

Abstract
Cancer vaccine application is limited to specific cancer types because few cancer-associated antigens are known to induce tumor rejection. Accordingly, we assessed the utility of Ad881, an oncolytic adenovirus in which viral replication was strictly regulated by the cancer-specific midkine promoter, as a cancer vaccine in a murine colorectal cancer model lacking specific cancer-associated antigens. In CT26 and CMT93 cells, Ad881 (multiplicity of infection: 100 or 1,000) showed stronger cytotoxicity and oncolysis in vitro than its equivalent replication-defective adenovirus, Ad884. CT26 cells (1 × 104) infected with Ad881 (multiplicity of infection: 1,000) for 24 hours were suitable as vaccine antigens without tumor formation in our model. Repeated vaccinations, but not single vaccination, induced a greater prophylactic immune response. The percentage of mice that rejected the tumor challenge was 0, 4, and 38% after no vaccination, single vaccination, and repeated vaccinations, respectively. Immunogenic cell death marker high-mobility group box 1 protein (HMGB1) and adenosine triphosphate in culture medium were higher after Ad881 infection (24.3 ng/ml and 48.2 nmol/l, respectively) than after Ad884 infection (8.6 ng/ml and 15.4 nmol/l, respectively) or oxaliplatin treatment (3.7 ng/ml and 1.8 nmol/l, respectively). These results indicate that repeated whole cell vaccination using an oncolytic adenovirus may be a potent approach to evoke immunogenic cell death.
Introduction
Cancer vaccines can be developed based on cancer-associated antigens (CAAs).1,2 However, most common malignancies bear no effective CAAs that induce rejection of tumors. Therefore, common cancers have been considered unsuitable targets for cancer vaccines, although some cancer-specific proteins, such as telomerase reverse transcriptase, have been proposed as universal CAAs.3 Colorectal cancer (CRC) is one of the most common malignancies worldwide.4 However, there are no promising vaccine protocols for clinical use, because there is no specific immunogenic antigen in CRC.5,6
Midkine is highly expressed in malignancies including CRC, but not in normal tissues.7 The utility of midkine promoter-driven gene therapy in a CRC model has been reported in the context of a replication-defective adenoviral vector.8 Therefore, we used a midkine promoter-driven oncolytic adenoviral vector in this study.
Oncolysis following oncolytic virus infection induces immune responses and increases the immunogenicity of cancer cells for a long time.9–11 Therefore, the application of oncolytic viruses has been proposed for cancer immunotherapy, including cancer vaccines.12–16 Immunogenic cell death (ICD) was found to be a novel mechanism of cell death induced by doxorubicin in 2005.17 Subsequently, other anticancer drugs have been shown to induce ICD, including oxaliplatin used for advanced CRC patients, as well as other anticancer treatments such as radiotherapy and virotherapy.18 Therefore, ICD is considered to be one of the favorable responses induced by oncolytic virotherapy.15,16
Oncolytic adenoviruses have been applied in many clinical trials of cancer therapy.19 However, the clinical efficacy is often hampered by immunity against adenoviruses.20,21 Although immune responses against infected cancer cells have been the focus, experimental designs have not been developed because of low cytotoxicity and oncolysis in murine models compared with those in humans.22,23
Repeated treatments using the same oncolytic virus are usually ineffective to directly kill cancer cells because of the immune response against the virus. However, repeated vaccination enhances immune responses against cancer cells.24
Here, we assessed a novel experimental model using an oncolytic adenovirus as a cancer vaccine against a CRC model in mice. We evaluated ICD induced by oncolytic adenovirus infection and the utility of repeated vaccinations of infected cancer cells to enhance tumor immunity.
Results
Assessment of cytotoxicity and oncolysis in vitro
First, we investigated the cytotoxic efficacy of adenoviral vectors in CRC cells. We tested two adenoviral vectors illustrated in Figure 1a.25,26 The replication-defective vector Ad884 did not induce apparent cell death at a multiplicity of infection (MOI) of up to 100 in human DLD-1 cells or up to 1,000 in murine CRC cell lines (CMT93 and CT26) and fibroblasts (Figure 1b). In contrast, a conditionally replicating oncolytic vector, Ad881, in which the adenoviral E1 gene is driven by the midkine promoter, showed dose-dependent cytotoxicity in DLD-1, CT26, and CMT93 cells, but not in fibroblasts (Figure 1c). In human DLD-1 cells, Ad881 induced cytotoxicity at an MOI of 0.1–1. In contrast, in murine CMT93 and CT26 cells, Ad881 induced cytotoxicity at a higher MOI of 100–1,000. These data were consistent with previous reports indicating that murine tumor cells are less permissive to infection by human adenoviruses compared with human tumor cells.22,23
Figure 1.
