

ABSTRACT
The cytokine gamma interferon (IFN-γ) induces cell-autonomous immunity to combat infections with intracellular pathogens, such as the bacterium Chlamydia trachomatis. The present study demonstrates that IFN-γ-primed human cells ubiquitinate and eliminate intracellular Chlamydia-containing vacuoles, so-called inclusions. We previously described how IFN-γ-inducible immunity-related GTPases (IRGs) employ ubiquitin systems to mark inclusions for destruction in mouse cells and, furthermore, showed that the rodent pathogen Chlamydia muridarum blocks ubiquitination of its inclusions by interfering with mouse IRG function. Here, we report that ubiquitination of inclusions in human cells is independent of IRG and thus distinct from the murine pathway. We show that C. muridarum is susceptible to inclusion ubiquitination in human cells, while the closely related human pathogen C. trachomatis is resistant. C. muridarum, but not C. trachomatis, inclusions attract several markers of cell-autonomous immunity, including the ubiquitin-binding protein p62, the ubiquitin-like protein LC3, and guanylate-binding protein 1. Consequently, we find that IFN-γ priming of human epithelial cells triggers the elimination of C. muridarum, but not C. trachomatis, inclusions. This newly described defense pathway is independent of indole-2,3-dioxygenase, a known IFN-γ-inducible anti-Chlamydia resistance factor. Collectively, our observations indicate that C. trachomatis evolved mechanisms to avoid a human-specific, ubiquitin-mediated response as part of its unique adaptation to its human host.
IMPORTANCE
Chlamydia trachomatis is the leading cause of sexually transmitted bacterial infections and responsible for significant morbidity, including pelvic inflammatory disease, infertility, and ectopic pregnancies in women. As an obligate intracellular pathogen, C. trachomatis is in perpetual conflict with cell-intrinsic defense programs executed by its human host. Our study defines a novel anti-Chlamydia host resistance pathway active in human epithelial cells. This defense program promotes the deposition of the small antimicrobial protein ubiquitin on vacuoles containing Chlamydia. We show that this ubiquitin-based resistance pathway of human cells is highly effective against a Chlamydia species adapted to rodents but ineffective against human-adapted C. trachomatis. This observation indicates that C. trachomatis evolved strategies to avoid entrapment within ubiquitin-labeled vacuoles as part of its adaptation to the human innate immune system.
INTRODUCTION
The intracellular bacterial pathogen Chlamydia trachomatis is among the most common causative agents of sexually transmitted infections. According to the World Health Organization, an estimated 100 million individuals are infected per annum (1). Many of these infections lead to disease and irreparable pathologies; C. trachomatis infections frequently result in urethritis in men and pelvic inflammatory disease, tubal factor infertility, and ectopic pregnancies in women (2–4). C. trachomatis-associated diseases in women are due to extensive pathogen exposure stemming from chronic, recurring, or repeat infections, all indicative of an inability of the human immune system to promptly sterilize C. trachomatis infections or to establish effective immune memory. The failure of our immune system to protect against C. trachomatis infections is likely the consequence of active or passive immune evasion by this stealth pathogen (2–8).
C. trachomatis is an obligate intracellular pathogen that resides and replicates within the confines of specialized intracellular vacuoles termed “inclusions” (9). C. trachomatis establishes an infection by taking primary residency inside epithelial cells. C. trachomatis enters epithelial cells in its infectious form known as the elementary body (EB) and then differentiates into the replicative reticulate body (RB). Following several rounds of binary fission within the inclusion, RBs begin to differentiate back into EBs, which then exit the spent host cell (9, 10). While naive epithelial cells are permissive for intracellular C. trachomatis growth, priming of human cells with the proinflammatory cytokine gamma interferon (IFN-γ) inhibits the ability of C. trachomatis to complete its developmental cycle (11).
IFN-γ is predominantly produced by lymphocytes in response to an infection, yet its cognate receptor is expressed in virtually all cell types (12). Priming of cells with IFN-γ induces the expression of hundreds of IFN-stimulated genes (ISGs), which control an extensive network of cell-autonomous defense programs (8, 12, 13). In human epithelial cells, IFN-γ-activated cell-autonomous immunity to C. trachomatis is mediated by the enzyme indole-2,3-dioxygenase (IDO). IDO metabolizes host cell tryptophan and thereby depletes intracellular tryptophan stores. Because C. trachomatis is a tryptophan auxotroph, tryptophan depletion restricts intracellular replication of C. trachomatis (14–16). In response to tryptophan starvation, C. trachomatis scavenges extracellular indole from its surrounding microbial community and thereby counteracts IDO-mediated nutritional immunity (6, 8, 17, 18). However, it has remained unknown whether and how C. trachomatis resists immunity executed by any human ISGs other than IDO.
In mice, the human-restricted pathogen C. trachomatis is quickly eliminated through IFN-γ-mediated immune responses that are independent of IDO (19–22). A forward genetic screen approach identified IFN-γ-inducible immunity-related GTPases (IRGs) as critical host resistance factors that execute sterilizing immunity against C. trachomatis in mice (20, 23). Members of the IRG protein family function cooperatively to detect the locations of inclusions within host cells (24). Following binding to inclusions, IRG proteins recruit E3 ligases, such as tumor necrosis factor receptor-associated factor 6 (TRAF6) and tripartite motif-containing protein 21 (TRIM21) and thereby promote the deposition of ubiquitin on unknown substrates associated with inclusion membranes (25). Ubiquitinated C. trachomatis inclusions become targets for the ubiquitin-binding protein p62, which escorts antimicrobial guanylate-binding proteins (GBPs) to inclusions. The IRG-dependent ubiquitination of inclusions ultimately results in inclusion rupture, the release of bacteria into the host cell cytosol (25), and the engulfment of the ejected bacteria inside degradative autolysosomes (26).
Mouse IRG proteins can be placed into two subgroups that are defined by the amino acid sequence of their GTP binding pockets and by their subcellular localization. The majority of IRG proteins feature a canonical GXXXXGKS sequence in the P-loop of the GTP binding site and are accordingly referred to as “GKS” proteins (24, 27). GKS proteins are predominantly found in the host cell cytosol yet are able to translocate to inclusion membranes upon infection with C. trachomatis (28). This inclusion targeting event is regulated by the second subgroup of the IRG protein family, the IRG family M (IRGM) proteins (29), which are defined by their noncanonical GXXXXGMS P-loop sequence (27). While GKS proteins bind to inclusions, IRGM proteins instead associate with host cell organelles and prevent GKS binding to these self-structures (24, 29). Membrane-bound IRGM proteins undergo transient interactions with GKS proteins and maintain GKS proteins in their inactive, monomeric GDP-bound state (30). GKS monomers acquire GTP, oligomerize, and bind to membranes once they come into contact with IRGM-depleted pathogen-containing vacuoles (29, 30). In IRGM-deficient mouse cells, cytoplasmic GKS proteins transition spontaneously into the GTP-bound state and form aggregates (30, 31). Aggregation depletes the pool of transportable GKS monomers, and IRGM-deficient mouse cells therefore fail to deliver GKS proteins, E3 ligases, and ubiquitin to C. trachomatis inclusions (25).
