system of the bacteria. Using this approach, we managed to protect immunized mice against aggressive B16 melanoma development in both a prophylactic and therapeutic setting. In this study, we further investigated the antigen-specific CD8+ T cell response, in terms of phenotypic and functional aspects, obtained after immunizations with a killed but metabolically active P. aeruginosa attenuated vector. We demonstrated that P. aeruginosa vaccine induces a highly functional pool of antigen-specific CD8+ T cell able to infiltrate the tumor. Furthermore, multiple immunizations allowed the development of a long-lasting immune response, represented by a pool of predominantly effector memory cells which protected mice against late tumor challenge. Overall, killed but metabolically active P. aeruginosa vector is a safe and promising approach for active and specific antitumor immunotherapy.
Introduction
For about 15 years, many cancer immunotherapy strategies have been evaluated in preclinical and clinical studies.1 The first active and specific immunotherapy approved by the FDA for the treatment of advanced prostate cancer in 2010 (sipuleucel-T),2 has paved the way for the development of new cancer vaccines.3 The aim of active and specific immunotherapy is to harness the immune system for the induction and/or the remobilization of specific cytotoxic T lymphocytes response(s) against tumor antigens. Nevertheless, we know that such approaches have to resolve several challenges.4 First, the stimulation of a robust proinflammatory innate immune response through a “Danger signal” is needed to mature APCs (antigen presenting cells). The type of signal will condition APCs maturation and their ability to both secrete stimulatory cytokines (i.e., interleukin (IL)-12) and present antigens. Second, tumor-derived antigens must be provided either into intracellular compartment or from extracellular compartment in order to be efficiently presented by major histocompatibility complex (MHC) I to CD8+ T cells, through direct or cross-presentation pathway, respectively. Third, antigen-specific lymphocytes must be able to infiltrate the tumor bed and overcome local immunosuppression. And finally, the generation of a memory pool of antigen specific T cell is critical for long term protection and relapse prevention.
One of the different innovative ways investigated to succeed in these four challenges is the use of live attenuated bacterial vectors. Their beneficial “natural adjuvant” properties and their particular easy molecular engineering allow tumor antigens delivery into the cytosol of APCs in vivo and therefore antigen presentation to CD8+ T lymphocytes. Two facultative intracellular bacteria Salmonella sp. and Listeria monocytogenes (Lm) are currently being evaluated in clinical trials and have been reported previously to raise strong antitumor antigen-specific CD8+ and CD4+ responses in mice.5,6 Pseudomonas aeruginosa (Pa) is also efficient at delivering proteins in vitro and in vivo into the cytosol of eukaryotic cells,7–12 using its needle-like device,13 the so called type 3 secretion system (T3SS). We have developed engineered and attenuated Pa vectors.14–16 OSTAB attenuated strain is deleted for major T3SS-exotoxins ExoS and ExoT and for two subunits (UvrA and UvrB) of the exonuclease ABC, which takes part in the nucleotide excision repair (NER) pathway that removes DNA damage induced by ultraviolet (UV) light. Following photo-inactivation, with long-wave UV light (UVA) radiation associated to the DNA crosslinking agent amotosalen (psoralen S-59), the bacteria become unable to replicate and are doomed to die, but are still immunologically and T3SS active.10 Thus, after photo-inactivation, the OSTAB bacteria turn into killed but metabolically active (KBMA). This strain is able to deliver any tumor-associated antigen of interest into host cells through T3SS and to drive cellular immune response against the tumor. We have previously shown that OSTAB attenuated and KBMA strains trigger antigen-specific CD8+ T cell priming, yielding to the protection of mice against aggressive melanoma development.9,10,17 In order to further characterize the Pa-induced antitumor responses, we have investigated the phenotype, the functionality, and the persistence of Pa-induced antigen specific CD8+ T cell response. We demonstrate that multiple immunizations with a safe attenuated KBMA P. aeruginosa vector induce a highly functional antigen-specific cellular immune response able to control B16 tumor growth in mice with a similar efficiency as the OSTAB attenuated Pa vector. Interestingly this was associated with a significant increase of antigen-specific tumor-infiltrating lymphocytes (TILS) and a benefit ratio antigen-specific effector CD8+ T cells/ regulatory T cells within the tumor bed. Finally, we show that a pool of predominantly effector-memory antigen-specific CD8+ T cells persists after immunizations and confers a long term protection against tumor development.
Results
KBMA vector based vaccine induces highly functional cellular immune response
We have previously shown the ability of KBMA Pa at inducing antigen-specific CD8+ immune response in vivo and to prolong survival of mice challenged with tumors.10 We aimed here at analyzing more deeply the efficiency of this vaccine. First, we focused on an accurate analysis of the functionality of the antigen-specific CD8+ T cells generated after immunizations. Thus the established immunizations schedule was applied (6 subcutaneous immunizations, 2×/week). We then assessed the frequency and the functionality of splenic Ova tumor-specific CD8+ T cell 10 days after last immunization with attenuated OSTAB S54-OVA or KBMA S54-OVA vaccine. Both approaches (OSTAB S54-OVA and KBMA S54-OVA) were able to induce the priming of tumor-specific T cells at similar frequencies 11.9 and 12.1% respectively (Figure 1a). To evaluate their functionality, cytokines secretion and expression of the degranulation marker (CD107a) were investigated after splenocytes stimulation with the immune-dominant MHC class I restricted OVA peptide (SIINFEKL epitope). Similar cytokine profiles were observed on tumor-specific CD8+ T cells following peptide stimulation in both vaccinated groups (Figure 1b–d). Tumor-specific CD8+ T cell response induced by OSTAB S54-OVA and KBMA S54-OVA vaccines displays a high degree of polyfunctionality, with respectively 12% and 11% of responding CD8+ T cells producing at the same time interferon (IFN)-γ, tumor necrosis factor (TNF)-α and IL-2 in association with degranulation and 58 and 52% respectively, producing at least two cytokines with degranulation (Figure 1c,d). Single functional specific CD8+ T cell represented a minority with only 13% in OSTAB S54-OVA and 12% in KBMA S54-OVA groups. These results are consistent with our previous observations that KBMA and OSTAB attenuated Pa-OVA vectors have similar ability at inducing potent cellular antitumor immune response.