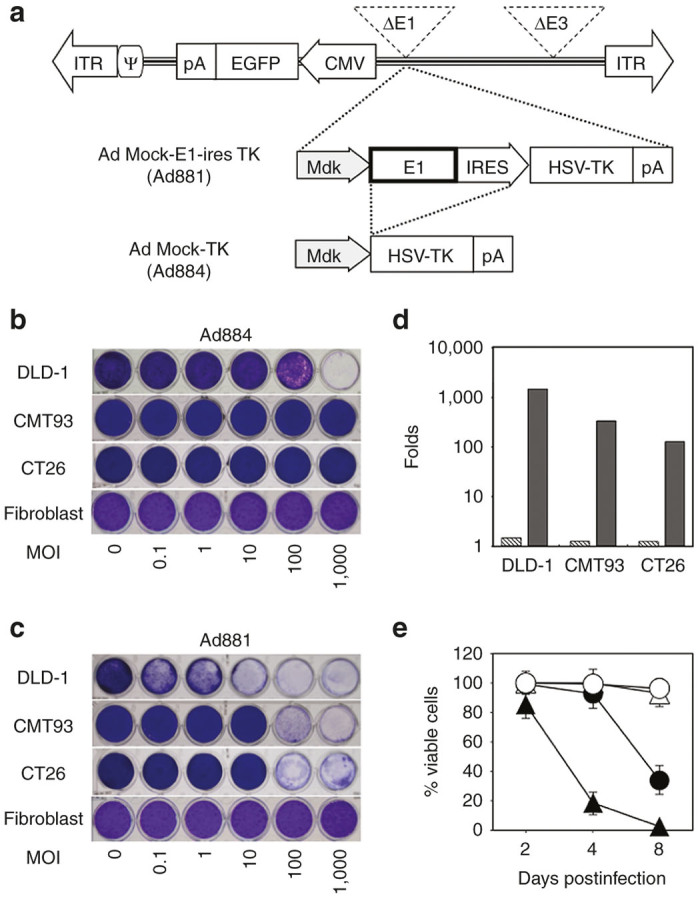
Schematic structure of adenoviral vectors and cytotoxic efficiencies of oncolytic adenoviruses in colorectal cancer (CRC) cells. (a) Schematic structure of adenoviral vectors. Schematic structures of Ad881 and Ad884 are shown. ITR, adenovirus inverted terminal repeat sequence; ψ, packaging signal; pA, polyadenylation signal; IRES, internal ribosome entry site; EGFP, enhanced green fluorescent protein; CMV, cytomegalovirus promoter; Mdk, midkine; HSV-TK, herpes simplex virus-thymidine kinase. (b)/(c) Cyotopathic assays using Ad884 in CRC cells and normal fibroblasts. CRC cell lines (DLD-1, CMT93, and CT26) and fibroblasts were infected with Ad884 (b) or Ad881 (c) at various multiplicity of infections (MOIs) (0, 0.1, 1, 10, 100, or 1,000). On day 8, cytotoxicity was assessed by the extent of crystal violet staining. (d) Virus progeny production in CRC cells. DLD-1, CMT93, and CT26 cells, as well as fibroblasts were infected with Ad881 or Ad884 at an MOI of 100. At 48 hours after infection, cells and media were harvested to determine the viral titer in transducing units by EGFP expression using flow cytometry. Virus production levels were normalized to the baseline value in fibroblasts. Data are representative of three independent experiments all yielding similar results. Ad881: black bar; Ad884: hatched bar. (e) Time-dependent cytotoxicity. DLD-1 (triangle) and CT26 (circle) cells (1 × 104/well) were cultured as multiple replicates in 96-well plates and infected with Ad881 (closed) or Ad884 (open) at an MOI of 100. On the indicated days, the number of surviving cells was analyzed by a colorimetric method using Alamar blue. Data shown are the mean ± SD of triplicates.
To assess oncolytic adenovirus propagation in murine CRC cells, both human and murine CRC cells were infected with Ad881 or Ad884 at an MOI of 100 for 3 hours and then cultured for 48 hours in virus-free medium. A viral progeny production assay was performed using the medium and cells as described in the “Materials and Methods”. When compared with the baseline value in fibroblasts and the values in Ad884-infected cells (DLD1 (1.6-fold), CMT93 (1.3-fold), and CT26 (1.2-fold)), Ad881 showed much more propagation in DLD-1 cells (1443.6-fold) and more propagation in murine CRC cells (CMT93 (351.5-fold) and CT26 (130.1 fold)) (Figure 1d).