The IRG gene family consists of approximately 20 members in the mouse but has undergone a dramatic collapse in the primate lineage. The human genome encodes a single IRGM ortholog and lacks GKS-encoding genes entirely (27, 32). Because the murine IRG system is essential for the delivery of ubiquitin to C. trachomatis inclusions (25), the question arose whether human cells could ubiquitinate inclusions in spite of their diminished IRG system. We report here that human cells possess an IRG-independent system for the attachment of ubiquitin to inclusions. We further report that human cells can deliver ubiquitin to inclusions formed by the rodent-adapted pathogen Chlamydia muridarum but that the human-adapted species C. trachomatis is resistant to ubiquitination in human cells. These observations indicate that C. trachomatis has evolved strategies to interfere with the human-specific ubiquitination machinery.
RESULTS
C. muridarum, but not C. trachomatis, inclusions are frequently associated with ubiquitin in human epithelial cells.
Because IFN-γ-primed mouse cells coat C. trachomatis inclusions with a layer of ubiquitin (25), we asked whether human cells were similarly able to deposit ubiquitin on C. trachomatis inclusions. To answer this question, we infected two commonly used human epithelial cell lines, A549 and HeLa cells, with C. trachomatis. When cells were stained with the antiubiquitin antibody FK2 at 20 h postinfection (hpi), ubiquitin-positive C. trachomatis inclusions were not detected in naive A549 cells and extremely rare in naive HeLa cells (less than 1% ubiquitin-positive inclusions [Fig. 1A and B]). IFN-γ priming resulted in a moderate increase in the number of ubiquitin-positive C. trachomatis inclusions yet did not exceed 5% in either cell line (Fig. 1B). We then monitored ubiquitin staining of inclusions formed by the rodent pathogen C. muridarum, which is resistant to ubiquitination in IFN-γ-primed mouse cells (25). Unexpectedly, we detected ubiquitin-positive C. muridarum inclusions at relatively high frequencies (up to 15%) compared to C. trachomatis inclusions in naive A549 and HeLa cells. IFN-γ priming further increased the frequency of ubiquitin-positive C. muridarum inclusions to approximately 50% (Fig. 1A and B). These data demonstrated that IFN-γ priming in human cells—similar to mouse cells—prompts robust ubiquitination of inclusions. Yet, in clear contrast to mouse cells, human cells fail to efficiently coat C. trachomatis inclusions with ubiquitin. The latter observations could be explained if the pathways leading to inclusion ubiquitination differed between mice and humans and if C. trachomatis was specifically adapted to the human, but not the mouse, pathway.
FIG 1 .
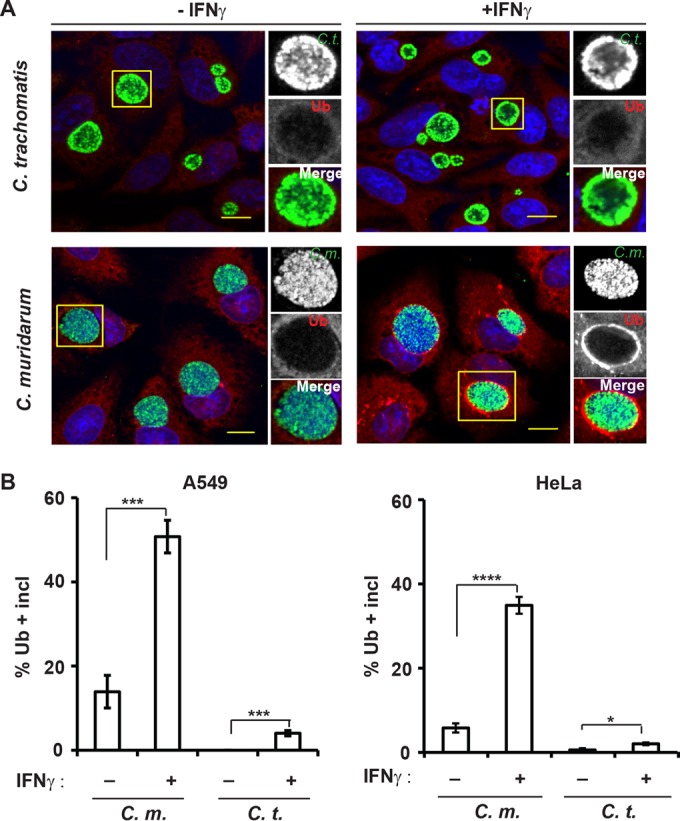
IFN-γ priming promotes ubiquitin deposition on inclusions in human cells. (A) A549 epithelial cells were infected with either C. muridarum (C.m.) or C. trachomatis (C.t.) and primed with IFN-γ (+IFNγ) (100 U/ml) at 3 hpi or left unprimed (−IFNγ). At 20 hpi, cells were stained for ubiquitin (Ub) with antibody FK2 (red), rabbit anti-Slc1 (green), and DNA (blue). Representative images of Ub-positive inclusions are shown. Bars = 10 μm. (B) Colocalization of ubiquitin (FK2) with inclusions (incl) in A549 cells and HeLa cells was quantified as described in Materials and Methods. At least 100 inclusions were counted for each condition. Data are representative of three independent experiments. Values are means ± standard deviations (SD) (error bars). Values that are statistically significantly different by two-tailed unpaired Student’s t test are indicated by a bar and asterisks as follows: *, P < 0.05; ***, P < 0.005; ****, P < 0.001.
Ubiquitination of C. muridarum inclusions is independent of human IRGM.
On the basis of the rationale outlined above, we asked whether the human and mouse pathways of inclusion ubiquitination were distinct. We showed previously that IRGM proteins were essential for the decoration of inclusions with ubiquitin and the killing of C. trachomatis in IFN-γ-primed mouse cells (25, 29). To determine whether the single human IRGM ortholog is required for the ubiquitination of inclusions, we generated A549 and HeLa clonal cell lines with large (~230-bp) deletions in the human IRGM coding region (Fig. 2A). The frequency at which C. muridarum inclusions stained positive for ubiquitin was not significantly altered in cell clones carrying deletions in the human IRGM gene (Fig. 2B), demonstrating that inclusion ubiquitination in human cells is IRGM independent and therefore mechanistically distinct from the murine pathway.
FIG 2 .
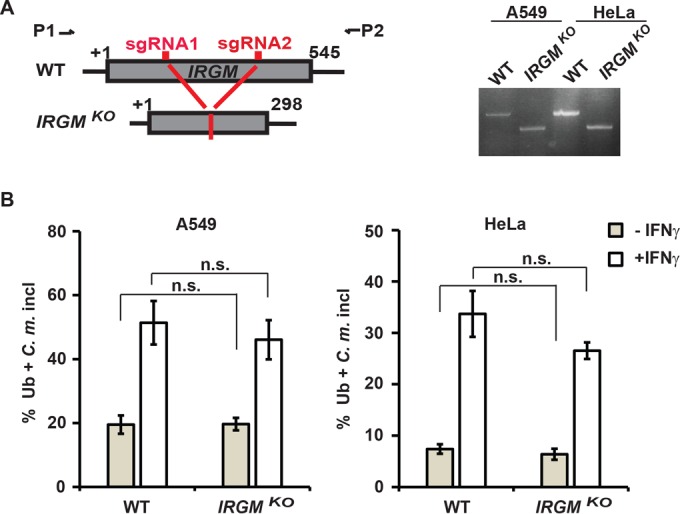
Human IRGM is dispensable for the ubiquitination of C. muridarum inclusions. (A) CRISPR/Cas9 genome editing was used to generate deletions of approximately 230 bp in the coding sequence of human IRGM. The locus was targeted using two small guide RNAs (sgRNA1 and sgRNA2), and individual cell clones were isolated from parental A549 and HeLa cell lines and confirmed for deletions in the IRGM gene by PCR. (B) Parental (wild type [WT]) and IRGMKO (KO stands for knockout) A549 and HeLa cells were infected with C. muridarum (C. m.) and primed with IFN-γ (100 U/ml) at 3 hpi or left unprimed. At 20 hpi, the cells were stained for ubiquitin (FK2) and C. muridarum (anti-Slc1). Colocalization of ubiquitin with inclusions in A549 cells and HeLa cells was quantified as described in Materials and Methods. At least 100 inclusions were counted for each condition. Values that are not statistically significantly different (n.s.) by two-tailed unpaired Student’s t test are indicated. Data are representative of three independent experiments. Values are means ± standard deviations (SD) (error bars).