Figure 1.

Functional characterization of splenic Ova-specific CD8+ T cells response 10 days after last immunization. (a) Representative flow cytometric profile (up) and frequency (down) of Ova-specific CD8+ T cells evaluated by H2Kb-SIINFEKL multimer staining on total CD8+-gated cells (b) Simultaneous production of the cytokines IFN-γ, TNF-α, and IL-2 by CD8+ T cells as well as degranulation (CD107a) marker were assessed during 4 hours OVA peptide (SIINFEKL) stimulation or in absence of stimulation (Medium). (c) Frequency of total CD8+ T cells allocated in each of the 15 analyzed combinations of IFN-γ, TNF-α, IL-2, and CD107a marker according to the diagram shown underneath the bar graphs. (d) Pie charts represent the spectrum of functions (cytokines and/or degranulation marker) among responding CD8+ T cells to OVA peptide stimulation, in KBMA and OSTAB groups. Each slice of pie represents the percentage of cells expressing between 1 and 4 effector functions. Mean ± SEM. IFN-γ, interferon-γ; IL-2, interleukin-2; KBMA, killed but metabolically active; TNF-α, tumor necrosis factor-α ; SEM, standard error of the mean.
Pa vectors based vaccines induce strong antigen-specific CD8+ T cells tumor infiltration and a positive shift between effector and immunosuppressive T cells
The efficacy of KBMA vaccine has been evaluated in a therapeutic setting. B16-OVA-bearing mice received up to five injections starting when tumors became palpable (Figure 2a). While mice that received control OSTAB S54-Ei (Pa strain without antigen) or heat-killed (HK) S54-OVA (dead strain unable to deliver antigen through T3SS) develop tumors over time, KMBA S54-OVA vaccine allows a significant control of tumor progression, comparable to live OSTAB S54-OVA strain (Figure 2b,c). More importantly, tumor growth curves reveal that KBMA S54-OVA treated mice have homogeneously responded to the treatment (Figure 2c). A mild therapeutic and heterogeneous effect was also observed with HK S54-OVA treatment compared with the OSTAB S54-Ei negative control. This result was expected since the HK S54-OVA immunization condition is closed to adjuvanted whole protein vaccination strategies that have shown such a mild protection effects in the clinic.
Figure 2.

Multiple immunizations with OSTAB S54-OVA or KBMA S54-OVA vaccines slow down tumor growth of aggressive B16-OVA melanoma. (a) Experimental setting. 2 × 105 B16-OVA cells have been injected s.c. at day 0. When tumors became palpable (at day 6 on the presented experiment), mice received s.c. injections of different treatment, 2×/week, up to 5 times. Tumor growth was monitored every 2 days and mice were killed when tumor surface reached 150 mm2. (b) Tumor growth curves (mean ± SEM) from one representative experiment out of three. n = 6 mice/group. Student’s unpaired t-test performed on the last tumor measurement (D21). *P < 0.05, ***P < 0.001, ****P < 0.0001. (c) Individual tumor growth curves of the same experiment. KBMA, killed but metabolically active; SEM, standard error of the mean.
Many studies have reported that high level of CD8+ TILs strongly predicts patient overall survival underlying the critical role of tumor-specific CD8+ T cells.18–20 However, one challenge in cancer vaccine development is not only to induce a strong CD8+ T cells effectors infiltration within the tumor, but also to overcome the immunosuppressive environment by shifting the balance between effector cells and regulatory cells.21 The analysis of TILs, 21 days posttumor implantation, revealed that both live and KBMA Pa vectors induce a significant increase in the Ova-specific CD8+-T cells within the tumor while empty or HK vectors failed to do so (Figure 3a,b). Importantly, both bacterial vectors induce a strong increase in the tumor-specific CD8+-T cells/T-regs ratio, thus positively shifting the balance between effector and immunosuppressive T cells (Figure 3c).
Figure 3.