The time course of cytotoxicity induced by adenoviral infection was assayed in DLD-1 and CT26 cells infected with Ad881 or Ad884 at an MOI of 100. Ad881 showed progressive cytotoxicity in both DLD-1 and CT26 cells, whereas Ad884 showed no significant cytotoxicity at any time point (Figure 1e).
We also assessed transduction efficiency in CT26 cells infected with Ad881 by enhanced green fluorescence protein (EGFP) expression, because Ad881 carried the EGFP gene as a marker. Following Ad881 infection, the percentage of CT26 cells positive for EGFP was increased in a virus dose-dependent manner and reached nearly 100% at an MOI of 1,000 (Figure 2).
Figure 2.
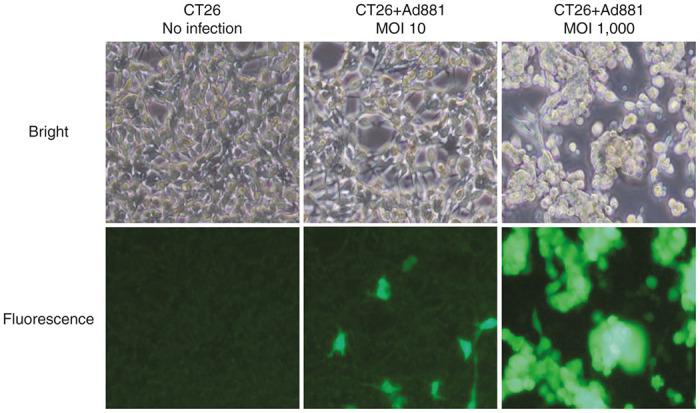
Infection efficacy of the oncolytic adenovirus Ad881 in CT26 cells. Representative bright field (top) and enhanced green fluorescence protein (EGFP) fluorescence (bottom) images of CT26 cells that were left uninfected (left) or infected with Ad881 at an multiplicity of infection (MOI) of 10 (middle) or 1,000 (right).
These data demonstrate that Ad881 infected, replicated, and produced viral progeny in mouse CRC cell lines CMT93 and CT26, although with less efficiency than that in human CRC cells (DLD1).
Assessment of tumor formation by infected cells in vivo
Next, we assessed tumor formation by infected cells in vivo. Cells infected with Ad881 should be eradicated by oncolysis and host immune responses in vivo, when tumor formation was inhibited. When mice (four per protocol) were inoculated with 1 × 105 CT26 cells that were infected with Ad881 for 10 hours at an MOI of 10, 100, or 1,000, they all developed tumors (Figure 3a). However, when 1 × 105 CT26 cells that were infected with Ad881 for 24 hours at an MOI of 1,000 were injected into mice, only one of the six inoculated mice developed tumors (Figure 3b). Similarly, when mice were inoculated with fewer (1 × 104) CT26 cells infected with Ad881 for 24 hours at an MOI of 1,000, none of the 24 inoculated mice developed tumors (Figure 3b). There was significant difference in tumor formation between MOI = 1,000 / 1 × 104 and MOI = 1,000 / 1 × 105 (P < 0.0001). Based on these findings, a longer incubation time (24 hours) and fewer tumor cells (1 × 104) were more appropriate for the vaccination protocol.
Figure 3.

Assessment of vaccine conditions. (a) The tumor formation rate in mice following subcutaneous injection with 1 × 105 Ad881-infected CT26 cells that had been infected for 10 hours at multiplicity of infections (MOIs) of 0, 10, 100, or 1,000. (b) The tumor formation rate in mice following subcutaneous injection of 1 × 104 or 1 × 105 Ad881-infected CT26 cells that had been infected for 24 hours at MOIs of 0 or 1,000.