Linear and branched ubiquitin, ubiquitin-binding proteins, and LC3 localize to C. muridarum inclusions in human cells.
We previously reported that K48- and K63-linked polyubiquitin and the ubiquitin-binding protein p62 associate with inclusions in IFN-γ-primed mouse cells (25). In order to determine whether a similar ubiquitination pattern is observed in human cells, we stained C. muridarum-infected A549 cells with linkage-specific antibodies. We found that K48- and K63-linked ubiquitin accumulated at C. muridarum, but not C. trachomatis, inclusions and that this association was exacerbated by IFN-γ priming (Fig. 3A). These results thus confirmed the data obtained with FK2 (Fig. 1), an antibody that detects K48- and K63-linked, but not linear, ubiquitin (33, 34). Linear ubiquitin polymers form through conjugation between the C-terminal α-carboxyl group of an incoming ubiquitin and the α-amino group of the N-terminal methionine (M1) of substrate-bound ubiquitin (35). Staining with anti-M1 ubiquitin antibody revealed that a high percentage of C. muridarum inclusions are decorated with M1 (Fig. 3A). In addition to these distinct types of ubiquitin conjugates, we also detected the presence of the ubiquitin-binding proteins p62 and NDP52 as well as the ubiquitin-like protein LC3 at C. muridarum, but not C. trachomatis, inclusions (Fig. 3B). Similar to the ubiquitin-staining pattern, we found that the association of p62, NDP52, and LC3 with C. muridarum inclusions was enhanced in IFN-γ-primed cells, indicating a functional link between inclusion ubiquitination and the recruitment of these additional host proteins.
FIG 3 .

Distinct ubiquitin species, ubiquitin-binding proteins, and LC3 associate with C. muridarum inclusions in human cells. Wild-type A549 cells were infected with C. muridarum or C. trachomatis and primed with IFN-γ (100 U/ml) at 3 hpi or left unprimed. (A and B) At 20 hpi, cells were stained with the linkage-specific antiubiquitin antibodies against K48, K63, or linear ubiquitin (M1) (A) or with antibodies against the host defense proteins (p62) NDP52 or LC3 as well as anti-chlamydial LPS and DNA (Hoechst) (B). (Left) Representative images are shown. Bars = 10 μm. (Right) Colocalization of different host markers with inclusions in A549 cells was quantified. At least 100 inclusions were counted for each condition. Data are representative of three independent experiments. Values are means ± SD. Values that are significantly different by two-tailed unpaired Student’s t test are indicated by a bar and asterisks as follows: **, P < 0.01; ***, P < 0.005.
Recruitment of host defense proteins p62, NDP52, and LC3 is restricted to ubiquitinated inclusions.
To address the question of whether inclusion ubiquitination could be functionally linked to the recruitment of additional host defense proteins, we first assessed whether ubiquitin, ubiquitin-binding proteins, and LC3 colocalized at inclusions. We observed that p62, NDP52, and LC3 localized exclusively to ubiquitin-positive inclusions, while more than half of ubiquitin-positive inclusions stained negative for p62, NDP52, or LC3 (Fig. 4A to C). These data indicated that ubiquitination occurs upstream of the recruitment of p62, NDP52, and LC3. We further recorded that many (~70%), but not all, NDP52-decorated inclusions stained positive for p62 and vice versa (Fig. 4D). Similarly, most, but not all, NDP52- or p62-positive inclusions (70 to 80%) colocalized with LC3 (Fig. 4E and F). These data suggested that the recruitment of p62, NDP52, and LC3 to inclusions is largely independent of each other, resembling previous observations made for the association of p62 and NDP52 with Salmonella-containing vacuoles (36).
FIG 4 .

NDP52, p62, and LC3 colocalize with ubiquitin on C. muridarum inclusions in A549 cells. (A to F) A549 cells expressing p62-GFP (A and D) or LC3-GFP (C, E, and F) or untransfected cells (B) were infected with C. muridarum, primed with IFN-γ (100 U/ml) at 3 hpi, fixed at 20 hpi, and stained with antibodies against the indicated host proteins. Hoechst staining was used to visualize bacterial DNA of inclusions. Representative images of colocalization between indicated host markers at C. muridarum inclusions are shown. At least 50 inclusions were counted for each condition. Data are representative of three independent experiments. The values in the graphs are means ± SD (error bars). Ub −, ubiquitin negative; Ub +, ubiquitin positive.
We then examined whether members of the GBP family of host resistance factor colocalized with Chlamydia based on our previous finding that murine GBPs bind to ubiquitinated C. trachomatis inclusions in mouse cells (25). To assess whether human GBPs could similarly target ubiquitinated inclusions in human cells, we monitored the subcellular localization of human GBP1 by ectopically expressing mCherry-labeled human GBP1 (mCherry-hGBP1) fusion protein in A549 cells. We found mCherry-hGBP1 localized to C. muridarum, but not C. trachomatis, inclusions (Fig. 5A). Colocalization of mCherry-hGBP1 with C. muridarum increased from approximately 5% in naive cells to about 40% in IFN-γ-primed cells (Fig. 5A), mirroring the IFN-γ-induced increase in the number of ubiquitin-positive inclusions. Most hGBP1-positive C. muridarum inclusions (>90%) also stained with antiubiquitin antibody FK2 (Fig. 5B), suggesting that hGBP1 specifically targets ubiquitin-positive inclusions. As an alternative explanation, we considered the possibility that inclusion-resident hGBP1 itself was required for the attachment of ubiquitin to inclusions. Refuting this model, we found that the frequency at which inclusions were decorated with ubiquitin remained the same between wild-type and hGBP1-defiicent A549 cells (Fig. 5C), demonstrating that hGBP1 itself is not required for labeling inclusions with ubiquitin.
FIG 5 .

Human GBP1 targets ubiquitin-positive inclusions. (A) A549 cells were transfected with an mCherry-hGBP1 expression construct and 24 h later infected with C. muridarum or C. trachomatis at an MOI of 2 and primed with IFN-γ (100 U/ml) at 3 hpi or left unprimed. At 20 hpi, the cells were fixed and stained with Hoechst. (Left) Representative images are shown. Bars = 10 μm. (Right) Percentage of mCherry-GBP1-positive inclusions in mCherry-expressing cells was quantified. Data are representative of three independent experiments. Values are means ± SD (error bars) (n = 3). Values that are significantly different (P < 0.001) by Student’s t test are indicated by a bar and four asterisks. (B) Experiment was performed as described above for panel A, except for staining cells additionally with anti-ubiquitin (FK2) and anti-Chlamydia (Slc1) antibodies. (Left) Representative images are shown. (Right) Ubiquitin staining of hGBP1-positive inclusions was quantified. Data are representative of two independent experiments. Values are means ± SD (n = 3). (C) Parental (WT) and GBP1KO A549 cells were infected with C. muridarum and primed with IFN-γ (100 U/ml) at 3 hpi or left unprimed. At 20 hpi, colocalization of ubiquitin (FK2) with inclusions was quantified. Data are representative of three independent experiments. Values are means ± SD. Values that are not significantly different (n.s.) are indicated.