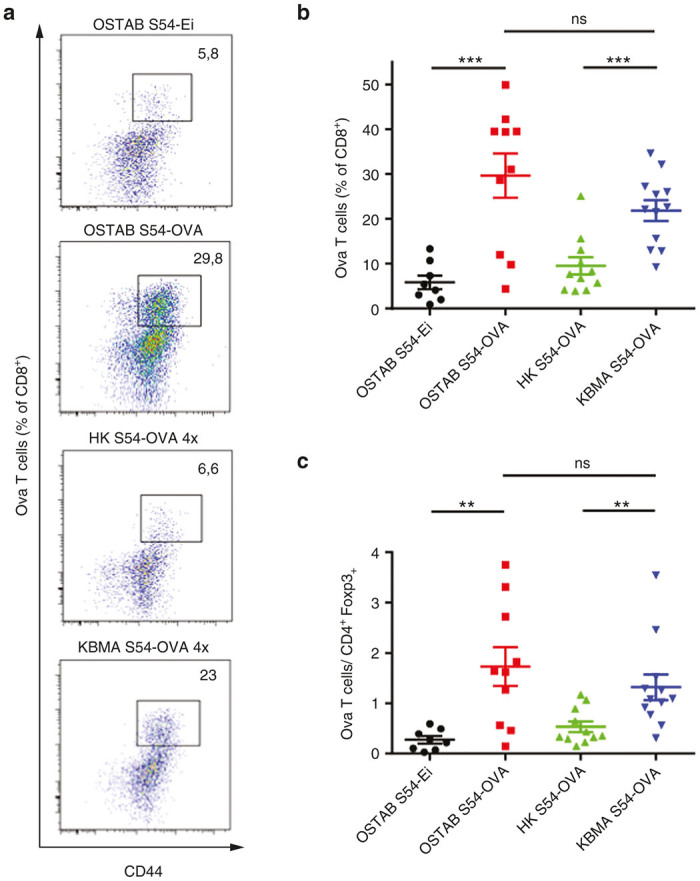
Immunizations with OSTAB S54-OVA or KBMA S54-OVA vaccines allow Ova-specific CD8+ T cells tumor infiltration and change the balance between effector and regulatory T cells within the tumor. Mice were immunized according to the schedule depicted in Figure 2a. Tumors were harvested at day 21. (a and b) Ova-specific CD8+ T cell tumor infiltration quantified by flow cytometry in dissociated tumors. (a) Representative flow-cytometric profile of each group indicating percentage of Ova CD8+ T cells among CD8+ TILs. (b) Mean percentage of Ova CD8+ T cells through CD8+ TILs (c) Ratio between Ova CD8+ T cells (gated on CD8+ TILs) and TREG cells (% FoxP3+ cells gated on tumor CD4+ cells). n = 4–6 mice/group. Data are representative of two pooled experiments. Mean ± SEM. **P < 0.01, ***P < 0.001 determined by unpaired Student’s t-test. KBMA, killed but metabolically active; TILs, tumor-infiltrating lymphocytes; SEM, standard error of the mean.
Pa vectors elicit a stable pool of tumor-specific CD8+ T memory cells
A key feature of the efficacy of a cancer vaccine lies on its ability to induce a long-lasting tumor-specific CD8+ T cells response and subsequent protection against relapse and/or metastasis. Thus, we analyzed the potency of Pa vaccination by the evaluation of the development of tumor-specific CD8+ memory response during and after six consecutive immunizations (2×/week). As soon as 6 days after the first immunization, tumor-specific CD8+ T cells in the blood were detected in groups treated with OSTAB S54-OVA or KBMA S54-OVA, reaching 2.2 and 1.8% respectively among CD8+-T cells as measured by multimers whereas in control groups HK S54-OVA or OSTAB S54-Ei empty vectors only 0.1% tetramer positive cells among CD8+ T cells corresponding to background signal (Figure 4a). In both, OSTAB S54-OVA or KBMA S54-OVA treated groups we observed an increase of tumor-specific CD8+ T cells number during the immunization period, albeit in a lesser extent in KBMA treated group (Figure 4a). The maximal number of tumor-specific CD8+ T cells was reached around 1 week after the last vaccination. This phase was followed by a biphasic contraction period, where the number of tumor-specific CD8+ effector T cells decreased in the two groups during 2 weeks after the last vaccination and then stabilized during next month. At 70 days, a substantial persisting pool of tumor-specific memory CD8+ T cells in both treated groups was still observed.
Figure 4.

Kinetic of Ova CD8+ T cells response and memory markers induced by Pa-OVA vaccines analyzed by flow cytometry in spleen and blood. Mice received 6 immunizations with Pa-OVA vaccines or controls (first immunization at D0) and blood or spleens were harvested at different time point. (a) Representative flow-cytometry profiles of Ova-specific CD8+ T cells in blood for treated and control groups at day 6 (up). Kinetic profile of Ova-specific CD8+ T cells (absolute number) in the blood (down). n = 5 or 6 mice per group per time point. Mean ± SEM. (b) Expression of CD62-L, CD127, and KLRG-1 markers assessed over the time by gating on Ova-specific CD8+ T cells in blood. (c) Representative profile of CD62-L and CD127 markers coexpression by gating on Ova-specific CD8+ T cells in splenocytes at effector phase (day 29) and memory phase (day 90). Numbers indicate percentage within each quadrant (mean ± SEM). n = 5 or 6 mice (left). Mean percentage of Memory (CD62-Llow CD127hi or CD62-LhiCD127hi) and effector (CD62-LlowCD127low) Ova-specific CD8+ T cells subpopulations at 90 days in splenocytes upon immunization with Pa-OVA vaccines. TEM: effector memory Ova-specific CD8+ T cells; TCM: central memory Ova-specific CD8+ T cells; TEFF: OVA-specific effector CD8+ T cells. Mean ± SEM. *P < 0.05, ***P < 0.001 determined by unpaired Student’s t-test. Pa, Pseudomonas aeruginosa; SEM, standard error of the mean.