Efficiency of the vaccine
We assessed the efficiency of the vaccination protocol by further inoculation of uninfected CT26 cells after vaccination (Figure 4a). A single vaccination with 1 × 104 Ad881-infected CT26 cells was relatively effective when the vaccine efficacy was evaluated by later challenging the mice with the same number of CT26 cells that was originally used for vaccination (Figure 4b). The tumor rejection rate (63%; 15 of 24 mice) of the MOI 1,000 group was significantly higher than that (19%; 3 of 16 mice) of the control group (P = 0.005) and relatively higher than that (33%: 6 of 18 mice) of the MOI 0 group (P = 0.06). In the control group, some mice rejected inoculation of 1 × 104 CT26 cells. In contrast, no mouse rejected inoculation of 1 × 105 CT26 cells among the 26 control mice. When mice that had received a single vaccination of 1 × 104 Ad881-infected CT26 cells were challenged with 1 × 105 CT26 cells, tumor growth was prevented in only 3.9% of mice (1 of 26 mice), which was much lower compared with inoculation of 1 × 104 CT26 cells. In contrast, repeated vaccinations prevented tumor growth in 38.5% of mice (10 of 26 mice) (Figure 4c). The incidence of tumor rejection following repeated vaccinations was significantly higher than that following a single vaccination (P = 0.0001) or no vaccination (P < 0.0001).
Figure 4.

Assessment of vaccine efficiency. (a) Schema of the vaccination protocol used in this study. (b) The tumor rejection rate in mice that had been previously vaccinated with 1 × 104 of Ad881-infected (MOI: 0 or 1,000) CT26 cells or no previous treatment when these mice were injected with 1 × 104 uninfected CT26 as a challenge. (c) The tumor rejection rate following challenge with 1 × 105 uninfected CT26 cells in mice that were vaccinated with 1 × 104 Ad881-infected CT26 cells once or twice (repeatedly). MOI, multiplicity of infection.
ICD
We evaluated the level of ICD by measuring high-mobility group box 1 protein (HMGB1) and adenosine triphosphate (ATP) in the culture medium. Without treatment, the culture medium contained 3.6 ng/ml HMGB1. In contrast, the culture medium of cells infected with replication-deficient adenovirus Ad884 (MOI: 1,000) contained 8.6 ng/ml HMGB1, that of cells infected with oncolytic adenovirus Ad881 (MOI: 1,000) contained 24.3 ng/ml HMGB1, and that of cells treated with 100 µmol/l oxaliplatin, which is the main anticancer drug for advanced CRC and recognized as an inducer of ICD, contained 3.7 ng/ml HMGB1 (Figure 5a).27 The level of HMGB induced by Ad881 (MOI: 1,000) was significantly higher than that induced by the other treatments (P < 0.0001). Without treatment, the culture medium contained 2.1 nmol/l ATP. In contrast, the culture medium of cells infected with Ad884 (MOI: 1,000) contained 15.4 nmol/l ATP, that of cells infected with the oncolytic adenovirus (MOI: 1,000) contained 48.2 nmol/l ATP, and that of cells treated with 100 µmol/l of oxaliplatin contained 1.8 nmol/l ATP (Figure 5b). The level of ATP induced by Ad881 (MOI: 1,000) was significantly higher than that induced by the other treatments (P < 0.0001 except for Ad884 (MOI: 1,000), P = 0.0008). These data indicate that virus infection can induce immunogenic changes in cancer cells, but these changes are greater when the virus has oncolytic properties.
Figure 5.
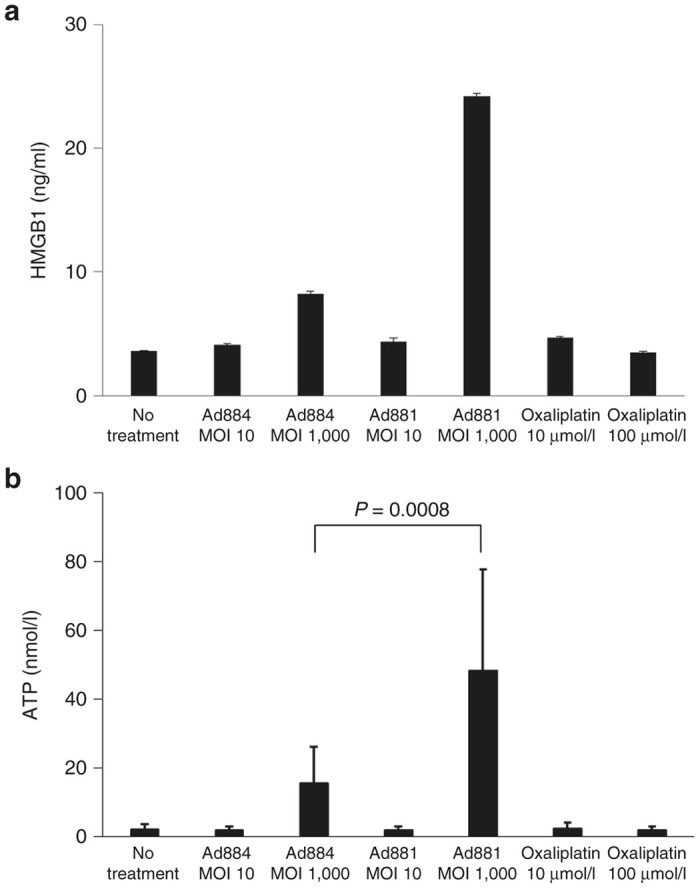
Immunogenic cell death (ICD). ICD was assessed by measuring HMGB1 (a) and ATP (b) in culture media after various treatments: no treatment, Ad884 (MOI: 10 or 1,000), Ad881 (MOI: 10 or 1,000), or oxaliplatin treatment (10 or 100 µmol/l). ATP, adenosine triphosphate; HMGB1, high-mobility group box 1 protein; MOI, multiplicity of infection.