Fusion with C. trachomatis inclusions reduces ubiquitination of C. muridarum inclusions.
Our data so far demonstrated that IFN-γ priming of human cells leads to the ubiquitination of C. muridarum inclusions and the subsequent recruitment of host defense proteins such as NDP52 and hGBP1. Two distinct models could explain why human host cells effectively ubiquitinate C. muridarum, but not C. trachomatis, inclusions. In the first model, a C. muridarum-derived factor X is directly recognized by the human host. This factor X would be predicted to be absent from C. trachomatis inclusions, which therefore remained largely devoid of ubiquitin. In the second model, both C. muridarum and C. trachomatis inclusions are bona fide targets of the host immune system, but only C. trachomatis is able to interfere with host-mediated ubiquitination through the activity of a C. trachomatis-derived factor Y (Fig. 6A). In order to differentiate between these two models, we took advantage of the ability of inclusions to fuse with each other in coinfected cells. To discern fused from unfused inclusions, we coinfected A549 cells with green fluorescent protein (GFP)-expressing C. muridarum and mCherry-expressing C. trachomatis. Next, we stained cells with antiubiquitin antibody and quantified the percentage of ubiquitin-positive inclusions containing GFP-positive (GFP+) or mCherry-positive (mCherry+) or a mixed population of both GFP+ and mCherry+ bacteria. We observed that the presence of C. trachomatis reduced the percentage of ubiquitin-positive C. muridarum inclusions from ~50% to ~10% (Fig. 6B) and also significantly reduced the percentage of p62-positive C. muridarum inclusions (Fig. 6C). While these data do not entirely dismiss the first model, they are more readily reconciled with the second model, in which a C. trachomatis-derived factor Y protects inclusions against host-mediated ubiquitination.
FIG 6 .

Fusion with C. trachomatis inclusions diminishes ubiquitination of C. muridarum-containing inclusions. (A) Two competing models to account for the preferential ubiquitin (Ub) targeting of C. muridarum (CM) versus C. trachomatis (CT) inclusions are depicted. N, nucleus. (B and C) A549 cells were coinfected with GFP+ C. muridarum (C.m.) (MOI of 3) and mCherry+ C. trachomatis (C.t.) (MOI of 5), primed with IFN-γ at 3 hpi, and fixed and stained with Hoechst and either antiubiquitin (FK2) (B) or anti-p62 (C) antibodies at 20 hpi. (Right) Representative images are shown. White arrowheads indicate ubiquitin- or p62-positive inclusions exclusively occupied by GFP+ C. muridarum. White arrows indicate inclusions containing a mixed population of GFP+ C. muridarum and mCherry+ C. trachomatis. (Left) For quantification, inclusions were categorized into three groups based on the type of bacteria they contained: GFP+ only (C.m. only), mCherry+ only (C.t. only), and GFP+ mCherry+ (C.m. + C.t.). Three independently infected wells were analyzed per data point and per well at least 30 inclusions of each category were scored for colocalization with ubiquitin or p62. Data are represented as means ± SD. Values that are significantly different by one-way ANOVA are indicated by a bar and asterisks as follows: *, P < 0.05; ***, P < 0.005; ****, P < 0.001.
IFN-γ priming activates a human host defense pathway that is effective against C. muridarum, but not C. trachomatis.
Our observations hitherto demonstrated that human epithelial cells mark C. muridarum inclusions with ubiquitin and a set of additional host defense proteins that include p62, LC3, and GBP1. We further showed that this process was significantly augmented by IFN-γ priming and that C. trachomatis can interfere with the recruitment of these antimicrobial host factors to inclusions. These observations thus led to the hypothesis that IFN-γ-primed human cells executed a cell-autonomous defense program that is effective against C. muridarum but ineffective against C. trachomatis. To test this hypothesis, we infected naive or IFN-γ-primed A549 cells with either C. muridarum or C. trachomatis and assessed bacterial burden by measuring infectious Chlamydia progeny (Fig. 7A) or by quantitative PCR (qPCR) quantification of Chlamydia DNA content (Fig. 7B). In agreement with previous reports (15, 16), we observed a greater than 1-log-unit reduction in chlamydial burden in IFN-γ-primed cells compared to naive cells (Fig. 7A and B). Because growth of C. muridarum and C. trachomatis was restricted equally in IFN-γ-primed A549 cells, we considered that the putative ubiquitin-dependent host defense pathway directed specifically against C. muridarum was masked by the IDO response.
FIG 7 .

IFN-γ promotes the clearance of C. muridarum from human A549 cells in an IDO-independent manner. A549 cells were primed overnight with IFN-γ (100 U/ml) or left unprimed. Cells were infected with C. muridarum or C. trachomatis at an MOI of 1 in the presence (+) or absence (−) of excess tryptophan (Trp) (100 µg/ml). (A) The number of IFUs per microliter were measured from each culture condition at 27 hpi. (B) Bacterial burden was assessed by qPCR at 27 hpi. Values that were significantly different by two-tailed unpaired Student’s t test are indicated by a bar and asterisks as follows: ***, P < 0.005; ****, P < 0.001. Values that were not significantly different (n.s.) by two-tailed unpaired Student’s t test are also indicated. (C and D) Cells were infected as described above, fixed at 27 hpi, and stained with anti-LPS (green) and Hoechst (blue). ImageJ software was used to measure inclusion size (area in square micrometer in panel C) and inclusion numbers per randomly acquired image field (D). Representative images are shown in panel C (bars = 10 μm). Data are representative of two independent experiments. Values are means ± SD. Values that are significantly different by one-way ANOVA are indicated by a bar and asterisks as follows: ***, P < 0.005; ****, P < 0.001. Values that were not significantly different (n.s.) by one-way ANOVA are also indicated.
IFN-γ priming of human epithelial cells induces the expression of IDO, an enzyme that depletes intracellular tryptophan stores and thereby restricts growth of the tryptophan auxotroph Chlamydia. Supplying excess tryptophan to the culture media is known to reverse the anti-Chlamydia effect of IDO (14–16). In agreement with these previous observations, we observed complete restoration of C. trachomatis burden in IFN-γ-primed A549 cells cultured in tryptophan-enriched media (Fig. 7A and B). However, tryptophan addition had only a limited effect on the number of inclusion-forming units (IFUs) generated by C. muridarum inside IFN-γ-primed A549 cells (Fig. 7A) and had no impact on C. muridarum burden as measured by qPCR (Fig. 7B). These data therefore demonstrated that IFN-γ-primed A549 cells repress growth of C. muridarum, but not C. trachomatis, independent of IDO-mediated tryptophan depletion.