To study the phenotypic changes of blood circulating tumor-specific CD8+ T cells over time, we followed the expression of CD127 (IL-7Rα) memory marker,22 homing receptor CD62-L and the killer cell lectin-like receptor G1 (KLRG-1) on antigen-experienced T cells23 (OVA specific) (Figure 4b). After the first immunizations (at day 6), tumor-specific CD8+ T cells from OSTAB S54-OVA and KBMA S54-OVA group had a typical effector phenotype CD127low CD62-Llow KLRG-1low (TEFF). Then, specific CD8+ T cells increased CD127 memory marker expression (from day 21) but kept a predominant effector phenotype (CD62-Llow), with a progressive increase of KLRG-1 expression (day 21, 28, and 56). Remarkably expression of this T cell senescent marker is corroborated with the drop of tumor-specific blood CD8+ T cells numbers from day 28 (Figure 4a). At 71 days after the first immunization a circulating pool persists comprising both effector (CD62-Llow CD127hi) and central-memory cells (CD62-Lhi CD127hi) in OSTAB S54-OVA and KBMA S54-OVA group with KLRG1hi and KLRG-1low cells.
Finally, we also investigated the tumor-specific CD8+ T cell response in splenocytes at effector phase (day 29) and during memory phase far after the last vaccination (day 90). Subsequent to the immunization period, a majority of tumor-specific CD8+ T cells seemed to take on the CD127hi memory phenotype with ~61% in OSTAB S54-OVA and 54% in KBMA S54-OVA group (day 29, Figure 4c left). During the memory phase, this tumor-specific CD8+ T cells evolved to a predominant CD62-Llow CD127hi in groups treated with OSTAB S54-OVA or KBMA S54-OVA, reaching 51.8 and 68.2%, which evokes the development of a major effector-memory response TEM (day 90, Figure 4c left and right). Surprisingly, in the OSTAB S54-OVA group, a significantly higher tumor-specific effector T cell proportion was still measured (26.1% TEFF, CD62-Llow CD127low) as compared with the KBMA S54-OVA (9%, P < 0.001). As a consequence a lower percentage of TEM cells were measured with OSTAB S54-OVA group versus the KBMA S54-OVA group (51.8% versus 68.2%, P < 0.05). This means that even 70 days after the last immunization, a T cell memory response is present with a slightly different signature for both groups. However, the presence of a similar central-memory TCM pool in each group was observed with ~15.7–20.5% of tumor-specific CD8+ T cells in OSTAB S54-OVA group and KBMA S54-OVA group respectively.
Tumor-specific CD8+ T memory cells induced by Pa-antigen vaccines promote protective long-term antitumor response
Since a remarkable stable memory CD8+ T cell response was reached ~50 days after the last immunization in mice, we challenged immunized mice by B16-OVA tumor cell implantation 100 days after the last immunization with P. aeruginosa vectors (Figure 5a). Mice immunized by OSTAB S54-OVA and KBMA S54-OVA vector remained tumor free beyond 150 days post-tumor challenge in 67% and 57% respectively of all individuals. At the opposite, mice from control groups HK S54-OVA and OSTAB S54-Ei develop tumors and had a median survival of 29 and 22 days, respectively (Figure 5b). We observed significantly better survival in mice vaccinated with OSTABS54-OVA and KBMA S54-OVA compared with control mice (strain OSTAB S54-Ei) (The P-value for the log-rank test are 0.0096 and 0.0005 respectively) whereas there is no statistical difference between OSTAB S54-OVA and KBMA S54-OVA (P = 0.9375). Thus, vaccination with P. aeruginosa vectors induces a long term protection and confers a great benefit to efficiently immunized mice.
Figure 5.
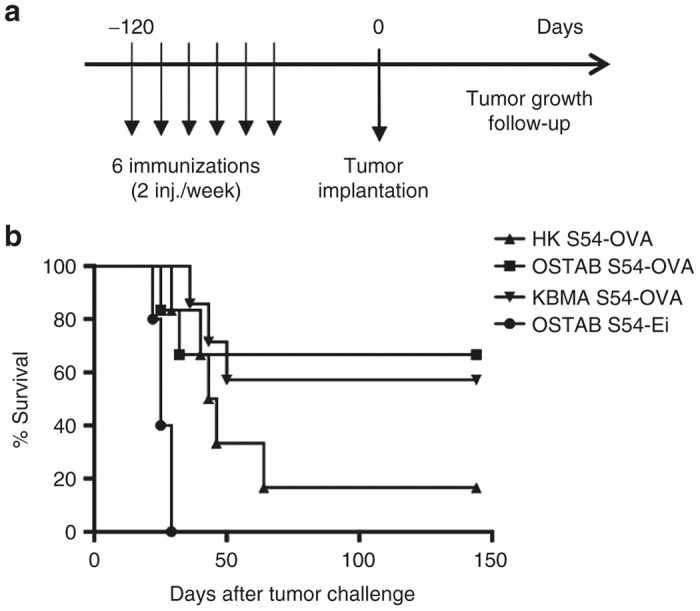
Long-term protection induced by Pa-OVA vaccines against B16-OVA melanoma. (a) Experimental setting. Mice have been injected 6 times (2×/week) and challenged with 2 × 105 B16-OVA cells 100 days after the last immunization. Tumor growth was monitored every 2 days and mice were killed when tumor reached 150 mm2. (b) Percent survival (based on 150 mm2 tumor surface) of mice challenged with tumors at memory phase. n = 5–7 mice per group. Log-rank test OSTAB S54-OVA and KBMA S54-OVA compared with OSTAB S54-Ei P = 0.0096 and P = 0.0005 respectively). Pa, Pseudomonas aeruginosa.
