

Gigaxonin is an E3 ubiquitin ligase adaptor that facilitates proteasomal degradation of GFAP in astrocytes. Gigaxonin is similarly involved in the degradation of some but not all disease-linked forms of GFAP. This degradation is mediated by a direct interaction of gigaxonin with GFAP and involves the proteasome.
Abstract
Alexander disease (AxD) is a primary genetic disorder of astrocytes caused by dominant mutations in the gene encoding the intermediate filament (IF) protein GFAP. This disease is characterized by excessive accumulation of GFAP, known as Rosenthal fibers, within astrocytes. Abnormal GFAP aggregation also occurs in giant axon neuropathy (GAN), which is caused by recessive mutations in the gene encoding gigaxonin. Given that one of the functions of gigaxonin is to facilitate proteasomal degradation of several IF proteins, we sought to determine whether gigaxonin is involved in the degradation of GFAP. Using a lentiviral transduction system, we demonstrated that gigaxonin levels influence the degradation of GFAP in primary astrocytes and in cell lines that express this IF protein. Gigaxonin was similarly involved in the degradation of some but not all AxD-associated GFAP mutants. In addition, gigaxonin directly bound to GFAP, and inhibition of proteasome reversed the clearance of GFAP in cells achieved by overexpressing gigaxonin. These studies identify gigaxonin as an important factor that targets GFAP for degradation through the proteasome pathway. Our findings provide a critical foundation for future studies aimed at reducing or reversing pathological accumulation of GFAP as a potential therapeutic strategy for AxD and related diseases.
INTRODUCTION
Intermediate filaments (IFs) are versatile cytoskeletal scaffolds that maintain mechanical strength and shape of the cell and provide dynamic platforms for the organization of the cytoplasm on a structural and functional level (Kim and Coulombe, 2007). Pathological inclusions composed of IF proteins are a common feature of neurodegenerative diseases, including neuronal IF aggregates in the neurons of patients with amyotrophic lateral sclerosis (Blokhuis et al., 2013), Alzheimer’s disease (Rudrabhatla et al., 2011), Parkinson’s disease, and dementia with Lewy bodies (Wakabayashi et al., 2013) and Rosenthal fibers (RFs) in the astrocytes of patients with Alexander disease (AxD; Alexander, 1949) and giant axonal neuropathy (GAN; Asbury et al., 1972; Berg et al., 1972; Pena, 1981). Although numerous mutant genes have been identified that cause these diseases, few have provided insights into the mechanisms responsible for IF aggregate formation. In none of these diseases is it understood what prompts aggregate formation in the first place. Nor is it even known whether it is the formation of the aggregate that compromises cell function or whether the aggregate formation is a protective detoxification mechanism of the cell (Ross and Poirier, 2005).
With respect to these IF aggregation diseases, AxD is unique in that it is a neurological disorder initiated by dysfunction in nonneuronal cells. The consistent pathological hallmark of AxD is the formation of cytoplasmic aggregates known as RFs that accumulate in the cell body and processes of astrocytes (Messing et al., 2012b). As with other IF aggregation disorders, the question of whether RFs per se cause astrocyte dysfunction and the precise trigger(s) for their formation are not clear. Using animal models, it has been shown that transgenic mice engineered to constitutively overexpress human wild-type glial fibrillary acidic protein (GFAP) in astrocytes developed RFs and died at an early age (Messing et al., 1998). In addition, AxD knock-in mice expressing disease-linked forms of mutant GFAP also formed RFs and spontaneously increased their levels of GFAP (Hagemann et al., 2006). Thus the genetic evidence suggests a critical role of elevations in total GFAP levels in the disease pathogenesis. Indeed, increased GFAP levels were consistently found in AxD patients both when measured in brain parenchyma (Walker et al., 2014) and in cerebrospinal fluid (Jany et al., 2015). The accumulation of GFAP that is found in AxD results in part from increased mRNA synthesis (Hagemann et al., 2005), and there is a spontaneous increase in the activity of the GFAP promoter (Jany et al., 2013). Cellular models and in vitro studies have provided additional evidence to suggest that alterations occur in the degradation pathways, including the ubiquitin-proteasome system (Tang et al., 2006, 2010) and autophagy (Tang et al., 2008). Together the alterations in GFAP synthesis and degradation could lead to positive feedback loops that further contribute to GFAP accumulation.
Abnormal aggregation of GFAP also occurs in several non-AxD conditions, one of which is GAN (Kretzschmar et al., 1987). GAN is an early-onset neurological disorder caused by mutations in the GAN gene, which encodes gigaxonin, a member of the BTB/Kelch family of E3 ligase adaptor proteins (Bomont et al., 2000; Furukawa et al., 2003; Xu et al., 2003; Pintard et al., 2004; Perez-Torrado et al., 2006). This rare disease is characterized by an extensive aggregation of several types of IFs. Although a broad range of cell types is affected in GAN, neurons display the most severe pathology, with accumulation and aggregation of neurofilaments and peripherin in the CNS and peripheral nervous system, which in turn causes the focal axonal enlargements for which the disease is named. Within the CNS, RFs have been described in several GAN patients (Kretzschmar et al., 1987; Thomas et al., 1987). The AxD-like pathology observed in GAN suggests that loss of gigaxonin also affects GFAP, causing it to accumulate and aggregate in a manner similar to its effects on other IF proteins. Although the mechanism by which loss-of-function mutations in gigaxonin interfere with the IF system is not known, recent studies suggested a functional role of gigaxonin in regulating the degradation of several IF proteins through the ubiquitin-proteasome pathway (Mahammad et al., 2013; Opal and Goldman, 2013; Israeli et al., 2016). In support of this role, studies of Gan−/− mice revealed pathological features similar to those found in GAN patients (Dequen et al., 2008; Ganay et al., 2011). These include an increase in IF protein levels, loss of peripheral axons, and formation of aggregates in several cell types throughout the CNS. Taken together, these studies show gigaxonin to be fundamentally important in sustaining IF organization and regulating degradation of IF proteins.
Because the expression of mutant GFAP is the root cause of AxD, and accumulation of GFAP above a toxic threshold is believed to be an essential element in pathogenesis (Messing et al., 2012b), reducing the expression or accumulation of GFAP could be one potential strategy for treatment. Considering the degradation pathway as a therapeutic target, one possibility to reduce GFAP aggregation is to increase its degradation. Small molecules that activate or enhance proteasome activity are therefore a theoretical possibility for treating AxD, but these are rare and not well studied (Huang and Chen, 2009). Given that gigaxonin is an E3 ubiquitin ligase adaptor that targets substrate proteins for proteasomal degradation and that its overexpression clears several IF proteins, including vimentin, peripherin, and neurofilaments (Mahammad et al., 2013), we carried out experiments to test whether gigaxonin is similarly involved in the clearance of GFAP, a type III IF protein expressed mainly in astrocytes.
