

Abstract
This perspective outlines strategies towards the development of MR imaging probes that our lab has explored over the last 15 years. Namely, we discuss methods to enhance the signal generating capacity of MR probes and how to achieve tissue specificity through protein targeting or probe activation within the tissue microenvironment.
Introduction
As modern medicine develops, the diagnosis, staging and treatment monitoring of disease is an increasingly less invasive endeavour. Planar X-ray imaging and computed tomography (CT), ultrasound, single photon emission computed tomography (SPECT), positron emission tomography (PET) and magnetic resonance imaging (MRI) are routinely used to identify and characterize disease in patients. Compared to these other modalities MRI is especially advantageous: it can image deep into tissue; there is no ionizing radiation; it provides 3D images with sub-millimeter spatial resolution; excellent soft tissue contrast; there are multiple types of contrast available; and the imaging is not operator dependent.1,2
MRI primarily involves detection of the 1H NMR signal of endogenous water. There are several ways to generate image contrast in order to delineate anatomy and pathology.1,2 Different tissues have differing water content and so-called proton density images reflect these differences. Water has different magnetic and physical properties in different tissues. For instance relaxation times (T1, T2, T2*, T1ρ) can vary by orders of magnitude in different tissues and data can be acquired that is weighted to these relaxation times in order to generate image contrast. One can also make images sensitive to differences in water diffusion rates. The movement of flowing blood can also be exploited to distinguish it from static tissue. One can also create contrast by transferring magnetization from the broad signal of water associated with macromolecules, or through saturation of exchangeable N-H or O-H protons.3 Finally there is contrast from chemical shift differences, mainly mobile lipid protons versus water.
Contrast can also be generated through the addition of an exogenous probe. Most commonly, this is a para- or superparamagnetic compound that shortens the relaxation times of water molecules it encounters, i.e. relaxation agents. Other probes termed chemical exchange saturation transfer (CEST) probes rely on the presence of exchangeable hydrogen atoms. These hydrogen resonances are magnetically saturated by a radiofrequency pulse and then chemical exchange with bulk water leads to a decrease of the bulk water signal. Relaxation agents and CEST probes are detected indirectly by their effect on bulk water. Other probes can be detected directly by virtue of their different magnetic resonance frequency (other 1H resonances,4,5 19F-based probes,6,7 hyperpolarized 13C, 3He, or 129Xe-based probes).8-10 Clinically, the only approved probes are relaxation agents based on discrete Gd(III) or Mn(II) complexes or iron oxide nanoparticles. Hydrophilic Gd(III) complexes are by far the most widely used with the Mn(II) and iron oxide – based probes now discontinued. Hyperpolarized 13C, 3He, and 129Xe-based and CEST probes have been used in clinical trials11-14
The clinical utility of MR imaging probes (contrast agents) is well established. At our institution just over 50% of all MRI procedures make use of a contrast agent. However, the full potential of contrast enhanced MRI has yet to be reached. The majority of clinical Gd(III)-based probes are small, hydrophilic, anionic or neutral complexes that freely distribute through extracellular spaces and are rapidly excreted via the kidneys. There is also an approved serum albumin-targeted probe used to image the blood pool,15 and a complex with substantial liver clearance used to image the hepatobiliary tree.16,17 The available imaging probes are highly useful in identifying vascular irregularities, detecting and delineating tumor boundaries, characterizing blood flow and tissue perfusion amongst numerous other purposes. However, the ability to target and report on a molecular level tissue could further the capacity to stage disease, guide treatment options and assess patient response.
In this Perspective we describe our efforts over the last 15 years in the design and application of MR probes. This covers work done with colleagues at the former Epix Medical, and since 2007 at Massachusetts General Hospital. This is not meant to be a thorough review of the subject, but a subjective personal account which details our rationale and efforts in this field. Other approaches are certainly with merit. Excellent comprehensive reviews of MR imaging probes exist.18-25
Our efforts have been motivated by the desire to bring new MR probes to the clinic. Clinical translation places several constraints on molecular design: the compound must be safe, must be completely excreted (if Gd-containing), must be reproducibly manufactured, must be highly soluble for formulation, and must provide diagnostic quality images in a short time frame after administration. A second consideration that has guided our efforts has been the generally low detection sensitivity of MR probes. Relaxation agents are detectable at low micromolar levels in vivo (per metal ion)26 while CEST and direct detection probes require at least millimolar concentrations for detection.27,28 For these reasons we have focused on discrete Gd(III)- and Mn(II)-based probes.
Gd(III) and high spin Mn(II) are very effective T1-relaxation agents. In a T1-weighted image, regions of short T1 appear bright. This is in contrast to iron oxide based probes which generally destroy signal and provide negative contrast.29,30 A T1-agent generates signal in an MR image and is conspicuous. On the other hand a T2-agent destroys signal and it is often difficult to identify areas of signal loss, especially since most artefacts in MRI cause signal loss.
We have also generally focused on discrete molecules containing Gd(III) or Mn(II) complexes instead of nanoparticles. While nanoparticles certainly deliver greater signal change per entity than a discrete chelate, nanoparticles have a number of pharmacokinetic limitations.31,32 They often circulate for very long times in the blood and target uptake can take hours to days. Nanoparticles are often retained in the body and this poses a risk for long-term toxicity. Such pharmacokinetic considerations are less important for preclinical applications, and indeed long blood residency times may be useful. However lower molecular weight (lt; 10 kDa) discrete molecules can be rapidly excreted into the urine, and these molecules are also small enough to rapidly extravasate from the blood into the interstitial space of most tissues and tumors.33,34 Rapid extravasation results in accumulation in target tissue while fast clearance results in high target:background ratios. In MRI where it is often necessary to acquire a baseline scan to understand the effect of the contrast enhanced image, rapid pharmacokinetics are desirable.
A limitation of our approach, compared to nanoparticles/macromolecules, is sensitivity. As mentioned above, single chelates can be detected at micromolar concentrations, whereas particles bringing thousands of paramagnetic ions can be detected at correspondingly lower concentrations. Our research has focused on 1) identifying relevant disease targets that are present at high concentrations; 2) utilizing amplification strategies to enhance sensitivity; 3) targeting alterations in tissue homeostasis such as pH or redox disregulation; 4) quantifying the molecular factors that influence relaxation and using this biophysical information to design better relaxation agents.
Protein Targeted Imaging Probes
One way to enhance the specificity of a molecular probe is to direct the probe to a specific target. This can be done by targeting a receptor which internalizes the probe into a cell, or by direct stoichiometric protein binding. The latter approach is straightforward in principle but challenging in practice. For discrete MR probes, one must identify abundant targets that can be detected using MR. The second challenge is to conjugate paramagnetic chelates to the targeting vector in a manner that does not destroy the affinity of the vector for its target.
Serum albumin
The serum albumin targeted probe MS-325 (gadofosveset, Ablavar, Figure 1) is an example of a protein-targeted probe. MS-325 is a clinically approved probe that was originally designed for imaging blood vessels.35,36 Present at 500-700 μM in serum, albumin is the most abundant blood plasma protein. MS-325 is a Gd(DTPA) derivative (Figure 1) that was designed by Lauffer and colleagues to have moderate affinity (Kd = 85 uM) for albumin which results in 85 - 90% of MS-325 being bound to albumin under steady state conditions.37-41 Albumin binding is promoted by a hydrophobic 4,4′-diphenylcyclohexyl moiety appended to the DTPA backbone via a phosphodiester linkage.15,38 The presence of the phosphodiester was shown to influence the pharmacokinetics by promoting renal clearance and extending the blood half-life. The unbound fraction of MS-325 is continuously filtered into the urine which leads to complete elimination of the molecule.
Figure 1.

Gadolinium and manganese complexes described and discussed in this perspective.
The proton relaxation enhancement (PRE) effect is a technique used in biochemistry to probe metal binding sites.42,43 There, an ion like Gd(III) is used to mimic Ca(II), and binding of the paramagnetic ion to the protein results in an increase of the solvent proton relaxation rates. This can be used to assess stoichiometry and protein dynamics. Lauffer recognized that reversible binding of Gd(III) complexes to proteins would also enhance relaxation,44 and the relaxivity of MS-325 is increased 5-fold when bound to albumin in plasma at the common imaging field strength of 1.5T.41 This concept is referred to as receptor induced magnetization enhancement (RIME). From an imaging standpoint, binding results in localization of the probe at the target, but it also results in a “turn-on” of signal when the probe is bound. This relaxivity turn-on provides greater target: background ratio and better delineation of the blood vessels.
Serum albumin possesses multiple binding pockets used in the transfer of hydrophilic substrates such as fatty acids, steroids, hormones and numerous drugs. Numerous Gd(III) and Mn(II) chelates have since been modified with hydrocarbon appendages to promote association with albumin through adventitious hydrophobic interactions.45-48
While albumin targeting was initially pursued for blood pool imaging, the presence of albumin in the lymphatics, tumor interstitium, and in atherosclerotic plaque have broadened the utility of these probes.49-51
Fibrin
Fibrin is another abundant protein suitable for detection with MRI. Fibrin is the polymeric protein network that serves to trap platelets and form a haemostatic plug at the site of vascular injury. The resultant thrombus, or blood clot, can be resolved by the action of endogenous proteases, a process referred to as fibrinolysis. However, uncontrolled clotting can block blood flow. Unstable thrombi can dissociate from the site of injury and deposit in smaller capillaries. Indeed, thrombus is often the culprit in ischemic stroke, myocardial infarction, pulmonary embolism, deep vein thrombosis etc. In this regard, thrombus detection is key to therapeutic intervention.52 Fibrin deposition is the result of the action of the protease enzyme thrombin on fibrinogen, a constituent blood protein that serves as the fibrin precursor, in response to insult to the vessel wall. The resultant fibrin protein is insoluble and precipitates at the site of injury. Cross-linking of strands via factor XIII fortifies the fibrin polymeric network. Circulating fibrinogen is present at about 7 μM in plasma and as polymerization and clotting occurs, the concentration of fibrin monomer increases to 10s-100s of μM.
Using phage display, we identified two families of cyclic peptides that showed specificity for fibrin over fibrinogen and other serum proteins.53 Significant medicinal chemistry effort was required to improve fibrin affinity, metabolic stability, and conjugate multiple Gd(III) chelates for robust thrombus detection.54-56 One compound, EP-2104R (Figure 2), emerged as a clinical development candidate.57,58 EP-2104R comprises an 11 amino acid peptide functionalized at the C- and N- termini with a total of four Gd(DOTA) reporters.59 Multiple chelates were appended to the targeting moiety in order to maximize signal response per binding event, and the DOTA chelator was chosen to minimize any release of Gd(III) in vivo. EP-2104R binds to two equivalent sites on the fibrin monomer (Kd = 1.7 μM), and this affinity is more than 100-fold higher than binding to fibrinogen or other blood proteins.59 The RIME effect is in play as binding to fibrin results in over a 2-fold increase in r1.59
Figure 2.