Discussion
In this study, we show that attenuated OSTAB S54-OVA and KBMA S54-OVA elicit a similar polyfunctional cellular antitumor immune response. Then, using B16-OVA tumor model, we further characterize T CD8+ antitumor immune response following immunization with both Pa-OVA vectors in terms of tumor infiltration and long-term protection. First, we demonstrate that we significantly inhibit tumor growth in a therapeutic setting, for both groups in this aggressive tumor model. This tumor growth control is correlated with a strong tumor infiltration by tumor-specific CD8+ T cells.
Furthermore, we show that the antigen-specific response is also accompanied by a significant rise of the proportion of antigen-specific CD8+ T cells among TILs and accordingly a shift in the balance T CD8+/CD4+Treg in favor to effector CD8+ cells. This observation is consistent with previous data from either mouse models or clinical trials describing that shifting the balance toward CD8+ T cells versus Treg is critical for effective cancer immunotherapy. Live-attenuated and KBMA Pa-OVA vectors, as for KBMA Lm, induce systemic T-cell responses including polyfunctional cytokine-secreting CD8+ T-cells.24
The contribution of different subsets of memory CD8+ T cells in recall responses following immunization with live attenuated bacterial vectors are poorly understood and still controversial. In this study, we explored the antigen-specific CD8+ memory response induced by Pa-OVA vectors and we show that antigen-specific CD8+ T cells evolve toward a pool of effector- and central-memory phenotype. This long-lasting response is correlated to a potent protection of mice against tumor challenge at the memory phase. The memory pool of CD8+ T cells, identified by its re-expression of CD127 (IL-7Rα) after effector phase,22 contains two major distinct subsets, the so-called T effector-memory (TEM) and T central-memory (TCM), that differ in their phenotype, trafficking patterns, tissue residence and functional characteristics.25 This division is based on CD62-L (homing receptor in lymphoid tissues) or C-C chemokine receptor type 7 (CCR7) expression. Typically, TCM cells express CD62-L and CCR7 and localize predominantly to lymphoid tissue, whereas TEM cells are negative for CD62-L and CCR7 and also prevail at peripheral sites. TCM have been also described as being better to proliferate following antigenic recall while TEM are highly cytolytic and proliferate slightly.26–28 We distinguished a difference in the memory phenotype of tumor-specific CD8+ T cells induced by the two vectors. It seems that KBMA Pa confers a strong memory response (TCM and TEM) whereas OSTAB-S54 OVA strain elicit an extended effector phenotype (~26% of cells versus 9% for KBMA bacteria), even at the memory phase. This could be explained by the presence of remaining live OSTAB S54-OVA bacteria in the organism that may maintain a pool of OVA antigen-specific CD8+ effector cells. Interestingly, a previous preclinical study has described distinct long-lasting tumor protection correlated to disparate CD8+ memory response following vaccination with recombinant OVA-expressing Salmonella enterica serovar Typhimurium (St) and Lm vectors. Although both of the recombinant vectors induced long-term Ova CD8+ T memory cells with comparative cytolytic functions, Lm led predominantly to central-memory CD8+ T cells (CD62-Lhigh CD127high) with strong proliferation and self-renewal capabilities, that conferred a better long-term tumor protection than St induced effector-memory CD8+ T cells (CD62-Llow CD127high) that were unable to expand and led to a lower tumor protection. Stark et al. concluded for the beneficial role of central memory.29 Nevertheless, in our work, we demonstrate a strong efficacy of TEM response in the same tumor model than Stark et al. after immunizations with Pa-specific tumor antigen vectors. Then the quality of the memory response may be strongly dependent on the vector used and the induced host response. The only observation of TCM or TEM phenotype could not be enough to conclude on the better efficacy of one or another vector.
Cancer vaccines aim at overcoming immunotolerance against tumor antigen(s) and at eliciting strong antitumor CD8+ T cell response(s). Several strategies, like adjuvanted peptide-based vaccine, DCs vaccine or in vivo delivery vectors had already achieved this fundamental step. Nevertheless, many of them induced only a limited response against the tumor growth. It seems that the modulation of the in vivo delivery approach constitute an important route of investigation to improve the quality of the antitumor immune response. During the last decade, bacterial vectors for in vivo tumor-antigen delivery demonstrated their attractive properties.30–32 They are natural adjuvants and their lifestyle contributes to intracellular delivery of full-length protein tumor antigen to immune host cells (in particular APCs), skirting the limits of MHC restriction and the choice of CD8+ and/or CD4+ epitope peptides. Pa is one of these recombinant bacterial vectors that had already proved to elicit protective response in tumor models. It can stimulate potently the innate immune system through TLRs activation and induce NLRC4 inflammasome leading to proinflammatory reaction and release of inflammatory cytokines IL-1, IL-6, IL-8, IL-18, and GM-CSF.33 Less is known about adaptive response following Pa injection but it was recently reported Pa-specific memory T helper responses Th1, Th17, and Th22 conferring protection to the bacteria.34,35 In further study, we need to explore more in details the CD4 response conferred by the bacterial vector and the antigen used for cancer immunotherapy knowing its important role in memory response36,37 and to realize deeper characterizations of the mechanism of action of this bacterial vector. Overall, the antitumor therapeutic efficacy of Pa vaccines (live and KBMA) looks promising and KBMA could be a safer approach that elicit poly-functional and long-lasting anticancer immune response.