RESULTS
Gigaxonin expression causes clearance of GFAP in primary astrocytes
Given the previous observations that gigaxonin clears several IF proteins, including vimentin, as well as peripherin and neurofilament light chain (Mahammad et al., 2013; Israeli et al., 2016), we tested whether gigaxonin could clear GFAP, a type III IF protein expressed predominantly in astrocytes. Primary astrocytes were infected with a lentivirus expressing FLAG-gigaxonin. Cells were fixed at 72 h postinfection and visualized by double-label immunofluorescence microscopy. Under these conditions, ∼80.2% ± 4.2% of cells (n = 300) were infected with gigaxonin lentiviruses. We found that expression of gigaxonin caused a nearly complete clearance of the GFAP IFs in primary astrocytes (Figure 1A). Despite the disappearance of GFAP IFs by 72 h, the organization of microtubules (Figure 1D) and microfilaments (Figure 1G) appeared normal in gigaxonin-expressing cells. Immunoblotting revealed that gigaxonin expression resulted in a decrease in GFAP to a level ∼8% of controls, and no significant changes in the levels of tubulin and actin (Figure 2A) were detected in these cells. Together these results demonstrated that gigaxonin has a substantial effect on GFAP IFs in astrocytes but not on the other major cytoskeletal proteins, tubulin and actin.
FIGURE 1:

Expression of gigaxonin caused clearance of GFAP IFs. Primary astrocytes were infected with lentiviruses containing Flag-gigaxonin. At 72 h after infection, cells were fixed and processed for double-label immunofluorescence microscopy using a polyclonal anti-gigaxonin antibody (B, E, and H) in combination with either anti-GFAP (A) or anti–β-tubulin (D). To label actin-containing microfilaments, cells immunostained for gigaxonin were costained with fluorescein isothiocyanate–phalloidin (G). The immunofluorescence for gigaxonin was in the red channel (B, E, and H), whereas the costaining for each of the cytoskeletal elements was in the green channel (A, D, and G). Right, merged images (C, F, and I). Under these conditions, transduction efficiency of gigaxonin ranged between 76 and 84%, as assessed by visual assessment of gigaxonin-positive cells in a population of 200–300 cells. Representative images were selected from three independent preparations that showed a gigaxonin-transduced cell surrounded by nontransduced cells. Scale bar, 10 μm.
FIGURE 2:
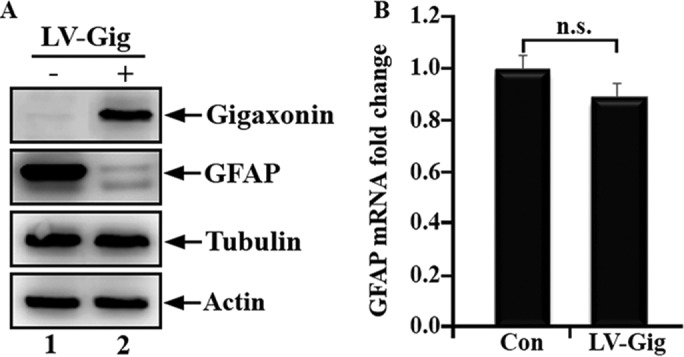
Clearance of GFAP was not associated with a corresponding decrease in GFAP mRNA level. Primary astrocytes were infected with lentiviruses containing either vector (A, lane 1) or gigaxonin (A, lane 2). At 72 h after infection, total cell lysates were prepared and analyzed by immunoblotting using antibodies specific to gigaxonin, GFAP, tubulin, and actin. GFAP levels decreased to 8.4 ± 4.1% compared with controls at 72 h postinfection. Representative blots were from three independent experiments. (B) RT-PCR was performed to determine GFAP mRNA levels in mock-infected (Con) and gigaxonin lentivirus-infected (LV-Gig) astrocytes. Levels of GFAP mRNA were normalized to the measurement of the housekeeping gene GAPDH, and fold increase relative to control was normalized to 1. Quantification results are shown as mean ± SD and presented as bar charts. Expression of gigaxonin (B, LV-Gig) had no significant effect on GFAP mRNA levels compared with mock-infected controls (B, Con).
Gigaxonin is predicted to be an E3 ligase adaptor, and its effect on the clearance of GFAP may involve the proteasomal degradation pathway. However, it is theoretically possible that the decrease in GFAP protein level is caused by a reduction in GFAP transcription. To test this hypothesis, we determined GFAP mRNA levels by quantitative PCR after expression of gigaxonin in primary astrocytes. As shown in Figure 2B, gigaxonin expression had no significant effect on GFAP mRNA levels compared with mock-infected control cells. These data provide further evidence in support of the role of gigaxonin in mediating the degradation of GFAP at the protein level.
Interaction of gigaxonin with GFAP
With respect to the localization of gigaxonin, immunofluorescence studies showed that it was evenly distributed throughout the cytoplasm when expressed in primary astrocytes (Figure 1, B, E, and H). We then extracted cells and assessed biochemically the solubility properties of gigaxonin. With use of a RIPA lysis buffer, we extracted gigaxonin into the soluble fraction from lentivirus-infected cells (Figure 3B, lane 1), conditions that also extracted GFAP (Figure 3A, lane 1). The presence of gigaxonin and GFAP in the soluble fractions raised the possibility that both were associated in a soluble complex. To investigate this hypothesis, we performed a series of coimmunoprecipitation experiments. The Flag-gigaxonin was transduced into primary astrocytes for 36 h, a time point at which GFAP had not been completely cleared by gigaxonin. The supernatant fractions prepared from lentivirus-infected cells were subjected to immunoprecipitation using monoclonal anti-Flag (Figure 3A, lane 3) and monoclonal anti-GFAP (Figure 3B, lane 1) antibodies. Immunoblotting analysis of the immunoprecipitates revealed that gigaxonin can be coprecipitated by anti-GFAP antibody (Figure 3B, lane 3), and GFAP can be coprecipitated by anti-Flag antibody (Figure 3A, lane 3).
FIGURE 3:
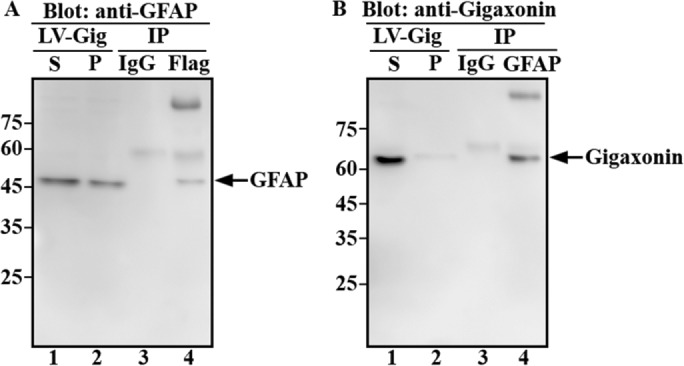
Gigaxonin interacted with GFAP. Primary astrocytes were infected with lentiviruses expressing Flag-gigaxonin for 36 h, a time point at which cells still contained GFAP. Cells were extracted with RIPA lysis buffer and the supernatant (A and B, lane 1) and pellet (A and B, lane 2) fractions were prepared as described in Materials and Methods. Supernatant fractions were subjected to immunoprecipitation using monoclonal anti-Flag (A, lane 4) or anti-GFAP (B, lane 4) antibodies. Mouse IgG was used as a control (A and B, lane 3). The supernatant (A and B, lane 1) and pellet (A and B, lane 2) fractions, as well as immunoprecipitates (A and B, lanes 3 and 4), were analyzed by immunoblotting using polyclonal anti-GFAP (A) and anti-gigaxonin (B) antibodies. Molecular mass markers are shown on the left. Representative blots from three independent experiments.