Fibrin-targeting probe EP-2104R and type I collagen targeting probe EP-3353. Charges omitted for clarity.
The lack of plasma protein binding results in a short blood half-life for EP-2104R, thus thrombus remains bright after the signal arising from blood and surrounding tissue return to near baseline levels. An example of EP-2104R imaging is shown in Figure 3. In this embolic stroke model, a blood clot is delivered to the middle cerebral artery of the brain.60 EP-2104R clearly delineates the location and extent of this thrombus (Figure 3B). In this model, the clot is delivered via a catheter into the internal carotid artery. EP-2104R enhanced imaging demonstrated that the introduction of the catheter damaged the vessel wall and resulted in microthrombi along the wall (Figure 3A, 3D). The diameter of the vessel in Figure 3D is 500 μm and the clear delineation of this mural thrombus could only be achieved with MRI; nuclear techniques lack the spatial resolution to distinguish mural (wall) thrombus from occlusive thrombus and optical techniques lack the depth penetration. Figure 3 is a powerful example of the potential of molecular MR imaging.
Figure 3.
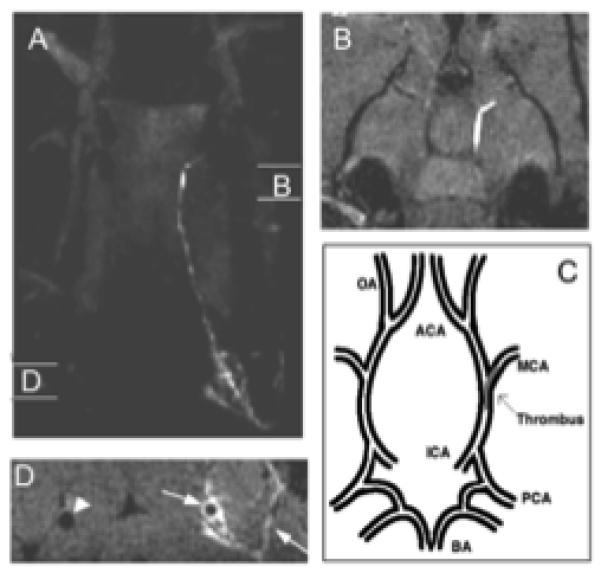
Panel A shows a maximum projection image of the head of a rat with an intracranial thrombus taken after injection of EP-2104R. The regions denoted “B” and “D” show the sites of the two dimensional images shown in panels B and D, respectively. Panel B shows a region of bright signal located in the internal carotid artery (ICA) and branching into the middle cerebral artery (MCA). Panel C shows a description of the vascular anatomy in the region of the brain represented in Panel B. Panel D shows cross sectional images of the common carotid arteries. The arrows denote mural thrombus along the vessel wall and of clotted side branches. The arrowhead shows the patent contralateral carotid artery. Adapted with permission from reference 60.
EP-2104R was validated in rodent, rabbit, and swine models of thrombosis and cancer.60-65 It was scaled up and studied in three human clinical trials. It showed a favourable safety profile and was effective at identifying thrombi in patients.57,58
Type I Collagen
Collagen is an abundant insoluble extracellular matrix protein. While there are over 20 different collagens known, type I is by far the most abundant and serves a structural role. Collagen is the most abundant protein constituent of connective tissue and thus a key biomarker for fibrosis, defined as the deposition of excess connective tissue. Fibrosis is a hallmark pathology of disease states ranging from cancers, cirrhosis, diabetic nephropathy, atherosclerosis, idiopathic pulmonary fibrosis, heart failure, and myocardial infarction. The resultant stiffening of the extracellular matrix can impede blood flow, disrupt organ function or hinder drug delivery. The present standard for evaluating most forms of fibrosis is biopsy, which is invasive, only samples a small fraction of the organ, and is associated with patient complications. A reliable method to non-invasively identify and evaluate fibrosis could be of great utility to diagnostic medicine.
Similar to our fibrin targeting approach, we identified a cyclic, disulphide containing polypeptide by phage display as the collagen specific substrate. Individual amino acids that are non-essential to collagen binding were then identified via systematic substitution with alanine. At these non-essential residues we introduced lysine functionalized at the ε-position with Gd(DTPA) through a thiourea linkage.66 This structure-activity approach led to the culmination of EP-3533 as the collagen-binding probe of choice (Figure 2). EP-3533 binds to multiple sites on type I collagen with low micromolar affinity (Kd = 1.8 μM) and incorporates 3 Gd(DTPA) moieties for signal enhancement.67 An analogous probe, EP-3600, utilizing the DOTA chelator has been reported and evaluated in a pig model of myocardial perfusion.68
EP-3533 exhibits rapid uptake in fibrotic tissue, but fast clearance from blood. Imaging at an appropriate delay after injection results in clear delineation of fibrotic tissue. In vivo specificity was confirmed by using the D-Cys isomer. This L-Cys to D-Cys modification decreases the affinity for collagen by over 2 orders of magnitude, but the relaxivities of the two isomers are equivalent.67 In vivo, the D-Cys isomer exhibits similar pharmacokinetics to EP-3533 but cannot bind collagen and acts as a useful negative control.
EP-3533 has been successfully applied to the imaging of fibrosis in numerous preclinical applications. It was first used to image myocardial scarring in mice.69 We have used EP-3533 to detect and stage liver fibrosis in two rodent models.70,71 Figure 4 shows examples of liver enhancement observed in a normal mouse compared to a mouse with fibrosis. Quantifying the contrast-to-noise (ΔCNR) ratios in T1-weighted MR images correlated well with histopathological scoring (Ishak scale, used to stage fibrosis72), and also to total liver collagen content as determined by ex vivo quantification of hydroxyproline. More recently, we have extended this work to the detection and quantification of pulmonary fibrosis.73
Figure 4.

Top: Axial T1-weighted image (4.7T) of liver in healthy control (Ishak 0, left) and severely fibrotic (Ishak 5, right) mice. False color scale depicts difference in liver to muscle contrast to noise (ΔCNR) ratio pre- and post-injection of EP-3353. Bottom Left: Correlation between ΔCNR and Ishak score. Bottom right: Correlation between ΔCNR and liver hydroxyproline concentration calculated ex vivo. Adapted with permission from reference 71.
Extracellular DNA
DNA is typically inaccessible to MR probes. However during necrotic cell death, DNA is released into the extracellular milieu for a period of hours before it is cleared. In these necrotic events, the concentrations of DNA released are in the micromolar range and detectable by a suitable probe. We previously reported a peptide-based DNA binding probe,74 but more recently Josephson and colleagues prepared a simple Gd-chelate conjugated to the vital dye thiazolium orange.75 This compound was shown to be effective in noninvasively documenting the cell death and remodelling process that occurs after myocardial infarction.
Strategies for increasing relaxivity
Relaxation agents are catalysts that are capable of changing the relaxation times of bulk solvent water (10′s of Molar in vivo) in a measurable way. By increasing the efficiency, i.e. the relaxivity, of these catalysts it is possible to detect them at much lower concentrations. Increased relaxivity opens the door to lower concentration targets, simplified probes (e.g. requiring only one Gd-chelate instead of 4 in EP-2104R), or stronger signal enhancement with existing targets. Since we are limited by detection sensitivity with discrete complexes, we have focused considerable effort to understanding the factors that influence relaxivity and used this knowledge to try to prepare higher relaxivity probes.
Like any catalyst, the effect will depend on the efficiency of the reaction (nuclear relaxation) when the substrate (water) encounters the catalyst, and off-rate for release of the substrate (water exchange rate). At a theoretical level these processes are understood, but in practice there are a number of molecular parameters involved which can make data interpretation challenging. We have employed an arsenal of magnetic resonance and optical techniques to interrogate hydration number,76-79 metal-hydrogen distance,80-82 rotational motion,83-87 electronic relaxation,88-90 water exchange kinetics,85,91-95 and how these are impacted by protein binding.78,85,87
The ability of T1 MR probes to relax the protons of solvent water is termed relaxivity r1 and is quantified by measurement of the change in relaxation rate (R1 = 1/T1) upon addition of probe, normalized to the concentration of the paramagnetic ion. There are 3 factors that affect relaxation: hydration, water exchange, and a correlation time that describes the fluctuating magnetic dipole created by the paramagnetic ion (also depicted in Figure 5). Hydration refers to water molecules in either the inner-coordination sphere, second coordination sphere, or in the outer-sphere. T1 relaxation is dipolar and the shorter the distance between the proton and the paramagnetic metal ion, the more efficient the relaxation. Protons of water molecules, that are coordinated directly to the metal ion, will contribute to inner-sphere relaxivity (r1IS). The number of directly coordinated water molecules is denoted as q. Water molecules in the second hydration sphere (q’) of the complex contribute to second sphere relaxivity (r1SS), outer sphere relaxivity (r1OS) refers to water molecules diffusing in the outer-sphere, equation 1.
| (1) |
Figure 5.

Molecular parameters that influence inner- and 2nd-sphere relaxivity.
Proton relaxation occurs by energy transfer from the fluctuating magnetic dipole created by the paramagnetic ion. This fluctuation can arise from the rotational diffusion of complex. It can also arise from the relaxation process of electrons in the ion (T1e). The rapid movement of water in and out of the second sphere also results in a fluctuating field for the water protons, as does the rapid diffusion of water in the outer-sphere. For water in the inner- or second-coordination sphere where the lifetime of the water is long enough to be correlated with the metal complex, the relaxation time of the bound water (T1m) is given by equation 2, which holds for fields where most imaging is performed, i.e. at Larmor frequencies above 10 MHz.
T1m is governed by a number of parameters (see also Figure 5): the gyromagnetic ratio of the proton (γH), the electronic g-factor (g), the Bohr magneton (μ0), the spin quantum number (S) of the corresponding metal ion, the proton Larmor frequency (ωH, as well as the metal-proton distance (rMH) and the correlation time (τc) that is the time constant for the fluctuating magnetic dipole.
| (2) |
| (3) |
The correlation time will be the fastest process that characterizes the magnetic dipole fluctuation, equation 3. This can be electronic relaxation (1/T1e), rotational motion of the complex (1/τR), or the water exchange rate (kex = 1/τm). Inspection of equation 2 reveals why we work with Gd(III) or high spin Mn(II) rather than other paramagnetic ions. Both have a large spin number, S=7/2 and 5/2, respectively. They also have symmetric S electronic ground states which results in relatively long T1e values.