We next asked whether this IDO-independent anti-Chlamydia defense pathway was bactericidal or bacteriostatic. IDO-mediated cell-autonomous immunity against Chlamydia acts in a bacteriostatic way, halting bacterial growth and thereby causing C. trachomatis inclusions to be smaller in size (Fig. 7C), while only minimally affecting the total number of inclusions under tryptophan-limiting culture conditions (Fig. 7D). In contrast to C. trachomatis infections, we found that the number of C. muridarum inclusions in IFN-γ-primed A549 cells was reduced by about 1 log unit, even under tryptophan-replete culture conditions (Fig. 7D). These data indicated that IFN-γ-primed A549 cells eliminated most C. muridarum inclusions and that the immune pathway described herein is independent of IDO and bactericidal in nature.
DISCUSSION
The human pathogen C. trachomatis is the most common cause of sexually transmitted bacterial infections. No vaccine is currently available, and recurring, repeat, or chronic infections are frequent. The inefficacy of our immune system to clear C. trachomatis infections and its deficiency in establishing protective immune memory demonstrate that C. trachomatis can undermine human immunity (2–4, 8). However, how C. trachomatis escapes clearance by both innate and adaptive immune responses of its human host is poorly understood. Here, we describe IFN-γ-inducible ubiquitination of inclusions and the associated eradication of intracellular bacteria in human cells as a novel anti-Chlamydia host defense pathway and further demonstrate that C. trachomatis is resistant to this newly described human immune response.
IFN-γ-primed human epithelial cells express high levels of the tryptophan-degrading enzyme IDO. Tryptophan depletion restricts growth of tryptophan-auxotrophic Chlamydia species and induces Chlamydia to undergo dramatic physiological and morphological changes (11, 17, 37, 38). In response to tryptophan starvation, C. trachomatis upregulates a partial trp operon, which allows C. trachomatis to consume exogenous indole and thereby produce tryptophan needed for survival in a tryptophan-depleted environment (11, 18). The fine-tuned interaction between host IDO and the C. trachomatis trp operon is absent from the commonly used animal model, in which mice are infected with the C. trachomatis-related pathogen C. muridarum. C. muridarum lacks a trp operon, and mouse genital epithelial cells express little to no IDO (5, 7, 15, 22). The relationship between the C. trachomatis trp operon and the human IDO response thus beautifully illustrates how C. trachomatis is specifically adapted to its human host. Our current study shows that IFN-γ-primed human epithelial cells not only express IDO as part of their anti-Chlamydia defense program but also execute a second resistance pathway to which C. trachomatis evolved an immune evasion strategy.
While IDO induction robustly restricts chlamydial growth, previous examinations of an array of human cell lines suggested that additional, uncharacterized IFN-γ-inducible anti-Chlamydia pathways may exist (15). Our study reports that IFN-γ priming triggers the deposition of ubiquitin on inclusions in human cells. The coating of intracellular pathogens with ubiquitin is a conserved defense mechanism found in host organisms as diverse as fruit flies and humans (39, 40). We recently demonstrated that IFN-γ priming of mouse cells leads to the ubiquitination of C. trachomatis inclusions (25). While the ubiquitination of C. trachomatis inclusions in mouse cells requires murine IRGM1 and IRGM3 proteins (25), we demonstrate here that ubiquitination of C. muridarum inclusions in human cells is independent of human IRGM (Fig. 2). These results reveal that mouse and human cells evolved separate mechanisms to encapsulate inclusions within a layer of ubiquitin (Fig. 8). Because the host machinery for inclusion ubiquitination is different in mice and humans, the corresponding microbial evasion mechanisms must be adapted to the host species. This argument is supported by our findings. We previously demonstrated that C. muridarum evades cell-autonomous immunity in mice through the inactivation of the murine IRG system (25, 28), a strategy that is ineffective in human cells. C. trachomatis on the other hand fails to inactivate the murine IRG systems, rendering it susceptible to ubiquitination in mouse cells (25), but it has the ability to escape from the IRG-independent ubiquitination response of human cells (Fig. 8). To understand how different Chlamydia species escape from inclusion ubiquitination in their respective hosts, we will need to identify the chlamydial factors that enable evasion of inclusion ubiquitination. Forward genetic screens using recently developed Chlamydia mutant libraries (41–43) provide one possible avenue to achieve this goal.
FIG 8 .

Distinct pathways in mice and humans control IFN-γ-inducible ubiquitination of inclusions. Inclusion ubiquitination in mouse cells is dependent on IFN-γ-inducible IRGs, which in their GTP-bound state form oligomers, bind to inclusions, and subsequently recruit E3 ubiquitin ligases such as TRAF6. By interfering with the recruitment of IRGs to its inclusion, the rodent pathogen C. muridarum (CM) blocks inclusion ubiquitination in mouse cells. Ubiquitinated inclusions undergo vacuolar lysis, leading to bacterial death (25). This current study demonstrates that IFN-γ priming also triggers inclusion ubiquitination in human cells, albeit by an IRG-independent mechanism. Whereas C. muridarum is susceptible to this IRG-independent pathway, the human pathogen C. trachomatis (CT) is resistant. Inclusion ubiquitination in human cells correlates with the elimination of inclusions from infected cells, but the underlying cellular mechanism is unknown. Ub, ubiquitin; UBP, ubiquitin-binding proteins; E1, ubiquitin-activating enzyme; E2, ubiquitin-conjugating enzyme.
A second area of future research will be focused on the identification and characterization of human factors that mediate inclusion ubiquitination and the cellular events that follow. Recent reports demonstrated that IFN-γ priming of human cells promote the ubiquitination of parasitophorous vacuoles (PVs) formed by the protozoan pathogen Toxoplasma gondii (44, 45), albeit by an unknown mechanism. IFN-γ-inducible cell-autonomous immune responses directed against Chlamydia and Toxoplasma have so far been found to be remarkably similar, as both types of pathogens are susceptible to the murine IRG and human IDO responses (46). On the basis of these precedents, it is tempting to speculate that the same molecular machinery drives the ubiquitination of both Toxoplasma PVs and inclusions in human cells, a hypothesis that will await future testing. Similar to our findings with C. muridarum, Selleck and colleagues showed that ubiquitinated Toxoplasma PVs corecruit ubiquitin-binding proteins and the autophagy marker LC3 (45). The same study demonstrated that parasites inside ubiquitin-positive PVs are moderately impeded for growth. However, in agreement with other work (47, 48), the IDO-independent effects of IFN-γ priming on overall parasitic burden remain negligible. In contrast to the moderate effects on Toxoplasma burden, we find the IFN-γ-inducible, IDO-independent anti-Chlamydia response to reduce bacterial burden by approximately 10-fold. This dramatic reduction in burden correlates with a 10-fold decrease in the number of inclusions, suggesting that IFN-γ-primed human cells destroy and remove inclusions, similar to what was observed in mouse cells (25). The precise mechanism by which the human host eliminates inclusions is currently unknown. Similar to the murine defense mechanism (25), inclusions inside IFN-γ-primed human epithelial cells may lyse and thereby release bacteria into the host cell cytosol for xenophagic destruction. Alternatively, inclusions may undergo acidification, as reported previously for inclusions formed inside IFN-γ-primed human macrophages (49). The study of Al-Zeer et al. (49) reported that hGBP1 and hGBP2 were required for C. trachomatis inclusion acidification in human macrophages, suggesting that C. trachomatis fails to counteract cell-autonomous host defense in human macrophages as it does in human epithelial cells. Future studies will have to determine whether inclusion acidification constitutes an effector pathway downstream of inclusion ubiquitination, or alternatively, a ubiquitination-independent pathway unique to macrophages.