Gigaxonin-mediated clearance of GFAP involves the proteasome
Because gigaxonin is a predicted substrate adaptor for the E3 ubiquitin ligase complex and facilitates proteasomal degradation of several IF proteins, we performed additional experiments to determine whether clearance of GFAP by gigaxonin involves the proteasome degradation pathway. Primary astrocytes that had been cleared of GFAP by expression of gigaxonin for 72 h were treated with MG-132 to inhibit proteasome for 12 h. Immunoblotting revealed that before the addition of MG-132, the level of GFAP was significantly reduced (Figure 4A, lane 2) compared with mock-infected control cells (Figure 4A, lane 1). Proteasome inhibition by treatment with MG132 resulted in the reappearance of GFAP (Figure 4A, lane 3). Longer exposure of the anti-GFAP immunoblot showed high–molecular mass laddering of GFAP, which would indicate ubiquitination (Figure 4A, top, lane 3). Immunofluorescence confirmed the reappearance of GFAP, which was mainly present as small aggregates (Figure 4C, arrows). These results suggest that gigaxonin targets GFAP for degradation via the proteasome pathway.
FIGURE 4:

Gigaxonin targeted GFAP for degradation through the proteasomal pathway. (A) Primary astrocytes were either mock infected (lane 1) or infected with lentiviruses containing gigaxonin (lanes 2 and 3). At 72 h after infection, gigaxonin-infected cells were treated with DMSO (lane 2) or 10 μM MG-132 (lane 3) for 12 h. Total lysates prepared from these cultures were analyzed by immunoblotting using antibodies to gigaxonin (Gig), GFAP, and actin, which was used as a loading control. Molecular mass markers (kilodaltons) are shown on the left. Note that increased exposure of the immunoblot was required to reveal the laddering of high–molecular weight GFAP (A, top, lane 3). Representative blots from three independent experiments. (B, C) Immunofluorescence showed the reappearance of GFAP. Primary astrocytes that had been cleared of GFAP by expression of gigaxonin for 72 h (B) were treated with MG-132 for 12 h (C). Cells were then double labeled with anti-GFAP (B and C, green channel) and anti-gigaxonin (B and C, red channel) antibodies. Merged images are shown. Note that GFAP reappeared as small aggregates (C, arrows) upon inhibition of the proteasome. Bar, 20 μm.
Gigaxonin clears some but not all GFAP mutants
To further confirm the role of gigaxonin in the degradation of GFAP, we established a cell line stably expressing human wild-type GFAP using the human adrenal carcinoma SW13 (vim-) cells. These cells, which do not express any endogenous cytoplasmic IFs (Hedberg and Chen, 1986), provide a useful experimental model system in which to explore the effect of gigaxonin on the clearance of this IF protein. Immunofluorescence studies revealed that when expressed in this line, wild-type GFAP formed extended filaments, as well as thick filament bundles dispersed throughout the cytoplasm (Figure 5A). Expression of gigaxonin resulted in clearance of GFAP IFs (Figure 5B) and caused a decrease in GFAP protein (Figure 5D, lane 2) to a level only ∼8% of those seen in controls (Figure 5D, lane 1). As in primary astrocytes, the decrease in the level of GFAP was not associated with a corresponding decrease in GFAP mRNAs (Supplemental Figure S1). By immunoblotting, no changes in tubulin and actin levels were observed, demonstrating a selective role of gigaxonin in IF clearance. In a parallel experiment, cells were infected with lentiviruses containing blue fluorescent protein (BFP)–tagged gigaxonin. BFP was introduced so that gigaxonin-infected cells could be identified and costained with both GFAP and tubulin. Immunofluorescence revealed that although GFAP disappeared by 72 h (Figure 6A), MT organization appeared normal in gigaxonin-expressing cells (Figure 6B).
FIGURE 5:

Gigaxonin cleared human wild-type GFAP. SW13 cells stably expressing human wild-type GFAP were infected with gigaxonin-containing lentiviruses for 72 h. Cells were fixed and processed for double-label immunofluorescence microscopy using anti-GFAP (A) and anti-gigaxonin (B) antibodies. (C) Merged image. Representative images from three independent preparations. Scale bar, 10 μm. (D) Total lysates prepared from mock-infected cells (lane 1) or cells infected with gigaxonin (lane 2) were analyzed by immunoblotting with antibodies to gigaxonin, GFAP, tubulin, and actin. The level of GFAP decreased to 8.2 ± 4.3%, of controls. Representative blots from three independent experiments.
FIGURE 6:

Distributions of GFAP and microtubules (MTs) in gigaxonin-expressing cells. SW13 cells stably expressing human wild-type GFAP were infected with lentiviruses containing BFP-gigaxonin. At 72 h after infection, cells were fixed and immunostained with anti-GFAP (A) and anti–β-tubulin (B) antibodies. BFP-gigaxonin accumulated in the puncta (C, arrows), which colocalized with the remnant of GFAP aggregates (A, arrows). (D) Merged image. Representative images from three independent preparations. Scale bar, 10 μm.
Because mutations in GFAP are associated with abnormal aggregation of IFs in astrocytes in patients with AxD, we sought to determine whether gigaxonin expression could also clear mutant forms of GFAP. Using SW13 cells, we established individual lines that stably expressed several disease-associated GFAP mutants. Immunofluorescence revealed that, like wild-type GFAP, R416W GFAP formed a well-dispersed IF network that distributed throughout the cytoplasm (Figure 7A). The IDF GFAP, resulting from deletion and insertion mutations that cause a frameshift (Flint et al., 2012), formed filamentous networks that tended to form bundles particularly at the cell periphery (Figure 7D, arrow). In contrast, the R88C GFAP produced a completely different staining pattern, characterized by diffuse background staining (Figure 7G). These lines were infected with gigaxonin-expressing lentiviruses for 72 h. Immunofluorescence revealed clearance of R416W (Figure 7A), IDF (Figure 7D), and R88C (Figure 7G) GFAP in cells expressing gigaxonin. Immunoblotting showed that expression of gigaxonin caused a decrease in GFAP levels to 18 ± 4.7% (Figure 7C, lane 2), 16.4 ± 3.7% (Figure 7F, lane 2), and 9.4 ± 4.5% (Figure 7I, lane 2) of controls (Figure 7, C, F, and I, lane 1) after lentiviral transduction into SW13 cells expressing R416W, IDF, and R88C, respectively. The decreased level of GFAP mutants was not due to decreased mRNA expression since real-time (RT)-PCR analysis showed no significant differences of transcript levels between gigaxonin-infected and mock-infected control cells (Supplemental Figure S1).
FIGURE 7:

Gigaxonin expression cleared mutant forms of GFAP. SW13 cells stably expressing R416W (A, B), IDF (D, E), or R88C (G, H) GFAP were infected with lentiviruses containing Flag-gigaxonin for 72 h. Cells were then fixed and processed for double-label immunofluorescence microscopy using anti-GFAP (A, D, G) and anti-gigaxonin (B, E, H) antibodies. Representative images from three independent preparations. Scale bar, 10 μm. Total cell lysates prepared from mock-infected cells (C, F, I, lane 1) or cells infected with gigaxonin lentiviruses (C, F, I, lane 2) were analyzed by immunoblotting using antibodies to gigaxonin, GFAP, and finally tubulin, which was used as a loading control. Representative blots from three independent experiments.