The lifetime of the water in the inner-sphere (τm) is usually long compared to the time constant for molecular rotation. For both Mn(II) and Gd(III), the electronic T1e is very short at very low fields (picoseconds) but increases with the square of the applied field. At commonly used imaging field strengths, 1.5 tesla and higher, T1e is long compared to τR. Thus for the majority of Gd- and Mn-based MR probes, the correlation time that describes relaxation for inner-sphere water is the rotational correlation time. To increase the relaxation of water in the inner-sphere, equation 2 teaches to increase τR up to the inverse of the proton Larmor frequency (ωH).
For water in the second-sphere, the mean residency time (τm’) can be very short (ps time scale), and can be shorter than, or comparable to the rotational correlation time of the complex. To increase the relaxivity contribution from water in the second-sphere, it is important to increase the lifetime of these water molecules.
Optimizing the relaxation of coordinated water ligands is only effective if those water ligands can undergo rapid exchange to transmit this relaxation effect to the bulk solvent. For 2-site water exchange, the inner-sphere relaxivity can be written as equation 4.96 Here relaxivity depends inversely on both the relaxation time of the coordinated water (T1m) and on its lifetime (τm). For rapidly tumbling small metal complexes like [Gd(DTPA)(H2O)]2−, T1m >> τm. A similar expression can be written for water in the second coordination sphere.
| (4) |
Smart complex design offers possibilities to carefully adjust all the parameters influencing r1. Below, we will discuss our corresponding efforts to modulate and optimize structure and dynamics to improve relaxivity.
Hydration
Equation 4 shows that r1IS is directly proportional to q (and r1SS is directly proportional to q’). Increasing q is an established method to increase relaxivity. However the strategy of increasing hydration number is offset by the requirement that the complex be thermodynamically stable and kinetically inert with respect to metal ion release in vivo. A further challenge is that opening up coordination sites for water ligands can instead lead to displacement of water ligands by coordinating anions. Yet, increasing q provides a directly proportional relaxivity boost across all field strength, and this has motivated us,77,97-99 and others,20,100-105 to repeatedly revisit the challenge of increasing q without considerable loss of kinetic inertness of the complex (Figure 5).
We have explored DO3A derivatives, i.e. where one carboxylate donor from DOTA is removed to make a q=2 complex. [Gd(DO3A)(H2O)2] displays fast water exchange kinetics,106 is reasonably inert with respect to Gd dissociation,107 but is known to bind endogenous anions like bicarbonate.108 We synthesized a DO3A analogue, with high affinity to HSA, and a pendant sulfonic acid moiety in the hope that increased negative charge would prevent anion coordination (compound [Gd(L2)(H2O)2]2− in Figure 1).76 In buffer, the corresponding Gd complex exhibited relaxivity typical for a q=2 complex, which was also confirmed by 1H ENDOR measurements. In the presence of HSA however, ENDOR studies revealed that the coordinated water ligands were displaced by a coordinating amino acid side chain from the protein.15 The resultant q=0 complex had a much lower relaxivity than originally anticipated.15
Parker and colleagues had shown that Gd(DO3A) derived from 3 adipic or glutaric acid moieties gave q=2 complexes with high relaxivity and resistance to anion binding.109 They had also shown that incorporating a sulphonamide donor off the forth nitrogen of cyclen (compound [Gd(L3)(H2O)2]3− in Figure 1) gave a complex that switched between q=2 at low pH (sulphonamide protonated, not coordinated) and q=0 at high pH when the sulphonamide was deprotonated and the anionic N donor could coordinate.110 We tried to amplify this pH switch by coupling it to the RIME effect by introducing a HSA binding moiety (compound [Gd(L4)(H2O)2]3− in Figure 1).98 While the relaxivity did show a pH dependent effect, the magnitude of the relaxivity change was not consistent with a q=2, HSA-bound species. Measurement of hydration number yet again confirmed displacement of both inner-sphere waters to form a q=0 complex. The moderate relaxivity enhancement in fact hailed from the relaxivity contribution of a long-lived second-sphere water.
Not all q=2 complexes show water displacement when bound to proteins. We showed that an N-methyl analog of MS-325 (compound [Gd(L1)(H2O)2]2− in Figure 1) was q=2 in both the presence and absence of HSA.97 However this complex also displayed much lower relaxivity than q=1 MS-325 when bound to HSA. In this instance, the rate of water exchange for the q=2 complex was much slower than for MS-325 and this slow exchange muted the relaxivity.
More recently we prepared the acyclic chelator CyPic3A. Picolinates are strong donors for lanthanides,111 and the cyclohexyl diethylamine backbone provides additional rigidity to the acyclic, hexacoordinate ligand design.112 Indeed, [Gd(CyPic3A)(H2O)2]− maintains q = 2 in water, even when challenged with high concentrations of various coordinating anions. The gadolinium complex has a kinetic inertness comparable to [Gd(DTPA)(H2O)]2− and may provide yet another attractive alternative for relaxivity enhancement via increase of hydration number.
It is known that second-sphere hydration can contribute significantly to relaxivity. In addition to the DO3A adipate derivatives mentioned above, the presence of phosphonate donors also results in increased second-sphere relaxivity. For instance the q=0 [Gd(DOTP)]5− complex shows higher than expected relaxivity due to this effect.113 In collaboration with the Sherry lab, we investigated the relaxivity of an albumin binding version of this complex [Gd(C11-DOTP)]5−.114 The relaxivity of this q=0 complex in the presence of HSA was considerably high (19.9 mM−1s−1 at 37 °C, 20 MHz). NMRD analysis revealed a long-lived (1 ns) second sphere water molecule.
More recently we found that acetamide donors with certain pendant groups showed enhanced relaxivity relative to size matched control complexes.90 We systematically explored this effect and found that pendant H-bond acceptors gave increased relaxivity provided there was one methylene unit between the amide N and the acid group. Thus amide pendants derived from glycine, iminodiacetic acid, aspartic acid, or aminomethylphosphonic acid gave a pronounced second sphere effect (Figure 6, complex type A). Variable temperature NMRD showed that this effect was due to water molecules with nanosecond lifetimes in the second coordination sphere. We also found that these second sphere effects were additive with relaxivity contributions from an inner-sphere water ligand.115
Figure 6.

Libraries of HSA-binding Gd complexes. Complex type A was a library where R1 and R2 groups were varied to enhance second sphere relaxivity, while the D1 donor group was varied to offset the slow water exchange effect of the two acetamide donors. Complex type B was a library that explored the effect of a single donor group D1 variation on water exchange kinetics, electronic relaxation, and how these affected relaxivity. See also Figure 8.
Rotational motion and multimeric probes
Altering rotational dynamics is one of the best levers to improve relaxivity. The targeted MR probes such as MS-325, EP-3533 and EP-2104R described in this perspective all experience a gain in relaxivity upon binding of the target due to an increase in τR. However controlling rotational dynamics is challenging. Equation 2 applies to isotropic rotational motion, but reality is more complex. There are various sources of internal motion (metal – O(water) rotation,116 rotation of the chelate about the linker to the targeting vector117,118 etc.), and all of these will reduce relaxivity.26,119 Even for a molecule like MS-325 where the protein-binding group is close to the chelator, the fairly high protein-bound relaxivity is still significantly lower than its theoretical maximum.85 As these molecules become more complex, it becomes challenging to design the compound in such a way that the correlation time is optimally increased upon binding to the target protein. A second consideration is the ideal correlation time since this will depend on the Larmor frequency as τR = 1/ωH. Thus at low fields (e.g. 1.5T), very slow motion associated with protein dynamics is desired, but at high fields (e.g. 7T) an intermediate correlation time is preferred.
We have sought to control rotational dynamics in several ways. First, we aim to utilize bifunctional chelators with the shortest practical linkage between the chelate and the targeting vector (see e.g. the chelates in Figure 8). A short linkage limits the degree of internal motion about the linker moiety. However care must be taken to avoid altering the affinity of the targeting vector or lowering the stability of the chelate itself.
Figure 8.
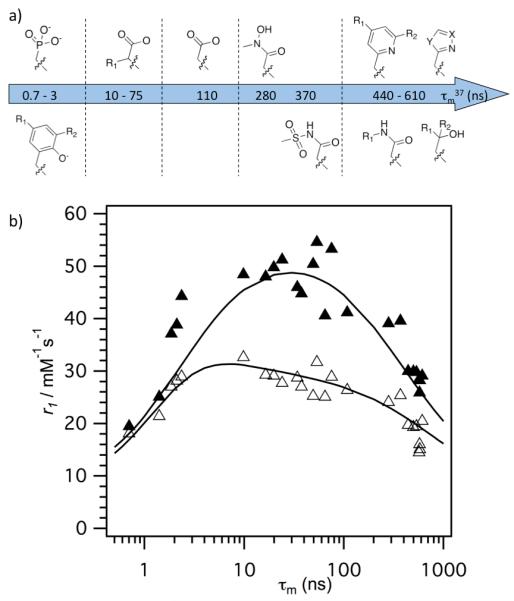
a) Varied donor group D1 from Figure 6, complex type B, resulting in q=1, HSA binding complexes with Gd. b) Relaxivity of these complexes in HSA solution at 37 °C plotted versus measured water residency time at 37 °C with data at 20 MHz (▴) and 60 MHz (▵). It is evident that τm limits relaxivity at a given field strength if it is either too long or too short, and that there is an optimal water exchange rate range for high relaxivity that becomes larger at higher fields. Reproduced with permission from reference 90.
Simply increasing the number of chelates within the molecule can be a powerful way to increase relaxivity. But to maintain the gain in per ion relaxivity, the rotational dynamics must be controlled.84 Figure 7 shows two strategies we have taken to this end. The first is to rigidify the probe upon binding to its target. Here we use two protein binding groups such that when both are bound, the rotation of the chelates is restricted. We have shown that relaxivity can be enhanced by screening small libraries to introduce secondary binding groups.120 The second approach comes from our work in developing probes with intermediate correlation times (0.5 < τR < 2 ns) for high fields.93,119
Figure 7.