In summary, our study describes a novel cell-autonomous immune response targeting Chlamydia inside human epithelial cells. The activation of this ubiquitin-based immune response is distinct from a similar ubiquitination pathway that we previously described in mouse cells (25). Therefore, mice and humans appear to have evolved distinct but functionally convergent systems to battle infections with intracellular Chlamydia. We further demonstrate that C. trachomatis is resistant to inclusion ubiquitination in human epithelial cells. As we will define mechanisms by which C. trachomatis can block the human-specific immune responses, it is hoped that our research will pave the way toward novel treatment options and improved vaccine strategies to minimize the impact of C. trachomatis infections on human health.
MATERIALS AND METHODS
Chlamydia strains, bacterial plasmids, and transformation.
C. trachomatis LGV-L2 and C. muridarum Nigg were propagated in Vero cells as described previously (29). C. trachomatis LGV-L2 was transformed with plasmid pGFP::SW2 or p2TK2-SW2-mCherry, and C. muridarum Nigg was transformed with plasmid pGFP::CM (50–52). Transformations were performed essentially as described previously (50). Briefly, purified elementary bodies (EBs) and plasmid were mixed in 200 µl CaCl2 buffer (10 mM Tris [pH 7.4] and 50 mM CaCl2) and incubated for 30 min at room temperature. One hundred microliters of this mixture was then added to a single well in a six-well plate together with 2 ml of Dulbecco’s modified Eagle’s medium (DMEM) plus 10% fetal calf serum (FCS). At 12 h postinfection (hpi), the medium was replaced with medium containing 1 µg/ml ampicillin. Passage 0 infections were harvested at 30 hpi and immediately used to infect a new six-well plate of McCoy cells. This process was repeated two more times, and at passage 4, fluorescent Chlamydia organisms were observed, plaque cloned twice, and expanded for future experiments. The presence of plasmid was confirmed by PCR.
Host cell culture, Chlamydia infections, and measurements of bacterial burden.
HeLa and A549 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 5% heat-inactivated fetal bovine serum (FBS). Infections with C. trachomatis and C. muridarum were performed at a nominal multiplicity of infection (MOI) of ~2 followed by treatment with 100 U/ml of IFN-γ at 3 hpi or as indicated, essentially as described previously (29). For coinfection experiments, A549 cells were infected with mCherry-expressing C. trachomatis and GFP-expressing C. muridarum at a MOI ratio of 5:3, followed by treatment with 100 U/ml of IFN-γ at 3 hpi. Burden was assessed by qPCR or inclusion-forming unit (IFU) assays. To assess chlamydial DNA content, total nucleic acid was prepared from trypsinized cell pellets using the QIAamp DNA minikit from Qiagen (Valencia, CA, USA). Samples were then subjected to SYBR green qPCR on an ABI 7000 sequence detection system to assess the amount of 16S Chlamydia and B2M host DNA in the sample. Chlamydia 16S DNA was detected through use of the following primers as described previously (20): 16S forward primer (5′ GGA GGC TGC AGT CGA GAA TCT 3′) and 16S reverse primer (5′ TTA CAA CCC TAG AGC CTT CAT CAC A 3′). Human B2M DNA was detected using the following primer sequences as described previously (53): B2M forward primer (5′ TGC TGT CTC CAT GTT TGA TGT ATC T 3′) and B2M reverse primer (5′ TCT CTG CTC CCC ACC TCT AAG T 3′). Standard curves were generated in parallel from known amounts of C. trachomatis and human DNA, and these curves were used to calculate the mass (in picograms) of Chlamydia DNA per unit mass (in micrograms) of human DNA per sample. IFU assays were performed essentially as described previously (20). Briefly, wild-type (WT) A549 and HeLa cells were primed or left unprimed overnight and infected with C. muridarum or C. trachomatis in 24-well plates. At 27 hpi, infected monolayers were lysed with water and physical dislodgement and suspended into 0.25 ml SPG buffer (220 mM sucrose, 12.5 mM phosphate, 4 mM l-glutamic acid, pH 7.5). Suspensions were transferred to sterile Eppendorf tubes and then sonicated. Samples were stored at −80°C. For quantification, samples were serially diluted, and dilutions were used to infect confluent monolayers of Vero cells in triplicate. The next day, samples were fixed with methanol and stained with mouse monoclonal anti-Chlamydia lipopolysaccharide (LPS), and host cell nuclei were labeled with Hoechst stain. Inclusions were visualized by using a microscope and counted in 10 fields per well for the calculation of infectious titer.
Immunocytochemistry.
Immunocytochemistry was performed essentially as described previously (29). The cells were washed three times with phosphate-buffered saline (PBS) (pH 7.4) prior to fixation. The cells were fixed either with methanol or with 4% paraformaldehyde (PFA) (wt/vol) for 20 min at room temperature (RT). In all experiments involving PFA fixation, fixed cells were permeabilized/blocked with 0.05% (wt/vol) saponin and 2% (wt/vol) bovine serum albumin (BSA) in PBS (SBP) for 30 min at RT. The cells were stained with the indicated primary antibodies: rabbit polyclonal anti-p62/SQSTM1 (PMO45; MBL International) at 1:500, mouse monoclonal anti-p62/SQSTM1 (Abnova) at 1:500, mouse antiubiquitin (FK2; ENZO) at 1:50, rabbit monoclonal antiubiquitin K48 (Millipore) at 1:500, rabbit monoclonal anti-K63 (Millipore) at 1:100, rabbit polyclonal anti-NDP52 (Calcoco2; Abnova) at 1:200, mouse monoclonal anti-chlamydial LPS (catalog no. 1681; Santa Cruz) at 1:50, rabbit anti-Slc1 (Ct043) at 1:100, rabbit monoclonal anti-linear ubiquitin clone 1E3 (Millipore) at 1:50, and rabbit polyclonal anti-LC3 (PMO36; MBL International) at 1:500. The cells were then stained with Alexa Fluor-conjugated secondary antibodies (Molecular Probes/Invitrogen). Nucleic DNA and bacterial DNA were stained with Hoechst 33258 according to the manufacturer’s protocol. Stained cells were washed with PBS, mounted on microscope slides with Mowiol (Sigma), and allowed to cure overnight. To monitor the subcellular localization of human GBP1, cells were transfected with a previously described mCherry-hGBP1 expression construct (54). Cells were imaged using either a Zeiss LSM 510 inverted confocal microscope or a Zeiss Axioskop 2 upright epifluorescence microscope. Colocalization of proteins with inclusions was quantified in at least three independent experiments. In each experiment, at least 10 randomly selected fields were imaged for each experimental condition and cell type. To determine the frequency with which ubiquitin (FK2, M1, K48, and K63), p62, NDP52, and LC3 proteins colocalize with inclusions, at least 100 inclusions were assessed for each experimental condition and cell type for every biological replicate. The fraction of ubiquitin-, p62-, NDP52- or GBP1-positive vacuoles was determined for each field by dividing the number of labeled vacuoles by the total number of vacuoles. To determine the number of inclusions per field, eight randomly selected microscopic fields were counted per condition. To determine inclusion size, inclusions in eight randomly selected fields were examined per condition, and area in square micrometers was calculated using ImageJ software.
CRISPR gene deletion cell lines.