Analysis of SW13 lines that stably express R239H (Figure 8A) and ∆4 (Figure 8D) GFAP revealed that both GFAP mutants were unable to assemble into IF networks. After lentiviral infection for 72 h, neither R239H (Figure 8B) nor ∆4 GFAP (Figure 8E) was cleared by gigaxonin, as determined by both immunofluorescence (Figure 8, A and D) and immunoblotting (Figure 8, C and F). These results suggest that gigaxonin can clear some but not all AxD-associated GFAP mutants.
FIGURE 8:

Gigaxonin was unable to clear R239H and ∆4 GFAP mutants. Expression of Flag-gigaxonin in SW13 cells stably expressing R239H or ∆4 GFAP was achieved by lentiviral infection for 72 h. Cells were processed for double-label immunofluorescence microscopy using anti-GFAP (A, D) and anti-gigaxonin (B, E) antibodies. Whereas ∆4 GFAP assembled into short filament pieces (D), R239H GFAP formed nonfilamentous structures distributed throughout the cytoplasm (A). Representative images from three independent preparations. Scale bar, 10 μm. Total lysates prepared from these cells (C, F, lane 2) were analyzed by immunoblotting with indicated antibodies and compared with mock-infected controls (C, F, lane 1). Representative blots from three independent experiments.
Disease-linked mutations in GFAP reduced its interaction with gigaxonin
Because the interaction of gigaxonin with GFAP is essential for its degradation by the proteasome, we determined whether mutations in GFAP affect its interaction with gigaxonin by coimmunoprecipitation experiments. Flag-gigaxonin was transduced into SW13 cells stably expressing either wild-type (Figure 9A) or mutant GFAP (Figure 9, B–F) for 36 h. The supernatant fractions (Figure 9, A–C, lane 1) prepared from these cultures were subjected to immunoprecipitation using a monoclonal anti-Flag antibody. Immunoblotting revealed that gigaxonin was enriched in Flag immunoprecipitates (Figure 9, A–F, lane 3). Like wild-type GFAP (Figure 9A, lane 3), R88C (Figure 9B, lane 3), R416W (Figure 9D, lane 3), and IDF (Figure 9E, lane 3) mutants were coprecipitated with gigaxonin (Figure 9A, lane 3). By contrast, comparatively little R239H (Figure 9B, lane 3) and no ∆4 (Figure 9C, lane 3) GFAP were coprecipitated with gigaxonin, suggesting that both R239H and ∆4 mutations in GFAP reduced their interactions with gigaxonin.
FIGURE 9:

Mutations in GFAP reduced its interaction with gigaxonin. SW13 cells stably expressing either wild-type (A) or mutant (B–F) GFAPs were infected with Flag-gigaxonin lentiviruses. At 36 h after infection, cells were extracted with a RIPA lysis buffer, and the supernatant (A–F, lane 1) and pellet (A–F, lane 2) fraction were prepared as described in Materials and Methods. The supernatant fractions were subjected to immunoprecipitation by using a mouse monoclonal anti-Flag antibody (A–F lane 4). Mouse IgG was used as a control (A–F, lane 3). The supernatant (A–F, lane 1) and pellet (A–F, lane 2) fractions, as well as the Flag (A–F, lane 4) and IgG (A–F, lane 3) immunoprecipitates, were analyzed by immunoblotting using rabbit polyclonal anti-gigaxonin (A–F, top) and anti-GFAP (A–F, bottom) antibodies. Molecular mass markers are shown on the left. R239H (C, lane 4) and ∆4 (F, lane 4) mutants that coprecipitate with gigaxonin were both decreased compared with wild-type (A, lane 4), R88C (B, lane 4), R416W (D, lane 4), and IDF (E, lane 4) GFAP. Full-length images of blots are shown in Supplemental Figure S2.
DISCUSSION
Gigaxonin, a predicted E3 ubiquitin ligase adaptor, is the first identified regulator that controls degradation of cytoskeletal IF proteins. Although the role of gigaxonin in the clearance of several IF proteins in neurons and fibroblasts is well established (Mahammad et al., 2013), its ability to clear the glial-specific IF protein GFAP in astrocytes has not previously been studied. Here, we showed that overexpression of gigaxonin using a lentivirus expression system induces a nearly complete clearance of the GFAP IFs not only in primary astrocytes, but also in IF-free SW13 cells stably expressing this IF protein. Of importance, the clearance of GFAP is not due to altered levels or stability of its mRNA. This was a specific effect on the GFAP IFs, since other cytoskeletal networks, such as microfilaments and microtubule networks, appeared normal. Furthermore, we showed that GFAP clearance by gigaxonin involves the proteasome degradation pathway, since proteasome inhibition by MG132 treatment partially restored GFAP expression.
How gigaxonin exerts its effect on GFAP
Our results clearly showed that gigaxonin targets GFAP for degradation via the proteasome pathway, although exactly how this occurs remains unknown. We believe the most likely mechanism is that GFAP IFs were first disassembled into nonfilamentous particles before being degraded by the ubiquitin proteasome system. This protein quality control targets misfolded or damaged proteins for proteasomal degradation by the addition of a ubiquitin chain through a multistep process involving an E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases. As an E3 ubiquitin ligase adaptor (Furukawa et al., 2003; Xu et al., 2003; Pintard et al., 2004, gigaxonin facilitates ubiquitination and confers substrate specificity through direct interaction with substrates through its C-terminal Kelch domain (Johnson-Kerner et al., 2015). Ubiquitinated substrates are directed into proteasomes and degraded in the inner core of 20S proteasome subunit, where the protease active sites of the catalytic subunits are located. However, only unfolded monomeric peptides are able to pass through the constraints of the proteolytic core, which functions by size exclusion (Glickman and Ciechanover, 2002). One implication from this model is that the GFAP IF will only be degraded after disassembling into its constituent proteins. Subsequently, in its role as an E3 ligase adaptor, gigaxonin could target the smallest disassembly units for degradation. In support of this mechanism, we found that GFAP degradation occurred coincidently with the disassembly of long, mature IFs into nonfilamentous particles and short filament pieces in cells expressing gigaxonin. However it is not known whether short IFs, unit length filaments, tetramers, dimers, monomers, or other disassembly intermediates are targeted for degradation. The mechanism by which gigaxonin expression leads to GFAP IF disassembly is also unknown.
It is possible that interactions between gigaxonin and GFAP might participate in GFAP IF disassembly, especially given that our results from immunoprecipitation experiments provide evidence to suggest that gigaxonin interacts with GFAP. However, we do not know the assembly state of GFAP involved in this interaction. In the immunoprecipitation experiments, GFAP was solubilized in the RIPA lysis buffer used to optimize conditions for GFAP and gigaxonin interactions. An alternative possibility is that phosphorylation of GFAP may be involved in the process of its degradation, since the major known regulator of IF disassembly is phosphorylation (Omary et al., 2006; Sihag et al., 2007). Support for this possibility stems from the finding that phosphorylation of GFAP plays an important role in the degradation and turnover of glial filaments (Takemura et al., 2002a, b). However, additional studies will be required to determine the phosphorylation state of GFAP in the presence of elevated gigaxonin.