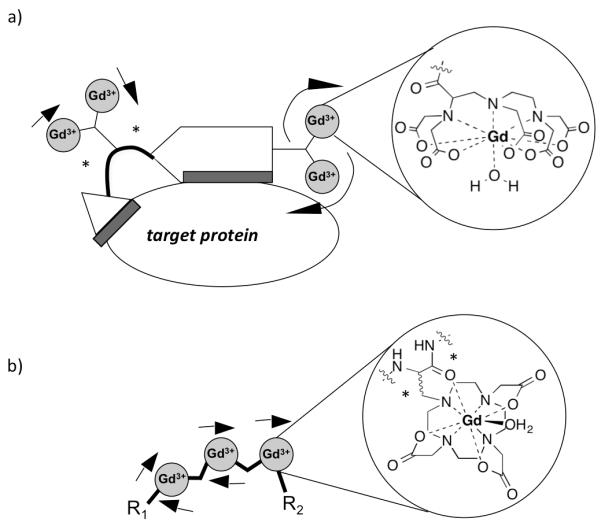
Strategies to closely control rotational dynamics. a) Binding of two protein binding groups to one targeting molecule results in restriction of the rotation of two of the chelates (as indicated by small arrows) by dual attachment (indicated in blue). b) A multimer of the single amino acid Gd chelate Gd(DOTAla) also provides dual attachment of the Gd complex through the α carbon, as well as the carbonyl of the peptide backbone (indicated by asterisks). It is also important to note that in both cases, the chelators were attached using short linkers to ensure maximum rigidity.
To reach an intermediate correlation time, we have constructed larger molecules using metal chelates as building blocks. To minimize internal motion we used the single amino-acid chelator DOTAla. The Gd complex experiences dual attachment through the α-carbon, as well as through coordination of the Gd to the carbonyl of the pepide backbone, minimizing the internal motion component. The amino acid is versatile enabling variation of Gd-payload, shape and size of the overall molecule using peptide synthesis.93 We predicted that a trimeric polypeptide structure based on DOTAla would provide the suitable intermediate τR for enhanced relaxivity at higher magnetic fields. Indeed, the obtained relaxivity at 4.7T and above exceeded the relaxivity of slowly tumbling (MS-325 bound to HSA) and rapidly tumbling complexes ([Gd(HP-DO3A)(H2O)].
Water exchange kinetics
For small molecular complexes, τR is short, generally resulting in the relaxation time of the coordinated water hydrogen atom being long compared with the water residency time (T1m > τm; eq 4). Thus many commercial Gd-based contrast agents have similar, low relaxivities despite having different water exchange kinetics. When τR is slowed via covalent or non-covalent association to a large molecule, τm can become a relaxivity-limiting or relaxivity-enhancing factor.90,115
Elegant coordination chemistry by a number of groups has demonstrated that the inner-sphere water residency time of Gd(III) complexes can range from 0.1 ns to 10’s of μs at 37 °C, and is dictated by the local coordination environment.121-123 Modification of just one donor group of polyamino-carboxylate Gd(III) complexes can lead to a change in τm of three orders of magnitude. Since the effect of water exchange on relaxivity is only revealed in a slow tumbling system (compared to simple chelates), we prepared a library of Gd(DOTA)-like chelates with the same HSA-binding group and measured variable temperature, variable field relaxivity in the presence and absence of HSA.90 We explored the effect of systematically replacing one acetate arm with a different donor and found the following reactivity, in order of increasing τm: phosphonate ~ phenolate < α-substituted acetate < acetate < hydroxamate ~ sulphonamide < acetamide ~ pyridyl ~ imidazole. For cyclen derived ligands, we found these effects to be additive (Figure 6, complex type B). For instance changing two acetates groups to acetamides slowed water exchange but changing a third acetate group to a phosphonate reversed this effect (complex type A, Figure 6).115 The effect of these τm changes on relaxivity is seen in Figure 8, which shows that there is an optimum water exchange range for slow tumbling chelates at low fields as Lauffer predicted.124 Note that the range of τm for optimal relaxivity is broader as one moves to higher fields.
The decrease in relaxivity on the right side of Figure 8 is because water exchange is too slow to efficiently transmit the effect of relaxation to bulk water (τm > T1m in eq 4). However the decrease in relaxivity as τm becomes very short is due to the fact that τm now becomes the dominant correlation time and causes T1m to increase (i.e. relaxation of the bound water is less efficient). We have begun to exploit this effect to create slow tumbling complexes that have high relaxivity at high fields. If a short τm becomes the correlation time for relaxation, it should be possible to identify a complex whose water exchange rate is matched to the Larmor frequency. We explored this hypothesis recently and confirmed that a monophosphonate functionalized DOTAla-type system (DOTAlaP, Figure 1) capable of HSA binding (short τm, long τR) exhibits higher relaxivity at high fields. However a challenge in accelerating water exchange by steric crowding is that the q=0 state may become more favourable and thus limit relaxivity.95
High spin Mn(II) as a relaxation agent
For MR probe development we have primarily focused on Gd(III) because of its high magnetic moment, slow electronic relaxation rate, and its ability to form very stable complexes with fast water exchange kinetics. On the other hand, only one Mn(II) complex was clinically approved for intravenous use, ([Mn(DPDP)]4−),125-127 and is no longer available. That complex is q=0, and slowly releases the metal ion in vivo which is what provides most of the relaxation enhancement. Therefore, kinetically inert Mn(II) complexes with one or two inner sphere water molecules provide a much safer alternative for targeted probe development. Mn(II) ions prefer to form 6- to 7-coordinate complexes in water, hence ligands that provide 6 donor atoms but yield a CN7 complex are preferred in order to provide q>0. In recent years, a myriad of chelators have emerged for the purpose of kinetically inert complexation of Mn(II). 128-132
Work from our lab included the synthesis and evaluation of the Mn(II) analogue of MS-325, where the octadentate DTPA moiety is substituted with hexadentate EDTA ([Mn(L5)(H2O)]3− in Figure 1).92 The corresponding q=1 Mn(II) complex indeed had sufficient kinetic inertness, maintained high affinity to HSA and produced relaxivity values that were comparable to MS-325. Although the S(S+1) term for Mn(II) in equation 2 is 5/9 that of Gd(III), this negative effect on relaxivity is offset by the shorter Mn-H distance. Much like MS-325, the relaxation enhancement upon binding to HSA relied on an increase in τR. This work provided some of the basis of our work on redox-active Mn probes discussed below.
Activatable Imaging Probes
MR relaxation probes are detected indirectly by their effect on bulk water. As we have seen, the magnitude of this effect is termed relaxivity and is dependent on a number of molecular factors. Relaxivity can change depending on the local environment. For instance the change in rotational correlation time upon protein binding can result in a large change in relaxivity. Thus MR probes that change their relaxivity can be viewed as sensors and are referred to as “activatable”, “responsive”, or “smart” probes.
The development of MR imaging probes that respond to physiological changes in tissue microenvironments is currently an area of intense research focus.19,22,133-135 Alterations in extracellular pH, pO2, redox potential, labile metal concentrations, expression of specific enzymes, etc. accompany cancers, ischemia, infection, vascular injury, inflammation and numerous other disease states. The ability to track dynamic changes in these microenvironments could offer the benefit of serial monitoring of tissue physiology during disease progression or therapeutic response, or inform drug delivery strategies in personalized medicine. This biomedical challenge affords a vast chemical space for activatable probe development. The last decade has seen the development of probe prototypes that respond to a range of enzymes, pH, pO2, and analytes such as Zn(II), Cu(II), and Ca(II).136-139
An elegant proof of principle was provided by Meade and co-workers in their seminal studies on a MR reporter gene. They designed the β-galactosidase (β-Gal) activated MR reporter EGad.140 β-Gal is the product of the LacZ reporter gene.141 Egad comprises a Gd(HPDO3A)-galactopyranose conjugate (Figure 1). β-Gal catalysed hydrolysis of the saccharide linkage effectively increases the accessibility of water to the chelate and results in higher relaxivity.142 Here the nanomolar enzyme catalytically converts micromolar concentrations of MR probe, which in turn is detected by its catalytic effect on molar concentrations of water.
Enzymatic activity can also be coupled to changes in molecular dynamics. For instance the myeloperoxidase probes developed by Bogdanov, Chen, Weissleder and colleagues undergo polymerization in the presence of the enzyme yielding increased relaxivity (see [Gd(L6)(H2O)]− in Figure 1).143-146
We coupled the RIME effect to enzymatic activation. Thrombin-activatable fibrinolysis inhibitor (TAFI) is an enzyme that inhibits thrombolysis through peptidase activity on C-terminal lysine residues that are recognized by fibrinolytic enzymes. The probe consisted of Gd(DTPA) conjugated to a NH2-X-Lys-Lys-Lys-COOH tetrapeptide through an amide linkage, where X is a hydrophobic amino acid (see [Gd(L7)(H2O)] in Figure 1).147 The positively charged Lys residues result in the complex having little affinity for serum albumin. TAFI cleaves the Lys residues giving a product with high affinity for HSA (and concomitant high relaxivity). The nature of the hydrophobic X group affects not only the magnitude of the RIME effect, but also the activation kinetics.
Non-invasive measurement of pH is another goal. Decreased extracellular pH is associated with many cancers, coronary artery disease and tissue ischemia. As mentioned above, pH-dependent protonation of a sulphonamide or phenolate donor can lead to the opening of a coordination site for a water ligand and associated increase in relaxivity. Sherry and colleagues have developed a different approach based on the Gd complex [Gd(DOTA-4AMP)(H2O)]−.86,148-151 This complex has four pendant non-coordinating phosphonate moieties. The protonation of these phosphonates and their prototropic exchange kinetics causes a large 2nd sphere pH-dependent relaxivity effect.151
A particular challenge in the in vivo translation of any of these activatable probes is in determining whether there is really a change in relaxivity. The change in MR signal is reflected in the T1 value but this depends on both relaxivity and concentration. In a test tube the concentration of the probe is known and fixed, but in vivo the concentration is not known and is changing with time. This has been addressed by administering two probes: first, a pH-independent probe and following MR signal change to estimate concentration; then the pH-dependent probe and using the first data set to assume its time-dependent concentration.149,150 This assumes identical pharmacokinetic behaviour between the two probes and also requires two injections and complex modelling.