Discussion
Our results demonstrated that cancer cells infected with an oncolytic adenovirus can be a source of tumor antigens for induction of ICD. Moreover, repeated vaccination induced a more potent antitumor effect than that of single vaccination.
Oncolytic viruses have been used for anticancer therapy through direct injection into cancer tissues or via intravenous injection.19–21,28 The targeting of oncolytic viruses to cancer cells is quite specific, and the safety of these viruses has been shown by previous clinical trials.20 However, oncolytic viruses have been insufficient to eradicate cancer cells because of rapid clearance by the host immune system, especially following repeated treatments. Therefore, while treatment with oncolytic viruses is an ideal approach to target cancer cells, improvements in delivering these viruses to cancer tissues are indispensable for their practical application.19–21,28 In this study, we performed ex vivo infection of murine cells with an oncolytic adenovirus, because oncolytic adenoviruses infect murine cells much less than human cells. We found that ex vivo infection with an oncolytic adenovirus induced cytotoxicity and oncolysis in murine CRC cells. Moreover, infected cells were suitable as tumor antigens for vaccination, although both a long incubation time of 24 hours and a higher MOI of 1,000 were necessary to eradicate inoculated cells in vivo. We have used fewer cells for vaccination to avoid tumor formation. This tumor formation showed that small number of cells survived against cytotoxic effect by oncolytic virus in vivo and these surviving cells should be killed by antitumor immunity when tumor formation was inhibited. This result also showed that the quality but not quantity of CAAs presentation was important for induction of antitumor immunity.
Recent studies have focused on effects of oncolytic viruses other than direct tumor killing and apoptosis.21,29,30 Specifically, oncolytic viruses may induce T cell and/or dendritic cell activation and stimulate innate and/or adaptive antitumor immune responses. Our data show that the oncolytic adenovirus induced ICD more strongly than the replication-deficient adenovirus or oxaliplatin treatment that was previously shown to induce ICD.27 These characteristics of oncolytic adenoviruses appear to be ideal for their application as a cancer vaccine.
Whole tumor antigens have been successfully used as cancer vaccines, and they are particularly useful for treating cancers without identified CAAs.31 Unlike vaccines that use specific CAAs, whole tumor antigen vaccines are available for all types of cancers. Furthermore, they target multiple epitopes that can induce an immune response. Lysates or RNAs of tumor and irradiated tumor cells have been used as whole tumor antigens.31 Tumor cells killed by oncolytic viruses are also likely to be suitable for antigen presentation because such viruses induce ICD. We used oxaliplatin for positive control for inducer of ICD because oxaliplatin is reported to induce ICD and used as one of the most important anticancer drug for colorectal cancer patients. 27 However, oxaliplatin did not induce ICD under the condition used in this study. We considered the cell density in this study should be different from that in the previous report. Adenoviruses used in this study induced stronger ICD than oxaliplatin in the same condition. Therefore, both oncolytic adenovirus and replicating deficient adenovirus are better inducer of ICD than oxaliplatin.
This study addresses the application and an experimental model of oncolytic adenoviruses for cancer therapy.21,29 The MOI of 1,000 used in this study appears to be very high, because the adenovirus used here was a human virus that infected human cells and not murine cells specifically, although we showed progeny production of adenoviruses and oncolysis in murine cells. However, our goal is clinical application for humans. Our results under a much lower transduction efficiency than that in human cells strongly suggest the utility of whole cell vaccination by oncolytic adenoviruses in humans, as well as in this model. The problem of the high MOI used in this study should be easily resolved in applications for humans.
For effective clinical use, enhancement of the anticancer immune response is necessary. Combinations with other immunotherapies, including granulocyte-macrophage colony-stimulating factor and immune checkpoint blockade appear to be promising.21 Although, many further experiments are needed before clinical use, our protocol provides a novel cancer vaccine that induces ICD by an oncolytic adenovirus.