Why some GFAP mutants are resistant to gigaxonin clearance
Given the dual potential of gigaxonin in clearing IF proteins and aggregates (Mahammad et al., 2013), it was of interest to know whether gigaxonin was similarly involved in the clearance of mutant forms of GFAP, a key component of the IF aggregates known as Rosenthal fibers in AxD. Initially, we considered using primary astrocytes derived from one of the existing knock-in mice carrying the R236H mutation in GFAP (Hagemann et al., 2006) as a model system, but these cells express both wild-type and mutant GFAPs, and only a small proportion (∼3.4%) of them contained GFAP aggregates (Cho and Messing, 2009). Instead, we established GFAP-expressing stable lines using SW13 (vim-) cells because they are readily transducible by lentiviruses, and they have no other endogenous cytoplasmic IFs that could complicate the interpretation. These cells provide a useful experimental model that allowed us to focus our attention on the role of gigaxonin in the clearance of mutant forms of GFAP. When expressed as stable transgenes, GFAP mutants formed a variety of IF phenotypes, including filamentous networks, filament bundles, and nonfilamentous structures that were distributed throughout the cytoplasm. However, no stable clones that contain GFAP aggregates have ever been selected. This most likely reflects the presence of GFAP aggregates being a potential stress that could lead to retarded cell growth (Cho and Messing, 2009), decreased cell viability (Chen et al., 2011), or even cell death (Mignot et al., 2007). Therefore aggregate-containing clones may not be able to survive through the isolation and expansion procedures.
Of interest, the R416W GFAP formed IFs that appeared indistinguishable from wild-type filaments, which contrasts with previous studies (Perng et al., 2006), in which the same mutation failed to assemble into filaments but instead formed clusters of cytoplasmic aggregates when transiently expressed in the same IF-free SW13 cells. This discrepancy might be related to differences between the transient transfection strategy that had been used and the stable expression of this mutant GFAP as we reported here. In contrast, R88C mutant failed to assemble into normal filaments. The observation that R88C GFAP was cleared by gigaxonin indicates that being incorporated into a filament is not essential for gigaxonin-mediated degradation, since the nonfilamentous forms of GFAP that contained the R88C mutation were efficiently degraded. Like R88C GFAP, both R239H and ∆4 GFAP (Flint et al., 2012) failed to self-assemble into extended IF networks, yet they were resistant to gigaxonin clearance. Given that both mutations lie within the 2A subdomain of GFAP, these results suggest that changing the precise sequence of this domain can have a dramatic effect on GFAP assembly, which is consistent with the results of previous studies (Hsiao et al., 2005; Flint et al., 2012; Messing et al., 2012a). However, the inability of gigaxonin to clear these GFAP mutants shows that not all GFAP mutants are the same with respect to their susceptibility to gigaxonin clearance.
Why are some GFAP mutants resistant to gigaxonin clearance? One possibility is that these mutations interfere with the interaction of GFAP with gigaxonin, leading to an impaired targeting of GFAP for proteasomal degradation. In support of this possibility, our data from immunoprecipitation experiments showed that R239H and ∆4 mutations in GFAP dramatically reduced their interactions with gigaxonin. The ∆4 splicing mutation resulted in a complete removal of the whole 2A subdomain (Flint et al., 2012), within which the R239 mutation is also located. These results suggest that, as a potential binding motif for gigaxonin, the 2A subdomain of GFAP is essential for its clearance by gigaxonin. A recent study on vimentin, a structurally related IF protein, provides additional evidence to support a role of the rod domain in IF protein degradation by gigaxonin (Mahammad et al., 2013). Another possibility is that accumulation of particular forms of mutant GFAP could impair proteasome function. Support for this possibility comes mainly from cell-based studies, which showed that accumulation of abnormal oligomers of mutant GFAP appeared to be the cause of proteasome dysfunction (Chen et al., 2011; Cho and Messing, 2009; Tang et al., 2006, 2010).
Therapeutic implication for AxD and related disorders
Our findings that gigaxonin targets GFAP for proteasomal degradation raise the possibility that one therapeutic approach for AxD and related disorders might be to deliver gigaxonin that could effectively reduce GFAP level and reverse GFAP aggregation. This approach is more attractive in light of the fact that complete absence of GFAP produces minimal phenotypes in the mouse (Gomi et al., 1995; Liedtke et al., 1996; McCall et al., 1996; Pekny et al., 1995). However, it is important to note that in addition to GFAP, astrocytes express several IF proteins, including vimentin, nestin, and synemin (Hol and Pekny, 2015). Although we did not test whether nestin and synemin are also cleared by gigaxonin, the clearance of GFAP, as well as vimentin, in astrocytes is likely to dramatically change the composition and organization of IF networks, leading to significant alterations in astrocyte function. Therefore determining the exact dosage of reintroduced gigaxonin that could promote dissolution of the aggregates without completely destroying the functional IF networks in the long term would be essential. Although the overexpression of gigaxonin has provided us with the most convincing evidence in support of its role in degrading IF proteins, it is possible that expression of gigaxonin in excess may have deleterious side effects that we have been unable to detect. Using the lentiviral transduction system, we were unable to control the level of gigaxonin expression, and this is a limitation of the present study. Although the mechanism by which gigaxonin is regulated in normal cells is not known, it was previously shown that it is normally expressed at extremely low levels (Cleveland et al., 2009). Better understanding of how gigaxonin is regulated is an essential element in understanding its role in the degradation of GFAP and other IF proteins.
In conclusion, we identified gigaxonin as an important factor that facilitates proteasomal degradation of GFAP. Our findings have important implications for the degradation and turnover of the glial filaments long believed to be one of the most stable cytoskeletal networks in astrocytes. Based on these findings, gigaxonin might ultimately prove valuable for treating AxD and possibly related diseases in which a decrease in GFAP levels may be beneficial.
MATERIALS AND METHODS
Plasmid construction and site-directed mutagenesis
GFAP mutations were introduced by site-directed mutagenesis (QuikChange; Stratagene, La Jolla, CA) using the human wild-type GFAP in the pcDNA 3.1(−) vector (Thermo Fisher Scientific, Waltham, MA) as a template (Perng et al., 2006). The mutagenic primers used for the construction of GFAP mutants were as follows:
R88C GFAP forward, 5′-ATCGAGAAGGTTTGCTTCCTGGAAC-3′
R88C GFAP reverse, 5′-GTTCCAGGAAGCAAACCTTCTCGAT -3′
∆4GFAP (a splice site mutation leading to skipping of exon 4) forward, 5′-AGGAAGATCCACGAGGAGTTTGCAGACCTGACAGACGCTGCT-3′
∆4GFAP reverse, 5′-AGCAGCGTCTGTCAGGTCTGCAAACTCCTCGTGGATCTTCCT-3′
IDF GFAP (deletion and insertion mutations leading to a frameshift of GFAP) forward, 5′-AGCAGGAGCACAAGGATGATCGGCAGGACCCACCTG-3′
IDF GFAP reverse, 5′-GATTTGGGTCCTGCCTCATGAGACGGGGCAGAGGCC-3′
All newly constructed GFAP mutants were verified by DNA sequencing before use. R239H GFAP (Hsiao et al., 2005) and R416W GFAP (Perng et al., 2006) were constructed as described previously. The lentiviral vector pLEX-MCS–FLAG-gigaxonin was previously described (Mahammad et al., 2013). BFP-gigaxonin (Lowery et al., 2016) was cloned into the lentiviral pLEX-MCS vector using the InFusion HD Cloning System (Clontech, Mountain View, CA).