We recently took advantage of new simultaneous MR-positron emission tomography (PET) technology to address the problem of decoupling concentration from relaxivity. PET is a sensitive technique that can independently provide probe concentration. We prepared a derivative of [Gd(DOTA-4AMP)(H2O)]− with an F-18 PET reporter and showed that it maintained its strong r1 pH dependence. By using simultaneous MR-PET imaging we demonstrated that pH could be determined without a priori knowledge of probe concentration.152
While quantitative imaging is desirable, in many cases unambiguous proof that probe activation occurred will yield diagnostically useful information. The ideal probe would have zero relaxivity in the off-state and a high relaxivity in the on-state. In practice, the contribution of second-sphere relaxivity results in some measurable relaxivity even for q=0 compounds. For instance the EGad example sees an increase in hydration upon activation (relaxivity goes up), but a decrease in molecular size and correlation time (relaxivity goes down) resulting in an overall modest relaxivity increase. Coupling activation to a large change in correlation time can result in more marked relaxivity turn on that can be readily detected in vivo. For example Sherry and co-workers coupled Zn(II) sensing to a RIME effect. Upon Zn(II) binding, [Gd(DOTA-bisBPEN)(H2O)]− binds to serum albumin and experiences a 3-fold increase in r1,153 resulting in ready detection of Zn(II) release from pancreatic β-cells in mouse models.154
We have recently explored developing activatable probes by switching between a T1e-dominated correlation time to a τR-dominated correlation time. At high fields (≥1.5T), rotation is the dominant correlation time for Mn(II), but for the S=2 Mn(III) ion T1e is short and dominates nuclear relaxation. Given that the Mn(II/III) redox couple is physiologically accessible,136 we sought to harness this mechanism of activation to create a redox sensitive probe. One requirement is a ligand capable of stabilizing both oxidation states and this is a challenge since ligands supporting each oxidation state tend to be mutually exclusive.
We used the ligand HBET (Figure 9) to support reversible conversion Mn(II) and Mn(III). HBET fuses elements of both EDTA and HBED, which support the di- and trivalent oxidations states, respectively.155 In addition to the correlation time switch, the Mn(II) complex is 7-coordinate, q=1 while the Mn(III) complex is likely 6-coordinate, q=0 which results in another lever to increase the relaxivity difference. Reduction of [MnIIIHBET]1− to [MnIIHBET]2− affords a 4-fold r1 increase at pH 7.4, RT and excellent contrast at 4.7T (Figure 9). Furthermore, we can reversibly toggle between either oxidation state using the biologically encountered reductants and oxidants glutathione and hydrogen peroxide, respectively. Since the relaxation mechanisms are different between Mn(II) and Mn(III), we anticipate that very large relaxivity differentials can be generated through judicious tuning of solution dynamics. We are currently exploring this possibility.156
Figure 9.

For [MnIIHBET]2−/1−, reversible switching between the Mn(II) and Mn(III) oxidation states can be achieved using glutathione (GSH) to increase relaxivity or H2O2 to decrease relaxivity. Adapted with permission from reference 155.
A final consideration in activatable probes is retention of the activated probe at the site of pathology. Thus either the probe must undergo rapid reversible activation to observe the effect at the site of disease, or the probe must somehow be trapped at the disease site after activation. The fibrin-targeted probes described above are effective for identifying blood clots, but are incapable of distinguishing whether the clot is freshly formed. To identify fresh thrombi, we targeted activated platelets which are present only in newly formed clots. We identified protein disulfide isomerase (PDI), an enzyme expressed on the surface of activated platelets, as a target for an activatable probe. PDI catalyzes the exchange of disulphide bonds in proteins, and in thrombosis PDI converts the integrin α2Bβ3 to an active form that allows the platelet to bind fibrinogen. From our fibrin-targeting work, we knew that the cyclic, disulphide bridged peptide (Figure 2) was absolutely critical for fibrin binding and that linear peptides of the same sequence did not bind fibrin. We hypothesized that a linear, mixed disulphide version of EP-2104R could act as a substrate for PDI. Thus in the presence of the enzyme, the peptide would undergo disulphide isomerization and cyclize, revealing a fibrin targeted probe. Since fibrin is also present in newly forming clots, the activated probe would then be retained in the fibrin mesh. However in older clots, where there is no PDI, there would be no activation and thus no binding.
To test this concept, we prepared a mixed disulphide prodrug of EP-2104R (Figure 10a).157 The mixed disulphide prodrug shows no appreciable affinity for the soluble fibrin fragment DD(E) whereas its activated form EP-2104R exhibits high nanomolar affinity for DD(E). Indeed, PDI efficiently catalyses the disulphide exchange and cyclization. In the presence of PDI and DTT (dithiothreitol, a sacrificial reductant that reactivates PDI after turnover), the mixed disulphide is rapidly activated. Figure 10b shows the competitive displacement of a fluorescent probe from DD(E) that occurs only in the presence of the enzyme. The relaxivity in the presence of fibrin or DD(E) is also increased by 72% at 1.4T upon activation and this is seen through changes in 1/T1 (Figure 10c). Figure 11c further illustrates the activatable nature of this probe. Note the signal increase upon activation in the presence of clotted fibrin; also note the localization of the activated probe within pelleted fibrin clot.
Figure 10.

(A) PDI activation of mixed disulphide prodrug to fibrin binding EP-2104R. (B) In the presence of PDI (filled symbols) but not the absence (open symbols), the prodrug activated and can displace a fluorescent probe bound to the soluble fibrin fragment DD(E). (C) PDI activation of the prodrug is also monitored by the increase in relaxivity upon binding to DD(E); filled symbols show 1/T1 in presence of PDI and open symbols show no change in T1 in absence of PDI. (D) T1-weighted images of pelleted fibrin at 1.4T. From left to right: fibrin alone, prodrug +PDI, prodrug −PDI, EP-2104R (positive fibrin binding control) +/−PDI, linear thioether (negative control) +/−PDI. Prodrug only shows binding to fibrin in presence of PDI. Adapted with permission from reference 157.
Conclusions
The above discussion provides examples of how we have played upon the well-established modular mechanisms controlling relaxivity to develop probes that interrogate both anatomical and biochemical aspects of disease. In particular, protein targeted and activatable probes provide opportunities to assess information previously inaccessible by MRI. Through rational design, we have designed probes that begin to approach the theoretically maximum relaxivity. The use of high-relaxivity probes is particularly important when endeavouring to target proteins or other analytes that are present in micromolar quantities. Chelates of higher relaxivity could simplify probe design by obviating the need for multimeric strategies and/or by making targets nearing the detection limit more conspicuous. Targets that present in nM quantities or below are poor candidates for direct targeting with discrete compounds, but strategies do exist to amplify sensitivity through catalytic accumulation of activated probes. This latter approach represents an excellent strategy for imaging enzymatic activity, provided there is a mechanism for probe retention at the site of pathology. Coupling an accumulation mechanism to a change in one or more of the modular parameters influencing relaxivity can be particularly advantageous in imaging targets present below the MR detection limit. It is our opinion that in most cases, the high thermodynamic stability, high kinetic inertness, and rapid clearance make hydrophilic Gd(III) chelates the best candidates for further optimization. Gd(III)-based probes are, and will likely remain, the workhorses of clinical MR contrast. However, there are circumstances where non Gd(III)-based probes may prove advantageous. For example, switching from Mn(III) to Mn(II) offers the potential for relaxivity turn-on differentials that supersede those theoretically possible for Gd(III). Thus, Mn-based probes merit further exploration in imaging redox dynamics. Similarly the high relaxivity potential of Mn(II) represents a useful strategy in developing probes for patients that would be contraindicated for Gd(III)-based compounds because of poor renal function.
Acknowledgements
This work was supported by grants from the National Cancer Institute (CA161221 to PC and a T32 post-doctoral fellowship to EMG, CA009502) and the National Institute for Biomedical Imaging and Bioengineering (EB009062). E.B. thanks the Swiss National Science Foundation (advanced.postdoc mobility fellowship) for support. The contributions of our co-authors to the papers cited is warmly acknowledged.