In order to make IRGM loss-of-function mutations, we utilized clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 technology with a pair of guide RNAs (gRNAs) that target IRGM near the 5′ and 3′ ends of the IRGM open reading frame (ORF). gRNAs were cloned into the human codon-optimized Streptococcus pyogenes (hSpCas9)-encoding vector pX330 (55), using BbsI restriction endonuclease in conjunction with preannealed or T4 polynucleotide kinase phosphorylated oligonucleotides containing compatible ends. The resulting constructs, pX330-IRGM guide 1 (target, 5′-ATCAGTGCCCTTCGAAACAC-3′) and pX330-IRGM guide 2 (target, 5′-GATGCTTGCCAAAACCGCTG-3′) were predicted to anneal at IRGM at positions +148 to 167 and positions +384 to 403, respectively. These constructs were cotransfected into A549 and HeLa cells using Lipofectamine LTX to introduce deletions of approximately 230 bp within the IRGM coding region. This deleted region encompasses a large portion of the predicted IRGM GTPase domain, including the G2 box/Switch I region and the G3 box/Switch II region (27, 32). Individual clones were isolated from the heterogeneous transfected population through serial dilution in 96-well plates. Wells containing single colonies (i.e., isolated clones) were identified by light microscopy. Genomic DNA was isolated from clonal populations using a DNeasy blood and tissue kit (Qiagen), and clones were screened for IRGM deletions through PCR with primers that flank the IRGM open reading frame. The screening primers used are listed as IRGM-Mutant-Screen-F (F stands for forward) (5′-GCCTCAGCCTCCTGTATTAGCTGG-3′) and IRGM-Mutant-Screen-R (R stands for reverse) (5′-GACAGGAATTAGTATTCACATAC-3′). A GBP1-deficient A549 cell line was previously reported (54).
Statistical analysis.
Where designated, statistical significance was determined using the unpaired Student’s t test or two-way analysis of variance (ANOVA), as appropriate. Values of P < 0.05 were considered significant. Results are represented as means ± standard deviations (SD).
ACKNOWLEDGMENTS
We thank Raphael Valdivia for sharing Chlamydia strains and anti-Slc1 antibody.
A.K.H. and J.C. designed research and analyzed data with feedback from all authors. A.K.H., A.S.P., R.F., S.T.E., and H.E.B. generated reagents and executed experiments. A.M.G., E.-M.F., and D.E.N. provided reagents. A.K.H. and J.C. wrote the paper with input from all authors. J.C. supervised the project.
Funding Statement
This work was supported by a National Science Foundation predoctoral award (to RF) and by National Institute Health grants R01AI099278 (to DEN) and R01AI103197 (to JC). EMF is supported by the Wellcome Trust (091664/B/10/Z) and the Francis Crick Institute (FC001076). JC holds an Investigator in the Pathogenesis of Infectious Disease Awards from the Burroughs Wellcome Fund. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Haldar AK, Piro AS, Finethy R, Espenschied ST, Brown HE, Giebel AM, Frickel E-M, Nelson DE, Coers J. 2016. Chlamydia trachomatis is resistant to inclusion ubiquitination and associated host defense in gamma interferon-primed human epithelial cells. mBio 7(6):e01417-16. doi:10.1128/mBio.01417-16.
REFERENCES
- 1.World Health Organization 2011. Global prevalence and incidence of selected curable sexually transmitted diseases: overview and estimates. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Redgrove KA, McLaughlin EA. 2014. The role of the immune response in Chlamydia trachomatis infection of the male genital tract: a double-edged sword. Front Immunol 5:534. doi: 10.3389/fimmu.2014.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darville T, Hiltke TJ. 2010. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis 201(Suppl 2):S114–S125. doi: 10.1086/652397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hafner LM. 2015. Pathogenesis of fallopian tube damage caused by Chlamydia trachomatis infections. Contraception 92:108–115. doi: 10.1016/j.contraception.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Abdelsamed H, Peters J, Byrne GI. 2013. Genetic variation in Chlamydia trachomatis and their hosts: impact on disease severity and tissue tropism. Future Microbiol 8:1129–1146. doi: 10.2217/fmb.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aiyar A, Quayle AJ, Buckner LR, Sherchand SP, Chang TL, Zea AH, Martin DH, Belland RJ. 2014. Influence of the tryptophan-indole-IFNgamma axis on human genital Chlamydia trachomatis infection: role of vaginal co-infections. Front Cell Infect Microbiol 4:72. doi: 10.3389/fcimb.2014.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coers J, Starnbach MN, Howard JC. 2009. Modeling infectious disease in mice: co-adaptation and the role of host-specific IFNgamma responses. PLoS Pathog 5:e1000333. doi: 10.1371/journal.ppat.1000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finethy R, Coers J 29 July 2016. Sensing the enemy, containing the threat: cell-autonomous immunity to Chlamydia trachomatis. FEMS Microbiol Rev doi: 10.1093/femsre/fuw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdelrahman YM, Belland RJ. 2005. The chlamydial developmental cycle. FEMS Microbiol Rev 29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wyrick PB. 2010. Chlamydia trachomatis persistence in vitro: an overview. J Infect Dis 201(Suppl 2):S88–S95. doi: 10.1086/652394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Randow F, MacMicking JD, James LC. 2013. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science 340:701–706. doi: 10.1126/science.1233028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beatty WL, Belanger TA, Desai AA, Morrison RP, Byrne GI. 1994. Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect Immun 62:3705–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roshick C, Wood H, Caldwell HD, McClarty G. 2006. Comparison of gamma interferon-mediated antichlamydial defense mechanisms in human and mouse cells. Infect Immun 74:225–238. doi: 10.1128/IAI.74.1.225-238.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrne GI, Lehmann LK, Landry GJ. 1986. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun 53:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beatty WL, Byrne GI, Morrison RP. 1993. Morphologic and antigenic characterization of interferon gamma-mediated persistent Chlamydia trachomatis infection in vitro. Proc Natl Acad Sci U S A 90:3998–4002. doi: 10.1073/pnas.90.9.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caldwell HD, Wood H, Crane D, Bailey R, Jones RB, Mabey D, Maclean I, Mohammed Z, Peeling R, Roshick C, Schachter J, Solomon AW, Stamm WE, Suchland RJ, Taylor L, West SK, Quinn TC, Belland RJ, McClarty G. 2003. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest 111:1757–1769. doi: 10.1172/JCI17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perry LL, Su H, Feilzer K, Messer R, Hughes S, Whitmire W, Caldwell HD. 1999. Differential sensitivity of distinct Chlamydia trachomatis isolates to IFN-gamma-mediated inhibition. J Immunol 162:3541–3548. [PubMed] [Google Scholar]
- 20.Bernstein-Hanley I, Balsara ZR, Ulmer W, Coers J, Starnbach MN, Dietrich WF. 2006. Genetic analysis of susceptibility to Chlamydia trachomatis in mouse. Genes Immun 7:122–129. doi: 10.1038/sj.gene.6364285. [DOI] [PubMed] [Google Scholar]