Primary cortical astrocyte cultures
All experiments involving animals were approved by the Institutional Animal Care and Use Committee of National Tsing Hua University. Primary cortical astrocytes were prepared from the fetal brain of Sprague Dawley rats at embryonic day 18 (E18). The cortices from individual pups were freed of meninges and dissected in Hanks balanced salt solution followed by incubation with 0.25% (wt/vol) trypsin (Thermo Fisher Scientific) at 37°C for 15 min. After incubation with DNase I (Sigma-Aldrich, St. Louis, MO) for an additional 5 min, the cortices were mechanically dispersed by triturating with a Pasture pipette. Dissociated cortical cells were collected by centrifugation at 1000 × g for 5 min and resuspended in plating medium containing MEM supplemented with 10% (vol/vol) horse serum (Thermo Fisher Scientific), 100 U/ml penicillin, and 100 μg/ml streptomycin. After filtration through a 70-μm nylon mesh (Greiner Bio-One, Frickenhausen Germany), cells were seeded onto poly-l-lysine–coated flasks at a density of 5 × 104 cells/cm2. Cells were maintained in a humidified 5% CO2 atmosphere at 37°C. Medium was changed every 3–4 d. At 9 d in vitro, the flasks were shaken overnight at 200 rpm to remove neurons, oligodendrocytes, and microglia. The adherent astrocyte population was detached by incubating with 0.25% (vol/vol) trypsin-ethylenediaminetetraacetic acid (EDTA), suspended in plating medium, and plated on 60-mm dishes, six-well plates, or 12-well plates as needed. The purity of these cultures is typically 95% GFAP-positive cells as determined by immunostaining.
Cell cultures and generation of stable cell lines
Human adrenal carcinoma SW13 (vim-) cells were kindly provided by Michael Brenner (University of Alabama at Birmingham, Birmingham, AL) and were maintained in DMEM (Thermo Fisher Scientific) supplemented with 10% (vol/vol) fetal bovine serum (Thermo Fisher Scientific) and 1% (vol/vol) penicillin/streptomycin (Thermo Fisher Scientific). SW13 cell lines stably expressing GFAP were established by first transfecting either wild-type or mutant GFAP in the mammalian expression vector pcDNA3.1 (Thermo Fisher Scientific; Chen et al., 2011). Selection of stable cell lines was initiated 2 d after transfection using 600 μg/ml G-418 (Thermo Fisher Scientific). Fourteen days after selection, GFAP-positive clones were isolated and cultured in 24-well plates and then transferred into six-well plates and finally into 100-mm Petri dishes. Stable cell lines were maintained in growth medium supplemented with 100 μg/ml G-418. Homogeneous expression of GFAP in stable cell lines was confirmed by immunofluorescence microscopy.
Cell transfection and treatments
For transient transfection studies, plasmid DNA was prepared using a Genopure Plasmid Midi Kit (Roche, Mannheim, Germany). Cells grown on 18-mm coverslips at a density of 50% confluency were transfected with BFP-gigaxonin (Lowery et al., 2016) using TansIT-LT1 Transfection Reagent (Mirus Bio, Madison, WI) according to the manufacturer’s instructions. Cells were allowed to recover for 48 h before further processing for immunofluorescence microscopy. In some experiments, cells were treated with 10 μM MG-132 (Calbiochem; 10 mM stock in dimethyl sulfoxide [DMSO]) for 12 h to inhibit proteasomal activity, and control cells were treated with a comparable concentration of DMSO (0.1% [vol/vol] in culture medium).
Immunofluorescence microscopy
Cells were processed for indirect immunofluorescence microscopy after fixation in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) as previously described (Perng et al., 2006). The primary antibodies used in this study were mouse anti-GFAP (SMI26; BioLegend, San Diego, CA), rabbit anti-GFAP (Z334; DakoCytomation, Glostrup, Denmark), mouse anti-vimentin (V9; Sigma-Aldrich), mouse anti–β-tubulin (Novus Biologicals), rabbit anti-gigaxonin (Sigma-Aldrich), and mouse monoclonal anti-Flag M2 (Sigma-Aldrich) antibodies. In some experiments, fluorescein isothiocyanate–conjugated phalloidin (1:50; Sigma-Aldrich) was used to label actin-containing microfilaments. Secondary antibodies used in this study included Alexa Fluor 488– and Alexa Fluor 594–conjugated goat anti-mouse and anti-rabbit secondary antibodies (Thermo Fisher Scientific). The nuclei were visualized by staining with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific). After staining, cells were mounted on glass slides and imaged using a Zeiss LSM510 laser scanning confocal microscope (Carl Zeiss, Jena, Germany) with a 40× (0.75 numerical aperture [NA]) Plan-Neofluar or 63× (1.40 NA) Apochromat objective lens. Images were collected in Multitrack mode by LSM510 software taking 1.0-μm optical sections and processed for figures using Photoshop CS6 (Adobe, San Jose, CA). Quantification of gigaxonin-expressing cells was performed by visual assessment of lentivirus-infected cells. For each lentiviral transduction, cells on three coverslips were counted, and ∼200–300 infected cells were assessed per coverslip.
Lentiviral production and transduction
Lentiviruses were produced by transiently cotransfecting pLEX-MCS–FLAG-gigaxon vector (Mahammad et al., 2013), along with the psPAX2 packaging (12260; Addgene, Cambridge, MA) and pMD2.G envelop (12259; Addgene) vectors at a ratio of 4:3:1 into 293T cells (Thermo Fisher Scientific) using the TransIT-LT1 Transfection Reagent (Mirus Bio). Culture supernatants containing virus particles were collected 48–72 h after infection, filtered through a 0.45-μm filter (Pall Corporation, Port Washington, NY), and concentrated by a Lenti-X concentrator (Clontech). Cells were infected by incubating with the viral supernatant supplemented with 8 μg/ml Polybrene (Sigma-Aldrich) for 4–8 h, after which the virus-containing medium was replaced with normal growth medium. Unless otherwise stated, cells infected with empty pLEX-MCS vector were used as controls. Cells were infected with lentivirus expressing different forms of GFAPs at a multiplicity of infection of 10 for 36–72 h. Under these conditions, ∼80% of cells were infected as determined by immunofluorescence microscopy.
Cell fractionation, immunoblotting, and immunoprecipitation
Cells grown on 60- or 100-mm Petri dishes were washed with phosphate-buffered saline and then lysed using RIPA lysis buffer (1% [vol/vol] Triton X-100, 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] SDS, 150 mM NaCl, 50 mM Tris-HCl, pH 8, 2 mM ethylene glycol tetraacetic acid, and 1 mM phenylmethylsulfonyl fluoride) supplemented with a cocktail of protease inhibitors (Roche) and phosphatase inhibitors (Calbiochem). Cell lysates were then homogenized on ice in a 1-ml Dounce homogenizer (Wheaton, Millville, NJ). After determination of protein concentration, total cell lysates were centrifuged at 16,000 × g for 10 min at 4°C in a bench-top centrifuge (Eppendorf). The resulting supernatants were collected as soluble fractions, and the remaining pellets, representing the insoluble fractions, were resuspended in Laemmli’s sample buffer (Laemmli, 1970) in a volume that was equivalent to that of the supernatant. In some experiments, an aliquot of the total cell lysates was mixed directly with appropriate volumes of Laemmli’s sample buffer and analyzed by SDS–PAGE and immunoblotting.