References
- (1).Young IR. Methods in Biomedical Magnetic Resonance Imaging and Spectroscopy. John Wiley & Sons Ldt.; Chichester: 2000. [Google Scholar]
- (2).Edelman GM, Hesselink JR, Zlatkin MB, Crues JV., III . Clinical Magneic Resonance Imaging. Volume 3. Elsevier Health; St. Louis: 2006. [Google Scholar]
- (3).Ward K, Aletras A, Balaban R. J. Magn. Res. 2000;143:79. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- (4).Yang Y, Schühle DT, Dai G, Alford JK, Caravan P. Contrast Media Mol. Imaging. 2012;7:276. doi: 10.1002/cmmi.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Harvey P, Blamire AM, Wilson JI, Finney K-LN, Funk AM, Senanayake PK, Parker D. Chem. Sci. 2013;4:4251. [Google Scholar]
- (6).Mizukami S, Takikawa R, Sugihara F, Hori Y, Tochio H, Wälchi M, Shirakawa M, Kikuchi K. J. Am. Chem. Soc. 2008;130:794. doi: 10.1021/ja077058z. [DOI] [PubMed] [Google Scholar]
- (7).Tirotta I, Mastropietro A, Cordiglieri C, Gazzera L, Baggi F, Baselli G, Bruzzone MG, Zucca I, Cavallo G, Terraneo G, Bombelli FB, Metrangolo P, Resnati G. J. Am. Chem. Soc. 2014;136:8524. doi: 10.1021/ja503270n. [DOI] [PubMed] [Google Scholar]
- (8).Rodrigues TB, Serrao EM, Kennedy BWC, Hu D-E, Kettunen MI, Brindle KM. Nat. Med. 2014;20:93. doi: 10.1038/nm.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu Z, Araki T, Okajima Y, Albert M, Hatabu H. Eur. J. Radiol. 2014;83:1282. doi: 10.1016/j.ejrad.2014.04.014. [DOI] [PubMed] [Google Scholar]
- (10).Garimella PD, Meldrum T, Witus LS, Smith M, Bajaj VS, Wemmer DE, Francis MB, Pines A. J. Am. Chem. Soc. 2014;136:164. doi: 10.1021/ja406760r. [DOI] [PubMed] [Google Scholar]
- (11).Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, M. F, van Criekinge M, Chang JW, Bok R, Park I, Reed G, Carvajal L, Small EJ, Munster P, Weinberg VK, Ardenkjaer-Larsen JH, Chen AP, Hurd RE, Odegardstuen LI, Robb FJ, Tropp J, Murray JA. Sci. Transl. Med. 2013;5:198ra108. doi: 10.1126/scitranslmed.3006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Svenningsen S, Kirby M, Starr D, Leary D, Wheatley A, Maksym GN, McCormack DG, Parraga G. J. Magn. Res. Imag. 2013;38:1521. doi: 10.1002/jmri.24111. [DOI] [PubMed] [Google Scholar]
- (13).Qing K, Ruppert K, Jiang Y, Mata JF, Miller GW, Shim YM, Wang C, Ruset IC, Hersman FW, Altes TA, Mugler JP., III J. Magn. Reson. Imaging. 2014;39:346. doi: 10.1002/jmri.24181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tietze A, Blicher J, Mikkelsen IK, Østergaard L, Stother MK, Smith SA, Donahue MJ. NMR Biomed. 2014;27:163. doi: 10.1002/nbm.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Caravan P. Acc. Chem. Res. 2009;42:851. doi: 10.1021/ar800220p. [DOI] [PubMed] [Google Scholar]
- (16).Junqiang L, Yinzhong W, Li Z, Shunlin G, Xiaohui W, Yanan Z, Kehu Y. J. Magn. Res. Imag. 2014;39:1079. doi: 10.1002/jmri.24354. [DOI] [PubMed] [Google Scholar]
- (17).Donati OF, Hunziker R, Fischer MA, Raptis DA, Breitenstein S, Patak MA. Eur. J. Radiol. 2014;83:1074. doi: 10.1016/j.ejrad.2014.03.020. [DOI] [PubMed] [Google Scholar]
- (18).Manus LM, Strauch RC, Hung AH, Eckermann AL, Meade TJ. Anal. Chem. 2012;84:6278. doi: 10.1021/ac300527z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Major JL, Meade TJ. Acc. Chem. Res. 2009;42:893. doi: 10.1021/ar800245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Datta A, Raymond KN. Acc. Chem. Res. 2009;42:938. doi: 10.1021/ar800250h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Merbach AE, Toth E. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging. John Wiley & Sons Ltd; New York: 2001. [Google Scholar]
- (22).De Leon-Rodriguez LM, Lubag AJM, Malloy CR, Martinez GV, Gillies RJ, Sherry AD. Acc. Chem. Res. 2009;42:948. doi: 10.1021/ar800237f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Terreno E, Castelli DD, Viale A, Aime S. Chem. Rev. 2010;110:3019. doi: 10.1021/cr100025t. [DOI] [PubMed] [Google Scholar]
- (24).Terreno E, Dastru W, Castelli DD, Gianolio E, Crich SG, Longo D, Aime S. Curr. Med. Chem. 2010;17:3684. doi: 10.2174/092986710793213823. [DOI] [PubMed] [Google Scholar]
- (25).Villaraza AJL, Bumb A, Brechbiel MW. Chem. Rev. 2010;110:2921. doi: 10.1021/cr900232t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Caravan P. Chem. Soc. Rev. 2006;35:512. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- (27).Aime S, Castelli DD, Crich SG, Gianolio E, Terreno E. Acc. Chem. Res. 2009;42:822. doi: 10.1021/ar800192p. [DOI] [PubMed] [Google Scholar]
- (28).Wu Y, Zhou Y, Ouari O, Woods M, Zhao P, Soesbe TC, Kiefer GE, Sherry AD. J. Am. Chem. Soc. 2008;130:13854. doi: 10.1021/ja805775u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pouliquen D, Le Jeune J, Perdrisot R, Ermias A, Jallet P. Magn. Reson. Im. 1991;9:275. doi: 10.1016/0730-725x(91)90412-f. [DOI] [PubMed] [Google Scholar]
- (30).Na HB, Song IC, Hyeon T. Adv. Mater. 2009;21:2133. [Google Scholar]
- (31).Li S-D, Huang L. Mol. Pharmaceutics. 2008;5:496. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- (32).Owens DE, III, Peppas NA. Int. J. Pharm. 2006;307:93. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- (33).Su MY, Mühler A, Lao X, Nalcioglu O. Magn. Reson. Med. 1998;39:259. doi: 10.1002/mrm.1910390213. [DOI] [PubMed] [Google Scholar]
- (34).Parmelee DJ, Walovitch RC, Ouellet HS, Lauffer RB. Investigative radiology. 1997;32:741. doi: 10.1097/00004424-199712000-00004. [DOI] [PubMed] [Google Scholar]
- (35).Rapp JH, Wolff SD, Quinn SF, Soto JA, Meranze SG, Muluk S, Blebea J, Johnson SP, Rofsky NM, Duerinckx A, Foster GS, Kent KC, Moneta G, Middlebrook MR, Narra MR, Toombs BD, Pollak J, Yucel EK, Shamsi K, Weisskoff RM. Radiology. 2005;236:71. doi: 10.1148/radiol.2361040148. [DOI] [PubMed] [Google Scholar]
- (36).Goyen M, Edelman M, Perreault P, O'Riordan E, Bertoni H, Taylor J, Siragusa D, Sharafuddin M, Mohlerm ER,I, Breger R, Yucel EK, Shamsi K, Weisskoff RM. Radiology. 2005;236:825. doi: 10.1148/radiol.2363040577. [DOI] [PubMed] [Google Scholar]
- (37).Lauffer RB, Parmelee DJ, Dunham SU, Oullet HS, Dolan RP, Witte S, McMurry TJ, Walovitch RC. Radiology. 1998;207:529. doi: 10.1148/radiology.207.2.9577506. [DOI] [PubMed] [Google Scholar]
- (38).McMurry TJ, Parmelee DJ, Sajiki H, Scott DM, Oullet HS, Walovitch RC, Tyeklár Z, Dumas S, Bernard P, Nadler S, Midelfort K, Greenfield MT, Troughton JS, Lauffer RB. J. Med. Chem. 2002;45:3465. doi: 10.1021/jm0102351. [DOI] [PubMed] [Google Scholar]
- (39).Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorg. Chem. 2007;46:6632. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- (40).Tyeklár Z, Dunham SU, Midelfort K, Scott DM, Sajiki H, Ong K, Lauffer RB, Caravan P, McMurry TJ. Inorg. Chem. 2007;46:6621. doi: 10.1021/ic7006843. [DOI] [PubMed] [Google Scholar]
- (41).Caravan P, Cluotier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JWM, Amedio JC, Jr., Looby RJ, Supkowski RM, Horrocks WD, Jr., McMurry TJ, Lauffer RB. J. Am. Chem. Soc. 2002;124:3152. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- (42).Haddy AE, Frasch WD, Sharp RR. Biochemistry. 1985;24:7926. [Google Scholar]
- (43).Fanali G, Cao Y, Ascenzi P, Fasano M. J. Inorg. Biochem. 2012;117:198. doi: 10.1016/j.jinorgbio.2012.08.013. [DOI] [PubMed] [Google Scholar]
- (44).Lauffer RB. Magn. Reson. Med. 1991;22:339. doi: 10.1002/mrm.1910220237. [DOI] [PubMed] [Google Scholar]
- (45).Aime S, Anelli PL, Botta M, Brocchetta M, Canton S, Fedeli F, Gianolio E, Terreno E. J. Biol. Inorg. Chem. 2002;7:58. doi: 10.1007/s007750100265. [DOI] [PubMed] [Google Scholar]
- (46).Gianolio E, Cabella C, Serra SC, Valbusa G, Arena F, Maiocchi A, Miragoli L, Tedoldi F, Uggeri F, Visigalli M, Bardini P, Aime S. J. Biol. Inorg. Chem. 2014;19:716. doi: 10.1007/s00775-014-1111-z. [DOI] [PubMed] [Google Scholar]
- (47).Cheng W, Ganesh T, Martinez F, Lam J, Yoon H, Macgregor RB, Jr., Scholl TJ, Cheng H-LM, Zhang X.-a. J. Biol. Inorg. Chem. 19:229. doi: 10.1007/s00775-013-1073-6. 2104. [DOI] [PubMed] [Google Scholar]
- (48).Aime S, Chiaussa M, Degilio G, Gianolio E, Terreno E. J. Biol. Inorg. Chem. 1999;4:766. doi: 10.1007/s007750050349. [DOI] [PubMed] [Google Scholar]
- (49).Lambregts DMJ, Heijnen LA, Maas M, Rutten IJG, Martens MH, Backes WH, Riedl RG, Bakers FCH, Cappendijk VC, Beets GL, Beets-Tan RGH. Abdom. Imaging. 2013;38:720. doi: 10.1007/s00261-012-9957-4. [DOI] [PubMed] [Google Scholar]
- (50).Essig M, Rohrer M, Giesel F, Tüttenberg J, Weber M-W, Michaely M, Gerigk L, Voth M. Eur. Radiol. 2010;20:218. doi: 10.1007/s00330-009-1530-3. [DOI] [PubMed] [Google Scholar]
- (51).Lobbes MB, Heeneman S, Passos VL, Welten R, Kwee RM, van der Geest RJ, Wiethoff AJ, Caravan P, Misselwitz B, Daeman MJ, van Engelshoven JM, Leiner T, Kooi ME. Invest. Radiol. 2010;45:275. doi: 10.1097/RLI.0b013e3181d5466b. [DOI] [PubMed] [Google Scholar]