Materials and Methods
Bacterial strains, cell line, and mice
The attenuated Pa OSTAB mutant susceptible to photochemical treatment and the corresponding OSTAB S54-Ei (“empty” plasmid with ExoS1-54 but without antigen) and OSTAB S54-OVA (chicken ovalbumin D248-A376 antigen fused to ExoS1-54) transformed strains were previously described.10 Transformed bacteria were grown in Lysogeny Broth (LB) (Sigma-Aldrich, Saint Louis, MO) medium in the presence of 300 mg/l carbenicillin at 37°C and 250g.
B16-OVA melanoma cells from C57BL/6 mice were cultivated in complete Dulbecco’s modified essential medium (DMEM) Glutamax containing 10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 mg/ml of streptomycin, and 500 µg/ml Geneticin. The cells were passaged using 0.05% trypsin/0.02% ethylenediaminetetraacetate (EDTA) solution and were cultivated at 37°C in a 5% CO2 humidified incubator. Medium and reagents were purchased from Thermo Fisher Scientific, Waltham, MA.
Female C57BL/6J mice were purchased at 5–6 weeks old from the Elevage Janvier (Le Genestet St. Isle, France) and were kept under specific pathogen-free conditions in an accredited establishment (PHTA animal facility, Plateforme de Haute Technologie Animale) according to the governmental guidelines N886/609/CEE. All animal experiments were approved by the Animal Experiment Committee of the Region (Cometh) and submitted to the French Ministry of Research and received Reference Number 00245.02 for the protocol entitled “Characterization of immune response induced by a bacterial vector for murine tumor”. Mice were anesthetized with isoflurane prior to subcutaneous (s.c.) bacteria injection and for retro-orbital blood sampling.
Photochemical treatment for KBMA vaccine
KBMA bacteria were prepared as previously described.10 Briefly, OSTAB S54-OVA strain was inoculated in LB at OD600 0.2 with 1 mmol/l of isopropyl-β-D-thiogalactoside (IPTG) (Sigma-Aldrich) and 300 mg/l of carbenicillin. When the OD600 reached 0.5, 10 µmol/l of photoactivatable amotosalen intercalant was added. Then cultures were grown until OD600 = 1. About 1 ml of culture was then transferred in a well (9.6 cm2) of a six-well cell culture plate for UVA irradiation at a dose of 7.2 J/cm2 with a Stratalinker 1800 device (Stratagen, La Jolla, CA). Viability of the photochemically inactivated cultures was assessed by plating bacteria on Pseudomonas Isolation Agar (PIA) and counting of the colony-forming units. Following the photochemical treatment with 10 μmol/l of amotosalen one live organism per 1.25 × 108 bacteria is able to replicate after OSTAB strain photo-inactivation.
Immunizations
For immunizations, KBMA bacteria were prepared as described above. Live OSTAB S54-OVA and OSTAB S54-Ei strains were grown overnight in LB containing 300 mg/l of carbenicillin at 37°C and 250rpm. The next day, after two washing steps in LB, bacteria were resuspended in LB supplemented with 1 mmol/l IPTG and 300 mg/l carbenicillin at an OD600 = 0.2 and cultured until an OD600 = 1.6 (~3 hours of culture). Then bacteria (KBMA and live OSTAB strains) were harvested and washed two times in phosphate buffered saline (PBS). HK S54-OVA bacteria were prepared from OSTAB S54-OVA strain by heating at 65°C for 1 hour.
Mice were s.c. injected in the left flank (single site vaccination, 100 µl each) with 5 × 106 OSTAB (S54-Ei or S54-OVA) bacteria or in the right and left flanks and the right and left shoulders (four sites vaccinations, 50 µl each) with 5 × 107 KBMA S54-OVA or HK S54-OVA bacteria/injection site every 4 days (5 or 6 times).
Tumor challenge
Therapeutic setting.
About 2 × 105 B16-OVA tumor cells were injected s.c. into the right flank at day 0 and when tumors became palpable (between 5–8 days), we began to immunize mice.
Long-term tumor prevention study.
About 6 mice per group were s.c. inoculated with different immunization conditions as previously described.17 One hundred days after the last immunization, 2 × 105 B16-OVA were injected into the right flank. In each experiment tumors were measured every 2 days (length × width in square millimeter). When tumor area reached 150 mm2 experiment was ended.
TILS analysis
B16-OVA tumors were harvested when significant difference in tumor size was observed between treated and control groups, cut into small pieces and resuspended in 3 ml of Roswell Park Memorial Institute (RPMI) 1640 medium containing Liberase TM 25 mg/ml (Roche Diagnostics, Indianapolis). A first dissociation with a gentle MACS Dissociator (Miltenyi Biotec, Cologne, Germany) was performed and samples were then incubated for 30 minutes at 37°C, before a second dissociation with the gentle MACS dissociator. After washing with PBS and passing through a 70 µm strainer, cell suspensions were used for subsequent immunostaining.