Immunoblotting was performed using either the semidry or the wet electrophoretic transfer system (both from Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. After transfer, the membranes were washed with Tris-buffered saline (TBS; 150 mM NaCl, 20 mM Tris-HCl, pH 7.4) three times, followed by incubation in the blocking buffer (3% [wt/vol] bovine serum albumin [BSA] in TBS containing 0.1% [vol/vol] Tween-20) for 1 h at room temperature. After blocking, the membranes were incubated at 4°C overnight with mouse anti-GFAP (SMI26; Biolegend), rabbit anti-GFAP (Z334; Dako), mouse anti–β-tubulin (DM-1A; Novus Biologicals, Littleton, CO), mouse anti–β-actin (AC-15; Novus Biologicals), mouse monoclonal anti-Flag M2 (Sigma-Aldrich), or rabbit anti-gigaxonin (Sigma-Aldrich) antibodies. After washing with TBS containing 0.1% (vol/vol) Tween-20 (TBST), membranes were incubated with horseradish peroxidase–conjugated goat anti-mouse or anti-rabbit secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. All antibodies were diluted in TBST containing 1% (wt/vol) BSA. Antibody labeling was detected by enhanced chemiluminescence (SuperSignal West Pico Substrate; Thermo Fisher Scientific) with use of a luminescent image analyzer (LAS 4000; GE Healthcare, Chicago, IL). The strength of signals was quantified using the ImageQuant software (GE Healthcare). To account for loading variability, all protein bands were normalized to either β-actin or β-tubulin.
For immunoprecipitation, soluble fractions prepared as described were precleared by incubation with a 50% (vol/vol) slurry of protein G Sepharose (GE Healthcare) for 1 h at 4°C. The clarified supernatant was incubated with either mouse monoclonal anti-Flag M2 (Sigma-Aldrich) or anti-GFAP SMI-26 antibody (BioLegend) for 3 h at 4°C, followed by the capture of immunocomplexes by incubation with protein G Sepharose beads (GE Healthcare) with gentle rocking overnight at 4°C. Purified mouse immunoglobulin (Ig) G1 (Sigma-Aldrich) was used as a specificity control. Immunoprecipitates were washed extensively with RIPA buffer without SDS, resuspended in Laemmli’s sample buffer, and analyzed by immunoblotting.
Determination of GFAP levels by quantitative real-time PCR
Total RNA was prepared from cells with the GeneJet RNA purification kit (Thermo Fisher Scientific). For reverse transcription, 1 μg of RNA was used to generate cDNA using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative real-time PCR was performed using a Maxima SYBR Green qPCR Master Mix kit (Thermo Fisher Scientific) on the CFS Connect Real-Time PCR Detection System (Bio-Rad Laboratories). Transcript expression levels of GFAP were determined and normalized to the internal reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the 2−ΔΔCT equation. The primer sets used in this study were as follows:
human GFAP forward, 5′-ATGGAGCGGAGACGTATCACC-3′
human GFAP reverse, 5′-GCGGAATGGTACCCAGGTGTC-3′
rat GFAP forward, 5′-ATGGAGCGGAGACTGATCACC-3′
rat GFAP reverse, 5′-GCGGAATGGTACCCAGGTGTC-3′
GAPDH forward, 5′-CAAGGTCATCCATGACAACTTTG-3′
GAPDH reverse, 5′-GTCCACCACCCTGTTGCTGTAG-3′
Supplementary Material
Acknowledgments
We thank Michael Brenner (Department of Neurology, University of Alabama at Birmingham, Birmingham, AL) for generously providing SW13 (Vim-) cells and Su-Chun Zhang (Waisman Center, University of Wisconsin–Madison, Madison, WI) for 293T cells. This work was supported by grants from the Ministry of Science and Technology (MOST), R.O.C. grants 103-2311-B-007-010 and 104-2918-I-007-008 (to N.S.L, Y.S.H, and M.D.P.), and the Juanma Fund and National Institute of Child Health and Human Development Grant HD03352 (to A.M.). The Goldman laboratory received funding from the Hannah’s Hope Fund and National Institute of General Medical Sciences Grant PO1GM096971. The Opal laboratory is funded by grants from the National Institutes of Health (R01NS062051 and R01NS082351).
Abbreviations used:
- GAN
giant axonal neuropathy
- GFAP
glial fibrillary acidic protein
- IF
intermediate filament.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-06-0362) on October 26, 2016.
REFERENCES
- Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373–381. doi: 10.1093/brain/72.3.373. [DOI] [PubMed] [Google Scholar]
- Asbury AK, Gale MK, Cox SC, Baringer JR, Berg BO. Giant axonal neuropathy–a unique case with segmental neurofilamentous masses. Acta Neuropathol. 1972;20:237–247. doi: 10.1007/BF00686905. [DOI] [PubMed] [Google Scholar]
- Berg BO, Rosenberg SH, Asbury AK. Giant axonal neuropathy. Pediatrics. 1972;49:894–899. [PubMed] [Google Scholar]
- Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:777–794. doi: 10.1007/s00401-013-1125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- Chen YS, Lim SC, Chen MH, Quinlan RA, Perng MD. Alexander disease causing mutations in the C-terminal domain of GFAP are deleterious both to assembly and network formation with the potential to both activate caspase 3 and decrease cell viability. Exp Cell Res. 2011;317:2252–2266. doi: 10.1016/j.yexcr.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W, Messing A. Properties of astrocytes cultured from GFAP over-expressing and GFAP mutant mice. Exp Cell Res. 2009;315:1260–1272. doi: 10.1016/j.yexcr.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Yamanaka K, Bomont P. Gigaxonin controls vimentin organization through a tubulin chaperone-independent pathway. Hum Mol Genet. 2009;18:1384–1394. doi: 10.1093/hmg/ddp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequen F, Bomont P, Gowing G, Cleveland DW, Julien JP. Modest loss of peripheral axons, muscle atrophy and formation of brain inclusions in mice with targeted deletion of gigaxonin exon 1. J Neurochem. 2008;107:253–264. doi: 10.1111/j.1471-4159.2008.05601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint D, Li R, Webster LS, Naidu S, Kolodny E, Percy A, van der Knaap M, Powers JM, Mantovani JF, Ekstein J, et al. Splice site, frameshift, and chimeric GFAP mutations in Alexander disease. Hum Mutat. 2012;33:1141–1148. doi: 10.1002/humu.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–1007. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- Ganay T, Boizot A, Burrer R, Chauvin JP, Bomont P. Sensory-motor deficits and neurofilament disorganization in gigaxonin-null mice. Mol Neurodegener. 2011;6:25. doi: 10.1186/1750-1326-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Gomi H, Yokoyama T, Fujimoto K, Ikeda T, Katoh A, Itoh T, Itohara S. Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron. 1995;14:29–41. doi: 10.1016/0896-6273(95)90238-4. [DOI] [PubMed] [Google Scholar]
- Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci. 2006;26:11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet. 2005;14:2443–2458. doi: 10.1093/hmg/ddi248. [DOI] [PubMed] [Google Scholar]
- Hedberg KK, Chen LB. Absence of intermediate filaments in a human adrenal cortex carcinoma-derived cell line. Exp Cell Res. 1986;163:509–517. doi: 10.1016/0014-4827(86)90081-9. [DOI] [PubMed] [Google Scholar]