- (52).Ciesienski KL, Caravan P. Curr. Cardiovasc. Imaging Rep. 2010;4:77. doi: 10.1007/s12410-010-9061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kolodziej AF, Nair SA, Graham P, McMurry TJ, Ladner RC, Wescott C, Sexton DJ, Caravan P. Bioconjugate Chem. 2012;53:548. doi: 10.1021/bc200613e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kolodziej AF, Zhang Z, Overoye-Chan K, Jacques V, Caravan P. Methods Mol. Biol. 2014;1088:185. doi: 10.1007/978-1-62703-673-3_13. [DOI] [PubMed] [Google Scholar]
- (55).Nair SA, Kolodziej AF, Bhole G, Greenfield MT, McMurry TJ, Caravan P. Angew. Chem. Int. Ed. 2008;47:4918. doi: 10.1002/anie.200800563. [DOI] [PubMed] [Google Scholar]
- (56).Zhang Z, Kolodziej AF, Qi J. N., Shrikumar A., Wang X, Case AW, Greenfield MT, Graham PB, McMurry TJ, Caravan P. New J. Chem. 2010;34:611. doi: 10.1039/b9nj00787c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Vymazal J, Spuentrup E, Cardenas-Molina G, Wiethoff AJ, Hartmann MG, Caravan P, Parsons EC., Jr. Invest. Radiol. 2009;44:697. doi: 10.1097/RLI.0b013e3181b092a7. [DOI] [PubMed] [Google Scholar]
- (58).Spuentrup E, Botnar RM, Wiethoff AJ, Ibrahim T, Kelle S, Katoh M, Ozgun M, Nagel E, Vymazal J, Graham PB, Günther RW, Maintz D. Eur. Radiol. 2008;18:1995. doi: 10.1007/s00330-008-0965-2. [DOI] [PubMed] [Google Scholar]
- (59).Overoye-Chan K, Koerner S, Looby RJ, Kolodziej AF, Zech SG, Deng Q, Chasse JM, McMurry TJ, Caravan P. J. Am. Chem. Soc. 2008;130:6025. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]
- (60).Uppal R, Ay I, Dai G, Kim YR, Sorenson AG, Caravan P. Stroke. 2010;41:1271. doi: 10.1161/STROKEAHA.109.575662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Spuentrup E, Katoh M, Buecker A, Fausten B, Wiethoff AJ, Wildberger JE, Haage P, Parsons EC, Jr., Botnar RM, Graham PB, Vettelschoss M, Günther RW. Invest. Radiol. 2007;42:586. doi: 10.1097/RLI.0b013e31804fa154. [DOI] [PubMed] [Google Scholar]
- (62).Spuentrup E, Fausten B, Kinzel S, Wiethoff AJ, Botnar RM, Graham PB, Haller S, Katoh M, Parsons EC, Jr., Manning WJ, Busch T, Günther RW, Buecker A. Circulation. 2005;112:396. doi: 10.1161/CIRCULATIONAHA.104.529941. [DOI] [PubMed] [Google Scholar]
- (63).Spuentrup E, Buecker A, Katoh M, Wiethoff AJ, Parsons EC, Jr., Botnar RM, Weisskoff RM, Graham PB, Manning WJ, Günther RW. Circulation. 2005;111:1377. doi: 10.1161/01.CIR.0000158478.29668.9B. [DOI] [PubMed] [Google Scholar]
- (64).Uppal R, Medarova Z, Farrar CT, Dai G, Moore A, Caravan P. Invest. Radiol. 2012;47:553. doi: 10.1097/RLI.0b013e31825dddfb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Uppal R, Catana C, Ay I, Benner T, Sorenson AG, Caravan P. Radiology. 2011;258:812. doi: 10.1148/radiol.10100881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Caravan P, Das B, Deng Q, Dumas S, Jacques V, Koerner S, Kolodziej AF, Looby RJ, Sun W-C, Zhang Z. Chem. Commun. 2009:430. doi: 10.1039/b819098d. [DOI] [PubMed] [Google Scholar]
- (67).Caravan P, Das B, Dumas S, Epstein FH, Helm PA, Jacques V, Koerner S, Kolodziej AF, Shen L, Sun W-C, Zhang Z. Angew. Chem. Int. Ed. 2007;46:8171. doi: 10.1002/anie.200700700. [DOI] [PubMed] [Google Scholar]
- (68).Spuentrup E, Ruhl KM, Botnar RM, Wiethoff AJ, Buhl A, Jacques V, Greenfield MT, Krombach GA, Günther RW, Vangel MG, Caravan P. Circulation. 2009;119:1768. doi: 10.1161/CIRCULATIONAHA.108.826388. [DOI] [PubMed] [Google Scholar]
- (69).Helm PA, Caravan P, French BA, Jacques V, Shen L, Xu Y, Beyers RJ, Roy RJ, Kramer CM, Epstein FH. Radiology. 2008;3:788. doi: 10.1148/radiol.2473070975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Polasek M, Fuchs BC, Uppal R, Schühle DT, Alford JK, Loving GS, Yamada Y, Wei L, Lauwers GY, Guimaraes AR, Tanabe KK, Caravan P. J. Hepatology. 2012;57:549. doi: 10.1016/j.jhep.2012.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Fuchs BC, Wang H, Yang Y, Wei L, Polasek M, Schühle DT, Lauwers GY, Parker A, Sinskey AJ, Tanabe KK, Caravan P. J. Hepatology. 2013;59:992. doi: 10.1016/j.jhep.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, Denk H, Desmet V, Korb G, MacSween RNM, Phillips MJ, Portmann BG, Poulsen H, Scheuer PJ, Schmid M, Thaler H. J. Hepatology. 1995;22:696. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- (73).Caravan P, Yang Y, Zachariah R, Schmitt A, Mino-Kenudson M, Chen HH, Sosnovik DE, Dai G, Fuchs BC, Lanuti M. Am. J. Respir. Cell Mol. Biol. 2013;49:1120. doi: 10.1165/rcmb.2013-0039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Caravan P, Greenwood JM, Welch JT, Franklin SJ. Chem. Commun. 2003;20:2574. doi: 10.1039/b307817e. [DOI] [PubMed] [Google Scholar]
- (75).Huang S, Chen HH, Yuan H, Dai G, Schühle DT, Mekkaoui C, Ngoy S, Liao R, Caravan P, Josephson L, Sosnovik DE. Circ. Cardiovasc. Imaging. 2011;4:729. doi: 10.1161/CIRCIMAGING.111.966374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zech SG, Sun WC, Jacques V, Caravan P, Astashkin AV, Raitsimring AM. Chem. Phys. Chem. 2005;6:2570. doi: 10.1002/cphc.200500250. [DOI] [PubMed] [Google Scholar]
- (77).Raitsimring AM, Astashkin AV, Baute D, Goldfarb D, Poluektov OG, Lowe MP, Zech SG, Caravan P. Chem. Phys. Chem. 2006;7:1590. doi: 10.1002/cphc.200600138. [DOI] [PubMed] [Google Scholar]
- (78).Eldredge HB, Spiller M, Chasse JM, Greenwood MT, Caravan P. Invest. Radiol. 2006;41:229. doi: 10.1097/01.rli.0000199293.86956.48. [DOI] [PubMed] [Google Scholar]
- (79).Gale EM, Zhu J, Caravan P. J. Am. Chem. Soc. 2013;135:18600. doi: 10.1021/ja4094132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Caravan P, Astashkin AV, Raitsimring AM. Inorg. Chem. 2003;42:3972. doi: 10.1021/ic034414f. [DOI] [PubMed] [Google Scholar]
- (81).Raitsimring AM, Astashkin AV, Baute D, Goldfarb D, Caravan P. J. Phys. Chem. A. 2004;108:7318. [Google Scholar]
- (82).Astashkin AV, Raitsimring AM, Caravan P. J. Phys. Chem. A. 2004;108:1990. [Google Scholar]
- (83).Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JWM, Amedio JC, Looby RJ, Supkowski RM, Horrocks WD, McMurry TJ, Lauffer RB. J. Am. Chem. Soc. 2002;124:3152. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- (84).Zhang Z, Greenfield MT, Spiller M, McMurry TJ, Lauffer RB, Caravan P. Angew. Chem. Int. Ed. 2005;44:6766. doi: 10.1002/anie.200502245. [DOI] [PubMed] [Google Scholar]
- (85).Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorg. Chem. 2007;46:6632. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- (86).Ali MM, Woods M, Caravan P, Opina AC, Spiller M, Fettinger JC, Sherry AD. Chem. Eur. J. 2008;14:7250. doi: 10.1002/chem.200800402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Zech SG, Eldredge HB, Lowe MP, Caravan P. Inorg. Chem. 2007;46:3576. doi: 10.1021/ic070011u. [DOI] [PubMed] [Google Scholar]
- (88).Zhou X, Caravan P, Clarkson R, Westlund P-O. J. Magn. Reson. 2004;167:147. doi: 10.1016/j.jmr.2003.12.006. [DOI] [PubMed] [Google Scholar]
- (89).Raitsimring A, Astashkin A, Poluektov O, Caravan P. Appl. Magn. Reson. 2005;28:281. [Google Scholar]
- (90).Dumas S, Jacques V, Sun W-C, Troughton JS, Welch JT, Chasse JM, Schmitt-Willich H, Caravan P. Invest Radiol. 2010;45:600. doi: 10.1097/RLI.0b013e3181ee5a9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Caravan P, Tóth É, Rockenbauer A, Merbach AE. J. Am. Chem. Soc. 1999;121:10403. [Google Scholar]
- (92).Troughton JS, Greenfield MT, Greenwood JM, Dumas S, Wiethoff AJ, Wang J, Spiller M, McMurry TJ, Caravan P. Inorg. Chem. 2004;43:6313. doi: 10.1021/ic049559g. [DOI] [PubMed] [Google Scholar]
- (93).Boros E, Polasek M, Zhang Z, Caravan P. J. Am. Chem. Soc. 2012;134:19858. doi: 10.1021/ja309187m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Polasek M, Caravan P. Inorg. Chem. 2013;52:4084. doi: 10.1021/ic400227k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Boros E, Karimi S, Kenton N, Helm L, Caravan P. Inorg. Chem. 2014;53:6985. doi: 10.1021/ic5008928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Swift T, Connick RE. J. Chem. Phys. 1962;37:307. [Google Scholar]
- (97).Caravan P, Amedio JC, Dunham SU, Greenfield MT, Cloutier NJ, McDermid SA, Spiller M, Zech SG, Looby RJ, Raitsimring AM. Chem. Eur. J. 2005;11:5866. doi: 10.1002/chem.200500338. [DOI] [PubMed] [Google Scholar]
- (98).Moriggi L, Yaseen MA, Helm L, Caravan P. Chem. Eur. J. 2012;18:3675. doi: 10.1002/chem.201103344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Gale EM, Kenton N, Caravan P. Chem. Commun. 2013;49:8060. doi: 10.1039/c3cc44116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Aime S, Calabi L, Cavallotti C, Gianolio E, Giovenzana GB, Losi P, Maiocchi A, Palmisano G, Sisti M. Inorg. Chem. 2004;43:7588. doi: 10.1021/ic0489692. [DOI] [PubMed] [Google Scholar]