Materials and Methods
Cell lines
The human CRC cell line DLD1 was purchased from the Japan Health Science Foundation (Osaka, Japan). Mouse CRC cell lines Colon-26 (CT26) and CMT93 were purchased from RIKEN BRC Cell Bank (Ibaraki, Japan) and DS Pharma Biomedical (Osaka, Japan), respectively. DLD1 and CT26 cells were cultured in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS). CMT93 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS and 2 mmol/l glutamine. Human dermal fibroblasts and their specific medium were purchased from Cell Systems (Kirkland, WA). Human embryonic kidney (HEK) 293 cells were purchased from Microbix (Toronto, Ontario, Canada). HEK293 cells were maintained in DMEM with 10% FBS. Culture media and FBS were obtained from Life Technologies Japan (Tokyo, Japan). All cells were grown at 37°C in a humidified incubator with 5% CO2.
Adenoviral vectors
In this study, we used two previously described adenoviruses (Figure 1a).25,26 Ad881 is a conditionally replicating oncolytic adenovirus in which the adenoviral E1 gene is driven by the midkine promoter. Ad884 is a replication-defective adenovirus that was used as a control nononcolytic adenovirus. Both vectors contain an independent cytomegalovirus promoter-driven EGFP marker gene in the adenoviral backbone. The vectors also contain a herpes simplex virus-thymidine kinase suicide gene that was not activated by a prodrug in this study. They were grown in HEK293 cells and purified by CsCl ultracentrifugation, followed by dialysis against 10 mmol/l Tris-HCL buffer (pH 8.0) with 10% glycerol. The titers of the vectors were assayed by conventional limiting dilution on HEK293 cells.
All experiments using oncolytic adenoviruses were approved by the Minister of Education, Culture, Sports, Science and Technology and by the Safety Committee for Recombinant DNA Experiments of Hyogo College of Medicine.
In vitro cytotoxicity assay
DLD1, CT26, and CMT 93 cells, as well as fibroblasts (1 × 105/well) were cultured on 24-well plates and infected with adenoviruses (Ad881 or Ad884) at various MOIs (0, 0.1, 1, 10, 100, or 1,000). Half of the medium was replaced with fresh medium every day. On day 8, the cells were fixed with 10% buffered formalin containing 1% crystal violet for 30 minutes. The cytotoxic effect of each virus was assessed by the extent of crystal violet staining.
To investigate time-dependent cytotoxic effects of each adenovirus, DLD-1, CMT93 and CT26 cells, as well as fibroblasts (1 × 104 cells/well) were seeded in triplicate into 96-well culture plates and infected with Ad881 or Ad884 at an MOI of 100. Half of the medium was replaced with fresh medium every day. On days 2, 4, and 8 after infection, viable cell numbers in the triplicate cultures were measured by the Alamar blue method according to the manufacturer’s instructions (Alamar Biosciences, Sacramento, CA). Briefly, the cells were incubated for 3 hours after addition of 40 μl Alamar blue. Then, fluorescence was measured by an ARVOX4 multilabel plate reader with 544 nm excitation and 590 nm emission wavelengths (PerkinElmer Japan, Tokyo, Japan). The ratio of viable cells was calculated by the fluorescence of each well divided by the fluorescence of untreated wells.
Viral progeny production assay
Cells (5 × 105/well) were seeded in six-well plates and infected with adenoviruses at an MOI of 100 for 3 hours. Then, the infection medium was replaced with fresh medium. Forty hours later, both cells and media were collected and freeze/thawed three times. Serial dilutions of the virus supernatants after centrifugation were tittered on HEK293 cells by EGFP expression using flow cytometry. Virus production levels of Ad881 and Ad884 were evaluated by the fold increases against the baseline level in fibroblasts. Data are representative of three independent experiments.
Mice
About four to six-week-old female BALB/cAJcl and C57/BL6JJcl mice were obtained from Japan Clea (Tokyo, Japan) and maintained in the Institute of Experimental Animal Science, Hyogo College of Medicine. Animal experiments were approved by the Animal Research Committee and carried out in accordance with institutional guidelines. The mice were maintained under standard environmental conditions with free access to food and water.