- Hol EM, Pekny M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol. 2015;32:121–130. doi: 10.1016/j.ceb.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Hsiao VC, Tian R, Long H, Perng MD, Brenner M, Quinlan RA, Goldman JE. Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J Cell Sci. 2005;118:2057–2065. doi: 10.1242/jcs.02339. [DOI] [PubMed] [Google Scholar]
- Huang L, Chen CH. Proteasome regulators: activators and inhibitors. Curr Med Chem. 2009;16:931–939. doi: 10.2174/092986709787581860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israeli E, Dryanovski DI, Schumacker PT, Chandel NS, Singer JD, Julien JP, Goldman RD, Opal P. Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy. Hum Mol Genet. 2016;25:2143–2157. doi: 10.1093/hmg/ddw081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jany PL, Agosta GE, Benko WS, Eickhoff JC, Keller SR, Köehler W, Koeller D, Mar S, Naidu S, Marie Ness J, et al. CSF and blood levels of GFAP in Alexander disease. eNeuro. 2015;2:1–11. doi: 10.1523/ENEURO.0080-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jany PL, Hagemann TL, Messing A. GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro. 2013;5:e00109. doi: 10.1042/AN20130003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Kerner BL, Garcia Diaz A, Ekins S, Wichterle H. Kelch domain of gigaxonin interacts with intermediate filament proteins affected in giant axonal neuropathy. PLoS One. 2015;10:e0140157. doi: 10.1371/journal.pone.0140157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Coulombe PA. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007;21:1581–1597. doi: 10.1101/gad.1552107. [DOI] [PubMed] [Google Scholar]
- Kretzschmar HA, Berg BO, Davis RL. Giant axonal neuropathy. A neuropathological study. Acta Neuropathol. 1987;73:138–144. doi: 10.1007/BF00693779. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 1996;17:607–615. doi: 10.1016/s0896-6273(00)80194-4. [DOI] [PubMed] [Google Scholar]
- Lowery J, Jain N, Kuczmarski ER, Mahammad S, Goldman A, Gelfand VI, Opal P, Goldman RD. Abnormal intermediate filament organization alters mitochondrial motility in giant axonal neuropathy fibroblasts. Mol Biol Cell. 2016;27:608–616. doi: 10.1091/mbc.E15-09-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahammad S, Murthy SN, Didonna A, Grin B, Israeli E, Perrot R, Bomont P, Julien JP, Kuczmarski E, Opal P, Goldman RD. Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest. 2013;123:1964–1975. doi: 10.1172/JCI66387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci USA. 1996;93:6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci. 2012b;32:5017–5023. doi: 10.1523/JNEUROSCI.5384-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- Messing A, Li R, Naidu S, Taylor JP, Silverman L, Flint D, van der Knaap MS, Brenner M. Archetypal and new families with Alexander disease and novel mutations in GFAP. Arch Neurol. 2012a;69:208–214. doi: 10.1001/archneurol.2011.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C, Delarasse C, Escaich S, Della Gaspera B, Noé E, Colucci-Guyon E, Babinet C, Pekny M, Vicart P, Boespflug-Tanguy O, et al. Dynamics of mutated GFAP aggregates revealed by real-time imaging of an astrocyte model of Alexander disease. Exp Cell Res. 2007;313:2766–2779. doi: 10.1016/j.yexcr.2007.04.035. [DOI] [PubMed] [Google Scholar]
- Omary MB, Ku NO, Tao GZ, Toivola DM, Liao J. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem Sci. 2006;31:383–394. doi: 10.1016/j.tibs.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Opal P, Goldman RD. Explaining intermediate filament accumulation in giant axonal neuropathy. Rare Diseases. 2013;1:e25378. doi: 10.4161/rdis.25378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Leveen P, Pekna M, Eliasson C, Berthold CH, Westermark B, Betsholtz C. Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J. 1995;14:1590–1598. doi: 10.1002/j.1460-2075.1995.tb07147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena SD. Giant axonal neuropathy: intermediate filament aggregates in cultured skin fibroblasts. Neurology. 1981;31:1470–1473. doi: 10.1212/wnl.31.11.1470. [DOI] [PubMed] [Google Scholar]
- Perez-Torrado R, Yamada D, Defossez PA. Born to bind: the BTB protein-protein interaction domain. Bioessays. 2006;28:1194–1202. doi: 10.1002/bies.20500. [DOI] [PubMed] [Google Scholar]
- Perng MD, Su M, Wen SF, Li R, Gibbon T, Prescott AR, Brenner M, Quinlan RA. The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and the association of alpha B-crystallin and HSP27. Am J Hum Genet. 2006;79:197–213. doi: 10.1086/504411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004;23:1681–1687. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration. Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla P, Jaffe H, Pant HC. Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer’s NFTs. FASEB J. 2011;25:3896–3905. doi: 10.1096/fj.11-181297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC. Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res. 2007;313:2098–2109. doi: 10.1016/j.yexcr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura M, Gomi H, Colucci-Guyon E, Itohara S. Protective role of phosphorylation in turnover of glial fibrillary acidic protein in mice. J Neurosci. 2002a;22:6972–6979. doi: 10.1523/JNEUROSCI.22-16-06972.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura M, Nishiyama H, Itohara S. Distribution of phosphorylated glial fibrillary acidic protein in the mouse central nervous system. Genes Cells. 2002b;7:295–307. doi: 10.1046/j.1365-2443.2002.00513.x. [DOI] [PubMed] [Google Scholar]
- Tang G, Perng MD, Wilk S, Quinlan RA, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) Inhibit the proteasome system in alexander disease astrocytes, and the small heat shock protein alphaB-crystallin reverses the inhibition. J Biol Chem. 2010;285:10527–10537. doi: 10.1074/jbc.M109.067975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Xu Z, Goldman JE. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem. 2006;281:38634–38643. doi: 10.1074/jbc.M604942200. [DOI] [PubMed] [Google Scholar]
- Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, Sulzer DL, Goldman JE. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17:1540–1555. doi: 10.1093/hmg/ddn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Love S, Powell HC, Schultz P, Lampert PW. Giant axonal neuropathy: correlation of clinical findings with postmortem neuropathology. Ann Neurol. 1987;22:79–84. doi: 10.1002/ana.410220118. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol. 2013;47:495–508. doi: 10.1007/s12035-012-8280-y. [DOI] [PubMed] [Google Scholar]
- Walker AK, Daniels CM, Goldman JE, Trojanowski JQ, Lee VM, Messing A. Astrocytic TDP-43 pathology in Alexander disease. J Neurosci. 2014;34:6448–6458. doi: 10.1523/JNEUROSCI.0248-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–321. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.