- (101).Baranyai Z, Uggeri F, Maiocchi A, Giovenzana GB, Cavallotti C, Takács A, Tóth I, Bányai I, Bényei A, Brucher E, Aime S. Eur. J. Inorg. Chem. 2013;2013:147. [Google Scholar]
- (102).Bretonnière Y, Mazzanti M, Pécaut J, Dunand FA, Merbach AE. Chem. Commun. 2001:621. doi: 10.1021/ic010591+. [DOI] [PubMed] [Google Scholar]
- (103).Tóth É, Vauthey S, Pubanz D, Merbach AE. Inorg. Chem. 1996;35:3375. doi: 10.1021/ic951492x. [DOI] [PubMed] [Google Scholar]
- (104).Pellegatti L, Zhang J, Drahos B, Villette S, Suzenet F, Guillaumet G, Petoud S, Toth E. Chem. Commun. 2008:6591. doi: 10.1039/b817343e. [DOI] [PubMed] [Google Scholar]
- (105).Messeri D, Lowe MP, Parker D, Botta M. Chem. Commun. 2001:2742. [Google Scholar]
- (106).Aime S, Botta M, Crich SG, Giovenzana G, Pagliarin R, Sisti M, Terreno E. Magn. Reson. Chem. 1998;36:S200. [Google Scholar]
- (107).Kumar K, Chang CA, Tweedle M. Inorg. Chem. 1993;32:587. [Google Scholar]
- (108).Supkowski RM, Horrocks WD. Inorg. Chem. 1999;38:5616. doi: 10.1021/ic990597n. [DOI] [PubMed] [Google Scholar]
- (109).Bruce JI, Dickins RS, Govenlock LJ, Gunnlaugsson T, Lopinski S, Lowe MP, Parker D, Peacock RD, Perry JJB, Aime S, Botta M. J. Am. Chem. Soc. 2000;122:9674. [Google Scholar]
- (110).Lowe MP, Parker D, Reany O, Aime S, Botta M, Castellano G, Gianolio E, Pagliarin R. J. Am. Chem. Soc. 2001;123:7601. doi: 10.1021/ja0103647. [DOI] [PubMed] [Google Scholar]
- (111).Pálinkás Z, Roca-Sabio A, Mato-Iglesias M, Esteban-Gómez D, Platas-Iglesias C, De Blas A, Rodriguez-Blas T, Tóth É. Inorg. Chem. 2009;48:8878. doi: 10.1021/ic9011197. [DOI] [PubMed] [Google Scholar]
- (112).Gale EM, Kenton N, Caravan P. Chem. Commun. 2013;49:8060. doi: 10.1039/c3cc44116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Sherry A, Ren J, Huskens J, Brücher E, Toth E, Geraldes C, Castro M, Cacheris W. Inorganic Chemistry. 1996;35:4604. [Google Scholar]
- (114).Caravan P, Greenfield MT, Li X, Sherry AD. Inorg. Chem. 2001;40:6580. doi: 10.1021/ic0102900. [DOI] [PubMed] [Google Scholar]
- (115).Jacques V, Dumas S, Sun W-C, Troughton JS, Greenfield MT, Caravan P. Inves. Radiol. 2010;45:613. doi: 10.1097/RLI.0b013e3181ee6a49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Dunand FA, Borel A, Merbach AE. J. Am. Chem. Soc. 2002;124:710. doi: 10.1021/ja016873q. [DOI] [PubMed] [Google Scholar]
- (117).Dunand FA, Tóth É, Hollister R, Merbach AE. J. Biol. Inorg. Chem. 2001;6:247. doi: 10.1007/s007750000193. [DOI] [PubMed] [Google Scholar]
- (118).Nicolle GM, Tóth É, Schmitt-Willich H, Radüchel B, Merbach AE. Chem. Eur. J. 2002;8:1040. doi: 10.1002/1521-3765(20020301)8:5<1040::aid-chem1040>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- (119).Caravan P, Farrar CT, Frullano L, Uppal R. Contrast Media Mol. Imaging. 2009;4:89. doi: 10.1002/cmmi.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Zhang Z, Kolodziej AF, Greenfield MT, Caravan P. Angew. Chem. Int. Ed. 2011;50:2621. doi: 10.1002/anie.201007689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Rudovský J, Kotek J, Hermann P, Lukes I, Mainero V, Aime S. Org. Biomol. Chem. 2005;3:112. doi: 10.1039/b410103k. [DOI] [PubMed] [Google Scholar]
- (122).Thompson MK, Botta M, Nicolle G, Helm L, Aime S, Merbach AE, Raymond KN. J. Am. Chem. Soc. 2003;125:14274. doi: 10.1021/ja037441d. [DOI] [PubMed] [Google Scholar]
- (123).Zhang S, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. Acc. Chem. Res. 2003;36:783. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- (124).Lauffer RB. Chem. Rev. 1987;87:901. [Google Scholar]
- (125).Rocklage SM, Cacheris WP, Quay SC, Hahn FE, Raymond KN. Inorg. Chem. 1989;28:477. [Google Scholar]
- (126).Lim K, Stark D, Leese P, Pfefferbaum A, Rocklage S, Quay S. Radiology. 1991;178:79. doi: 10.1148/radiology.178.1.1898539. [DOI] [PubMed] [Google Scholar]
- (127).Schmidt PP, Toft KG, Skotland T, Andersson KK. J. Biol. Inorg. Chem. 2002;7:241. doi: 10.1007/s007750100290. [DOI] [PubMed] [Google Scholar]
- (128).Drahoš B, Lukeš I, Toth E. Eur. J. Inorg. Chem. 2012;2012:1975. [Google Scholar]
- (129).Kálmán FK, Tircsó G. Inorg. Chem. 2012;51:10065. doi: 10.1021/ic300832e. [DOI] [PubMed] [Google Scholar]
- (130).Drahoš B, Kubíček V, Bonnet CS, Hermann P, Lukeš I, Tóth É. Dalton Trans. 2011;40:1945. doi: 10.1039/c0dt01328e. [DOI] [PubMed] [Google Scholar]
- (131).Drahoš B, Kotek J, Císařová I, Hermann P, Helm L, Lukeš I, Tóth E. v. Inorg. Chem. 2011;50:12785. doi: 10.1021/ic201935r. [DOI] [PubMed] [Google Scholar]
- (132).Kueny-Stotz M, Garofalo A, Felder-Flesch D. Eur. J. Inorg. Chem. 2012;2012:1987. [Google Scholar]
- (133).Tu C, Osborne EA, Louie AY. Ann. Biomed. Eng. 2011;39:1335. doi: 10.1007/s10439-011-0270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Tsitovich PB, Burns PJ, McKay AM, Morrow JR. J. Inorg. Biochem. 2014;133:143. doi: 10.1016/j.jinorgbio.2014.01.016. [DOI] [PubMed] [Google Scholar]
- (135).Do QN, Ratnakar SJ, Kovács Z, Sherry AD. ChemMedChem. 2014;9:1116. doi: 10.1002/cmdc.201402034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Aime S, Botta M, Gianolio E, Terreno E. Angew. Chem. Int. Ed. 2000;39:747. doi: 10.1002/(sici)1521-3773(20000218)39:4<747::aid-anie747>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- (137).Major JL, Bolteau RM, Meade TJ. Inorg. Chem. 2008;47:10788. doi: 10.1021/ic801458u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Que EL, Gianolio E, Baker SL, Wong AP, Aime S, Chang CJ. J. Am. Chem. Soc. 2009;131:8527. doi: 10.1021/ja900884j. [DOI] [PubMed] [Google Scholar]
- (139).Dhingra K, Fousková P, Angelovski G, Maier ME, Logothetis NK, Tóth E. J. Biol. Inorg. Chem. 2008;13:35. doi: 10.1007/s00775-007-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Moats RA, Fraser SE, Meade TJ. Angew. Chem. Int. Ed. 1997;36:726. [Google Scholar]
- (141).Louie AY, Hüber MM, Ahrens ET, Rothbächer U, Moats R, Jacobs RE, Fraser SE, Meade TJ. Nat. Biotechnol. 2000;18:321. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- (142).Urbanczyk-Pearson LM, Femia FJ, Smith J, Parigi G, Duimstra JA, Eckermann EL, Luchinat C, Meade TJ. Inorg. Chem. 2002;47:56. doi: 10.1021/ic700888w. [DOI] [PubMed] [Google Scholar]
- (143).Chen JW, Querol Sans M, Bogdanov A, Jr, Weissleder R. Radiology. 2006;240:473. doi: 10.1148/radiol.2402050994. [DOI] [PubMed] [Google Scholar]
- (144).Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S, Moskowitz MA, Weissleder R. Proc. Natl. Acad. Sci. 2008;105:18584. doi: 10.1073/pnas.0803945105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Ronald JA, Chen JW, Chen Y, Hamilton AM, Rodriguez E, Reynolds F, Hegele RA, Rogers KA, Querol M, Bogdanov A. Circulation. 2009;120:592. doi: 10.1161/CIRCULATIONAHA.108.813998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Rodríguez E, Nilges M, Weissleder R, Chen JW. J. Am. Chem. Soc. 2009;132:168. doi: 10.1021/ja905274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Nivorozhkin AL, Kolodziej AF, Caravan P, Greenfield MT, Lauffer RB, McMurry TJ. Angew. Chem. Int. Ed. 2001;40:2903. [PubMed] [Google Scholar]
- (148).Zhang S, Wu K, Sherry AD. Angew. Chem., Int. Ed. Engl. 1999;38:3192. [PubMed] [Google Scholar]
- (149).Garcia-Martin ML, Martinez GV, Raghunand N, Sherry AD, Zhang S, Gillies RJ. Magn. Reson. Med. 2006;55:309. doi: 10.1002/mrm.20773. [DOI] [PubMed] [Google Scholar]
- (150).Raghunand N, Howison C, Sherry AD, Zhang S, Gillies RJ. Magn. Reson. Med. 2003;49:249. doi: 10.1002/mrm.10347. [DOI] [PubMed] [Google Scholar]
- (151).Kálmán FK, Woods M, Caravan P, Jurek P, Spiller M, Tircsó G, Király R, Brücher E, Sherry AD. Inorg. Chem. 2007;46:5260. doi: 10.1021/ic0702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Frullano L, Catana C, Benner T, Sherry AD, Caravan P. Angew. Chem. Int. Ed. 2010;122:2432. doi: 10.1002/anie.201000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Esqueda AC, López JA, Andreu-de-Riqure G, Alvarado-Monzón JC, Ratnakar SJ, Lubag AJM, Sherry AD, De León-Rodríguez LM. J. Am. Chem. Soc. 2009;131:11387. doi: 10.1021/ja901875v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Lubag AJM, De León-Rodríguez LM, Burgess SC, Sherry AD. Proc. Natl. Acad. Sci., U.S.A. 2011;108:18400. doi: 10.1073/pnas.1109649108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Loving GS, Mukherjee S, Caravan P. J. Am. Chem. Soc. 2013;135:4623. doi: 10.1021/ja312610j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Gale EM, Mukherjee S, Liu C, Loving GS, Caravan P. Inorg. Chem. 2014;53:10748. doi: 10.1021/ic502005u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Loving GS, Caravan P. Angew. Chem. Int. Ed. 2014;53:1140. doi: 10.1002/anie.201308607. [DOI] [PMC free article] [PubMed] [Google Scholar]