Antibodies, MHC multimers, and flow cytometry
Antimouse antibodies for CD45 (30-F11), CD4 (GK1.5 or RM4-5), CD8a (53–6.7), CD44 (IM7), CD127 (SB/199), CD62-L (MEL-14), KLRG1 (2F1), IFNγ (XMG1.2), TNFα (MP6-XT22), IL-2 (JES6-5H4), CD107a (1D4B), FoxP3 (FJK-16s) were obtained from eBioscience (Paris, France) or BD Biosciences (San Jose, CA). H2Kb-SIINFEKL Dextramer, a multimer of H2Kb loaded with OVA257-264 antigen was purchased at Immudex (Copenhagen, Denmark). Staining was performed as described by manufacturer’s instruction. The Fixable Viability Dye (FVD eF780, eBioscience) was used for tumor-infiltrated study in order to exclude dead cells for flow cytometry analysis. Flow cytometry acquisition was done using a BD Biosciences FACSCanto II and analysis carried out with FlowJo software (Tree Star, Ashland, OR).
Phenotypic characterization of antigen-specific CD8+ T cells
Splenocytes or blood cells were harvested at various time intervals, stained first with H2Kb-SIINFEKL multimers and with antibodies against CD45 (only for blood cells), CD4, CD8, CD44, CD127, CD62-L, and KLRG1. For absolute counting in blood samples, we used Flow-Count Fluorospheres (Beckman Coulter, Miami, FL). Blood cells were fixed in fluorescence-activated cell sorting (FACS) lysing buffer (BD Biosciences) and acquired by flow cytometry.
For the tumor infiltration study, cells were stained first with H2Kb-SIINFEKL multimers, then with antibodies against CD45, CD4, CD8, CD44, and with the fixable viability dye. In a second step, intracellular staining of FoxP3 was performed with the FoxP3/Transcription Factor Staining Buffer Set (eBioscience).
CD8+ T cell function and intracellular staining
Intracellular cytokines production by antigen-specific CD8+ T cells was carried out by 4 hours restimulation with splenocytes in the presence of SIINFEKL peptide (OVA257-264, 10 µg/ml) or without peptide as a control. Monensin (0.7 µl/ml, GolgiStop from BD Biosciences) and anti-CD107a antibody to analyze degranulation by cytotoxic T lymphocytes were added before starting restimulation. Cells were then surface-labeled with anti-CD8, anti-CD4 antibodies and were subjected to intracellular cytokines staining (BD Biosciences). Intracellular cytokines were analyzed only on CD8+ T cells.
Statistical analysis
Results are expressed as the mean ± SEM unless stated otherwise. Statistical comparisons between two groups were evaluated by the Student’s t-test. A P-value < 0.05 was considered to indicate statistical significance. Unless otherwise indicated, all experiments were conducted at least twice. Log-rank tests (Mantel–Cox) were used to compare the Kaplan–Meier tumor survival curves. Statistical and graphical analyses were performed using GraphPad Prism Version 6 (GraphPad Software, La Jolla, CA).
Acknowledgments
We are grateful to Pierre Champelovier and Erwann Ventre for the flow cytometry analysis. This work was supported by the GEFLUC of Grenoble (Groupement des Entreprises Françaises dans la Lutte contre le Cancer), the Ligue contre le cancer (comité Isère) and the Canceropole Rhône alpes Auvergne (CLARA), Region Auvergne Rhône Alpes and Metropole de Lyon.
D.H., A.L.G., B.P., L.B., and B.T. declare competing financial interest. A.L.G., B.P., B.T., and L.B. are the cofounders of APCure, a start-up that produces immunotherapy treatments for patients with cancer.
References
- Galluzzi, L, Vacchelli, E, Bravo-San Pedro, JM, Buqué, A, Senovilla, L, Baracco, EE et al. (2014). Classification of current anticancer immunotherapies. Oncotarget 5: 12472–12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff, PW, Higano, CS, Shore, ND, Berger, ER, Small, EJ, Penson, DF et al. IMPACT Study Investigators. (2010). Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363: 411–422. [DOI] [PubMed] [Google Scholar]
- McCune, JS (2016). Immunotherapy to treat cancer. Clin Pharmacol Ther 100: 198–203. [DOI] [PubMed] [Google Scholar]
- Mellman, I, Coukos, G and Dranoff, G (2011). Cancer immunotherapy comes of age. Nature 480: 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman, J and Paterson, Y (2013). Live-attenuated Listeria-based immunotherapy. Expert Rev Vaccines 12: 493–504. [DOI] [PubMed] [Google Scholar]
- Toussaint, B, Chauchet, X, Wang, Y, Polack, B and Le Gouëllec, A (2013). Live-attenuated bacteria as a cancer vaccine vector. Expert Rev Vaccines 12: 1139–1154. [DOI] [PubMed] [Google Scholar]
- Bichsel, C, Neeld, DK, Hamazaki, T, Wu, D, Chang, LJ, Yang, L et al. (2011). Bacterial delivery of nuclear proteins into pluripotent and differentiated cells. PLoS One 6: e16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouazi, M, Toussaint, B, Quénée, L, Epaulard, O, Guillaume, M, Marlu, R et al. (2008). High-yield production of secreted active proteins by the Pseudomonas aeruginosa type III secretion system. Appl Environ Microbiol 74: 3601–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epaulard, O, Toussaint, B, Quenee, L, Derouazi, M, Bosco, N, Villiers, C et al. (2006). Anti-tumor immunotherapy via antigen delivery from a live attenuated genetically engineered Pseudomonas aeruginosa type III secretion system-based vector. Mol Ther 14: 656–661. [DOI] [PubMed] [Google Scholar]
- Le Gouëllec, A, Chauchet, X, Laurin, D, Aspord, C, Verove, J, Wang, Y et al. (2013). A safe bacterial microsyringe for in vivo antigen delivery and immunotherapy. Mol Ther 21: 1076–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack, B, Vergnaud, S, Paclet, MH, Lamotte, D, Toussaint, B and Morel, F (2000). Protein delivery by Pseudomonas type III secretion system: Ex vivo complementation of p67(phox)-deficient chronic granulomatous disease. Biochem Biophys Res Commun 275: 854–858. [DOI] [PubMed] [Google Scholar]