Inoculation of infected cells in vivo to assess the vaccination protocol
The tumor formation of infected cells was assessed initially to determine a suitable vaccine protocol. CT26 cells were infected with the Ad881 vector (MOI: 0, 10, 100, or 1,000) for 10 hours. Then, 1 × 105 infected cells were injected subcutaneously into the flanks of BALB/cAJcl mice. CT26 cells were alternately infected with the Ad881 vector (MOI: 0 or 1,000) for 24 hours. Then, 1 × 104 or 1 × 105 infected cells were injected subcutaneously into the flank of the mice. CMT93 cells were not used for in vivo experiments because they did not induce any tumor formation, even after the inoculation of 1 × 107 cells into 16 C57/BL6JJcl mice.
Vaccination and tumor challenge
Mice received a subcutaneous injection in their flank of 1 × 104 CT26 cells infected with Ad881 at an MOI of 0 or 1,000 as a prophylactic vaccine. One week after the vaccination, 1 × 104 or 1 × 105 uninfected CT26 cells were injected in the opposite flank of the mice to assess the effect of the vaccine (Figure 4a). To evaluate the utility of repeated vaccination, 1 × 104 CT26 cells infected with Ad881 (MOI: 1,000) were injected subcutaneously into the flank of mice twice with an interval of 1 week. Then, at 1 week after the last vaccination, 1 × 105 uninfected CT26 cells were injected into the opposite flank of the mice to assess the effect of the vaccine. The resulting data shown in this study are the sum of three independent experiments.
ICD assessment
ICD was assessed by HMGB1 and ATP release into culture medium after virus infection or oxaliplatin treatment. In a 12-well plate, 3 × 105 CT26 cells/well were infected with Ad881 at an MOI of 10 or 1,000 or with Ad884 at an MOI of 10 or 1,000 for 24 hours, or treated with 10 or 100 µmol/l oxaliplatin for 4 hours. The total volume was adjusted to 1 ml in each condition. The amount of HMGB1 and ATP in the culture medium was measured using an HMGB1 enzyme-linked immunosorbent assay II kit (SHINO-TEST, Tokyo, Japan) and a luciferin-based ENLITEN ATP assay (Promega, Madison, WI), respectively, according to each manufacturer’s instructions. These experiments were performed twice in two wells, and the results are presented as the mean + standard deviation (SD).
Statistical analysis
Statistical analyses were performed using JMP version 11 (SAS Japan, Tokyo, Japan). Data available for analysis are expressed as the mean with SD of the mean. The two-tailed Student’s t-test was performed to evaluate significance between treatments and values. Vaccine efficacy was assessed by comparing the tumor rejection rate among the vaccine types using the χ2-test. All differences with a P-value of <0.05 were considered statistically significant.
Acknowledgments
This work was supported by Japanese Society for the Promotion of Science Grants-in-Aid (23591973 and 25460484), a Ministry of Education, Culture, Sports, Science and Technology HAITEKU grant (S1291009), and Chugai Pharmaceuticals. The authors thank the joint-use research facilities and animal facilities of Hyogo College of Medicine for collecting data and Edanz Group Ltd for English editing.
We declare support from Chugai Pharmaceuticals to perform this study.
References
- van der Bruggen, P, Traversari, C, Chomez, P, Lurquin, C, De Plaen, E, Van den Eynde, B et al. (1991). A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254: 1643–1647. [DOI] [PubMed] [Google Scholar]
- Tagliamonte, M, Petrizzo, A, Tornesello, ML, Buonaguro, FM and Buonaguro, L (2014). Antigen-specific vaccines for cancer treatment. Hum Vaccin Immunother 10: 3332–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton, G, Silcocks, P, Cox, T, Valle, J, Wadsley, J, Propper, D et al. (2014). Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol 15: 829–840. [DOI] [PubMed] [Google Scholar]
- Torre, LA, Bray, F, Siegel, RL, Ferlay, J, Lortet-Tieulent, J and Jemal, A (2015). Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108. [DOI] [PubMed] [Google Scholar]
- Melero, I, Gaudernack, G, Gerritsen, W, Huber, C, Parmiani, G, Scholl, S et al. (2014). Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 11: 509–524. [DOI] [PubMed] [Google Scholar]
- Xiang, B, Snook, AE, Magee, MS and Waldman, SA (2013). Colorectal cancer immunotherapy. Discov Med 15: 301–308. [PMC free article] [PubMed] [Google Scholar]
- Tokuyama, W, Mikami, T, Fujiwara, M, Matsui, T and Okayasu, I (2007). Midkine expression in colorectal tumors: correlation with Ki-67 labeling in sporadic, but not ulcerative colitis-associated ones. Pathol Int 57: 260–267. [DOI] [PubMed] [Google Scholar]
- Hanari, N, Matsubara, H, Hoshino, I, Akutsu, Y, Nishimori, T, Murakami, K et al. (2007). Combinatory gene therapy with electrotransfer of midkine promoter-HSV-TK and interleukin-21. Anticancer Res 27(4B): 2305–2310. [PubMed] [Google Scholar]