- 21.Coers J, Gondek DC, Olive AJ, Rohlfing A, Taylor GA, Starnbach MN. 2011. Compensatory T cell responses in IRG-deficient mice prevent sustained Chlamydia trachomatis infections. PLoS Pathog 7:e1001346. doi: 10.1371/journal.ppat.1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson DE, Virok DP, Wood H, Roshick C, Johnson RM, Whitmire WM, Crane DD, Steele-Mortimer O, Kari L, McClarty G, Caldwell HD. 2005. Chlamydial IFN-gamma immune evasion is linked to host infection tropism. Proc Natl Acad Sci U S A 102:10658–10663. doi: 10.1073/pnas.0504198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein-Hanley I, Coers J, Balsara ZR, Taylor GA, Starnbach MN, Dietrich WF. 2006. The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc Natl Acad Sci U S A 103:14092–14097. doi: 10.1073/pnas.0603338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pilla-Moffett D, Barber MF, Taylor GA, Coers J. 2016. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol 428:3495–3513. doi: 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haldar AK, Foltz C, Finethy R, Piro AS, Feeley EM, Pilla-Moffett DM, Komatsu M, Frickel EM, Coers J. 2015. Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc Natl Acad Sci U S A 112:E5628–E5637. doi: 10.1073/pnas.1515966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Zeer MA, Al-Younes HM, Braun PR, Zerrahn J, Meyer TF. 2009. IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One 4:e4588. doi: 10.1371/journal.pone.0004588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, Glowalla E, Leptin M, Howard JC. 2005. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol 6:R92. doi: 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coers J, Bernstein-Hanley I, Grotsky D, Parvanova I, Howard JC, Taylor GA, Dietrich WF, Starnbach MN. 2008. Chlamydia muridarum evades growth restriction by the IFN-gamma-inducible host resistance factor Irgb10. J Immunol 180:6237–6245. doi: 10.4049/jimmunol.180.9.6237. [DOI] [PubMed] [Google Scholar]
- 29.Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, Frickel EM, Valdivia RH, Coers J. 2013. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog 9:e1003414. doi: 10.1371/journal.ppat.1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, Kaiser F, Zerrahn J, Martens S, Howard JC. 2008. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J 27:2495–2509. doi: 10.1038/emboj.2008.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traver MK, Henry SC, Cantillana V, Oliver T, Hunn JP, Howard JC, Beer S, Pfeffer K, Coers J, Taylor GA. 2011. Immunity-related GTPase M (IRGM) proteins influence the localization of guanylate-binding protein 2 (GBP2) by modulating macroautophagy. J Biol Chem 286:30471–30480. doi: 10.1074/jbc.M111.251967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bekpen C, Marques-Bonet T, Alkan C, Antonacci F, Leogrande MB, Ventura M, Kidd JM, Siswara P, Howard JC, Eichler EE. 2009. Death and resurrection of the human IRGM gene. PLoS Genet 5:e1000403. doi: 10.1371/journal.pgen.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujimuro M, Yokosawa H. 2005. Production of antipolyubiquitin monoclonal antibodies and their use for characterization and isolation of polyubiquitinated proteins. Methods Enzymol 399:75–86. doi: 10.1016/S0076-6879(05)99006-X. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto ML, Dong KC, Yu C, Phu L, Gao X, Hannoush RN, Hymowitz SG, Kirkpatrick DS, Dixit VM, Kelley RF. 2012. Engineering and structural characterization of a linear polyubiquitin-specific antibody. J Mol Biol 418:134–144. doi: 10.1016/j.jmb.2011.12.053. [DOI] [PubMed] [Google Scholar]
- 35.Rieser E, Cordier SM, Walczak H. 2013. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem Sci 38:94–102. doi: 10.1016/j.tibs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 36.Cemma M, Kim PK, Brumell JH. 2011. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy 7:341–345. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKenzie CR, Heseler K, Müller A, Däubener W. 2007. Role of indoleamine 2,3-dioxygenase in antimicrobial defence and immuno-regulation: tryptophan depletion versus production of toxic kynurenines. Curr Drug Metab 8:237–244. doi: 10.2174/138920007780362518. [DOI] [PubMed] [Google Scholar]
- 38.Kane CD, Vena RM, Ouellette SP, Byrne GI. 1999. Intracellular tryptophan pool sizes may account for differences in gamma interferon-mediated inhibition and persistence of chlamydial growth in polarized and nonpolarized cells. Infect Immun 67:1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyle KB, Randow F. 2013. The role of “eat-me” signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol 16:339–348. doi: 10.1016/j.mib.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. 2013. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kokes M, Dunn JD, Granek JA, Nguyen BD, Barker JR, Valdivia RH, Bastidas RJ. 2015. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17:716–725. doi: 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brothwell JA, Muramatsu MK, Toh E, Rockey DD, Putman TE, Barta ML, Hefty PS, Suchland RJ, Nelson DE. 2016. Interrogating genes that mediate Chlamydia trachomatis survival in cell culture using conditional mutants and recombination. J Bacteriol 198:2131–2139. doi: 10.1128/JB.00161-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajaram K, Giebel AM, Toh E, Hu S, Newman JH, Morrison SG, Kari L, Morrison RP, Nelson DE. 2015. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect Immun 83:2870–2881. doi: 10.1128/IAI.00106-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coers J, Haldar AK. 2015. Ubiquitination of pathogen-containing vacuoles promotes host defense to Chlamydia trachomatis and Toxoplasma gondii. Communicat Integr Biol 8:e1115163. doi: 10.1080/19420889.2015.1115163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selleck EM, Orchard RC, Lassen KG, Beatty WL, Xavier RJ, Levine B, Virgin HW, Sibley LD. 2015. A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain-specific manner in IFN-gamma-activated human cells. mBio 6:e01157-15. doi: 10.1128/mBio.01157-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howard JC, Hunn JP, Steinfeldt T. 2011. The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr Opin Microbiol 14:414–421. doi: 10.1016/j.mib.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 47.Ohshima J, Lee Y, Sasai M, Saitoh T, Su Ma J, Kamiyama N, Matsuura Y, Pann-Ghill S, Hayashi M, Ebisu S, Takeda K, Akira S, Yamamoto M. 2014. Role of mouse and human autophagy proteins in IFN-gamma-induced cell-autonomous responses against Toxoplasma gondii. J Immunol 192:3328–3335. doi: 10.4049/jimmunol.1302822. [DOI] [PubMed] [Google Scholar]
- 48.Pfefferkorn ER. 1984. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc Natl Acad Sci U S A 81:908–912. doi: 10.1073/pnas.81.3.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Zeer MA, Al-Younes HM, Lauster D, Abu Lubad M, Meyer TF. 2013. Autophagy restricts Chlamydia trachomatis growth in human macrophages via IFNG-inducible guanylate binding proteins. Autophagy 9:50–62. doi: 10.4161/auto.22482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agaisse H, Derré I. 2013. A C. trachomatis cloning vector and the generation of C. trachomatis strains expressing fluorescent proteins under the control of a C. trachomatis promoter. PLoS One 8:e57090. doi: 10.1371/journal.pone.0057090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Chen C, Gong S, Hou S, Qi M, Liu Q, Baseman J, Zhong G. 2014. Transformation of Chlamydia muridarum reveals a role for Pgp5 in suppression of plasmid-dependent gene expression. J Bacteriol 196:989–998. doi: 10.1128/JB.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toegel S, Huang W, Piana C, Unger FM, Wirth M, Goldring MB, Gabor F, Viernstein H. 2007. Selection of reliable reference genes for qPCR studies on chondroprotective action. BMC Mol Biol 8:13. doi: 10.1186/1471-2199-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnston AC, Piro A, Clough B, Siew M, Virreira Winter S, Coers J, Frickel EM. 2016. Human GBP1 does not localize to pathogen vacuoles but restricts Toxoplasma gondii. Cell Microbiol 18:1056–1064. doi: 10.1111/cmi.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]