- Bichsel, C, Neeld, D, Hamazaki, T, Chang, LJ, Yang, LJ, Terada, N et al. (2013). Direct reprogramming of fibroblasts to myocytes via bacterial injection of MyoD protein. Cell Reprogram 15: 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser, AR (2009). The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7: 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouazi, M, Wang, Y, Marlu, R, Epaulard, O, Mayol, JF, Pasqual, N et al. (2010). Optimal epitope composition after antigen screening using a live bacterial delivery vector: application to TRP-2. Bioeng Bugs 1: 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epaulard, O, Derouazi, M, Margerit, C, Marlu, R, Filopon, D, Polack, B et al. (2008). Optimization of a type III secretion system-based Pseudomonas aeruginosa live vector for antigen delivery. Clin Vaccine Immunol 15: 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gouëllec, A, Chauchet, X, Polack, B, Buffat, L and Toussaint, B (2012). Bacterial vectors for active immunotherapy reach clinical and industrial stages. Hum Vaccin Immunother 8: 1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y, Gouëllec, AL, Chaker, H, Asrih, H, Polack, B and Toussaint, B (2012). Optimization of antitumor immunotherapy mediated by type III secretion system-based live attenuated bacterial vectors. J Immunother 35: 223–234. [DOI] [PubMed] [Google Scholar]
- Galon, J, Costes, A, Sanchez-Cabo, F, Kirilovsky, A, Mlecnik, B, Lagorce-Pagès, C et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313: 1960–1964. [DOI] [PubMed] [Google Scholar]
- Sato, E, Olson, SH, Ahn, J, Bundy, B, Nishikawa, H, Qian, F et al. (2005). Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102: 18538–18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L, Conejo-Garcia, JR, Katsaros, D, Gimotty, PA, Massobrio, M, Regnani, G et al. (2003). Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348: 203–213. [DOI] [PubMed] [Google Scholar]
- Finn, OJ (2008). Cancer immunology. N Engl J Med 358: 2704–2715. [DOI] [PubMed] [Google Scholar]
- Kaech, SM, Tan, JT, Wherry, EJ, Konieczny, BT, Surh, CD and Ahmed, R (2003). Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4: 1191–1198. [DOI] [PubMed] [Google Scholar]
- Joshi, NS, Cui, W, Chandele, A, Lee, HK, Urso, DR, Hagman, J et al. (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27: 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, EA, Spadaccia, MR, Norton, T, Demmler, M, Gopal, R, O’Brien, M et al. (2015). Attenuated Listeria monocytogenes vectors overcome suppressive plasma factors during HIV infection to stimulate myeloid dendritic cells to promote adaptive immunity and reactivation of latent virus. AIDS Res Hum Retroviruses 31: 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, SN, Gebhardt, T, Carbone, FR and Heath, WR (2013). Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 31: 137–161. [DOI] [PubMed] [Google Scholar]
- Bachmann, MF, Wolint, P, Schwarz, K, Jäger, P and Oxenius, A (2005). Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J Immunol 175: 4686–4696. [DOI] [PubMed] [Google Scholar]
- Seder, RA, Darrah, PA and Roederer, M (2008). T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 8: 247–258. [DOI] [PubMed] [Google Scholar]
- Wherry, EJ, Teichgräber, V, Becker, TC, Masopust, D, Kaech, SM, Antia, R et al. (2003). Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 4: 225–234. [DOI] [PubMed] [Google Scholar]
- Stark, FC, Sad, S and Krishnan, L (2009). Intracellular bacterial vectors that induce CD8(+) T cells with similar cytolytic abilities but disparate memory phenotypes provide contrasting tumor protection. Cancer Res 69: 4327–4334. [DOI] [PubMed] [Google Scholar]
- Forbes, NS (2010). Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer 10: 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, IY, Van, TT and Smooker, PM (2015). Live-attenuated bacterial vectors: tools for vaccine and therapeutic agent delivery. Vaccines (Basel) 3: 940–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, LM and Paterson, Y (2014). Attenuated Listeria monocytogenes: a powerful and versatile vector for the future of tumor immunotherapy. Front Cell Infect Microbiol 4: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, TS and Prince, AS (2013). Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest 123: 1630–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes, HK, Bicknell, S, MacGregor, G and Evans, TJ (2014). T helper cell subsets specific for Pseudomonas aeruginosa in healthy individuals and patients with cystic fibrosis. PLoS One 9: e90263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, W, Huang, J, Duan, B, Traficante, DC, Hong, H, Risech, M et al. (2012). Th17-stimulating protein vaccines confer protection against Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 186: 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen, EM, Lemmens, EE, Wolfe, T, Christen, U, von Herrath, MG and Schoenberger, SP (2003). CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421: 852–856. [DOI] [PubMed] [Google Scholar]
- Kupz, A, Guarda, G, Gebhardt, T, Sander, LE, Short, KR, Diavatopoulos, DA et al. (2012). NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8⁺ T cells. Nat Immunol 13: 162–169. [DOI] [PubMed] [Google Scholar]