- Lindenmann, J and Klein, PA (1967). Viral oncolysis: increased immunogenicity of host cell antigen associated with influenza virus. J Exp Med 126: 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel, WA, Murray, DR and Phillips, HS (1983). A phase II study on the postsurgical management of stage II malignant melanoma with a Newcastle disease virus oncolysate. Cancer 52: 856–860. [DOI] [PubMed] [Google Scholar]
- Cassel, WA and Murray, DR (1992). A ten-year follow-up on stage II malignant melanoma patients treated postsurgically with Newcastle disease virus oncolysate. Med Oncol Tumor Pharmacother 9: 169–171. [DOI] [PubMed] [Google Scholar]
- Lemay, CG, Rintoul, JL, Kus, A, Paterson, JM, Garcia, V, Falls, TJ et al. (2012). Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol Ther 20: 1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett, DL, Liu, Z, Sathaiah, M, Ravindranathan, R, Guo, Z, He, Y et al. (2013). Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer 12: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woller, N, Gürlevik, E, Ureche, CI, Schumacher, A and Kühnel, F (2014). Oncolytic viruses as anticancer vaccines. Front Oncol 4: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, ZS, Liu, Z and Bartlett, DL (2014). Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol 4: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workenhe, ST and Mossman, KL (2014). Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther 22: 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casares, N, Pequignot, MO, Tesniere, A, Ghiringhelli, F, Roux, S, Chaput, N et al. (2005). Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 202: 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepp, O, Senovilla, L, Vitale, I, Vacchelli, E, Adjemian, S, Agostinis, P et al. (2014). Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3: e955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, M, Curie, DT (2010). Current issues and future directions of oncolytic adenoviruses. Mol Ther 18: 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf, B and Lauer, UM (2015). Assessment of current virotherapeutic application schemes: “hit hard and early” versus “killing softly”? Mol Ther Oncolytics 2: 15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusi-Kerttula, H, Hulin-Curtis, S, Davies, J and Parker, AL (2015). Oncolytic adenovirus: strategies and insights for vector design and immuno-oncolytic applications. Viruses 7: 6009–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldén, G, Hill, R, Wang, Y, Anand, A, Liu, TC, Lemoine, NR et al. (2003). Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol Ther 8: 412–424. [DOI] [PubMed] [Google Scholar]
- Zhang, L, Hedjran, F, Larson, C, Perez, GL and Reid, T (2015). A novel immunocompetent murine model for replicating oncolytic adenoviral therapy. Cancer Gene Ther 22: 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev, L, Ranki, T, Joensuu, T, Jäger, E, Karbach, J, Wahle, C et al. (2015). Repeated intratumoral administration of ONCOS-102 leads to systemic antitumor CD8(+) T-cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology 4: e1017702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, S, Kawasaki, Y, Yamaoka, N, Tagawa, M, Kasahara, N, Terada, N et al. (2010). Complete regression of human malignant mesothelioma xenografts following local injection of midkine promoter-driven oncolytic adenovirus. J Gene Med 12: 681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi-Kimura, M, Yamano, T, Tamamoto, A, Okamura, N, Okamura, H, Hashimoto-Tamaoki, T et al. (2013). Enhanced antitumor efficacy of fiber-modified, midkine promoter-regulated oncolytic adenovirus in human malignant mesothelioma. Cancer Sci 104: 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesniere, A, Schlemmer, F, Boige, V, Kepp, O, Martins, I, Ghiringhelli, F et al. (2010). Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 29: 482–491. [DOI] [PubMed] [Google Scholar]
- Turnbull, S, West, EJ, Scott, KJ, Appleton, E, Melcher, A and Ralph, C (2015). Evidence for oncolytic virotherapy: where have we got to and where are we going? Viruses 7: 6291–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, HL, Kohlhapp, FJ and Zloza, A (2015). Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 14: 642–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin, D and Pesonen, S (2015). Replication-competent viruses as cancer immunotherapeutics: emerging clinical data. Hum Gene Ther 26: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, CL, Coukos, G and Kandalaft, LE (2015). Whole tumor antigen vaccines: where are we? Vaccines (Basel) 3: 344–372. [DOI] [PMC free article] [PubMed] [Google Scholar]