

In budding yeast, the spindle position checkpoint ensures that cells exit from mitosis only when their spindle is properly aligned along the mother–bud axis. Exit from mitosis is controlled by both negative signals in the mother cell compartment and positive signals in the bud.
Abstract
In budding yeast, alignment of the anaphase spindle along the mother–bud axis is crucial for maintaining genome integrity. If the anaphase spindle becomes misaligned in the mother cell compartment, cells arrest in anaphase because the mitotic exit network (MEN), an essential Ras-like GTPase signaling cascade, is inhibited by the spindle position checkpoint (SPoC). Distinct localization patterns of MEN and SPoC components mediate MEN inhibition. Most components of the MEN localize to spindle pole bodies. If the spindle becomes mispositioned in the mother cell compartment, cells arrest in anaphase due to inhibition of the MEN by the mother cell–restricted SPoC kinase Kin4. Here we show that a bud-localized activating signal is necessary for full MEN activation. We identify Lte1 as this signal and show that Lte1 activates the MEN in at least two ways. It inhibits small amounts of Kin4 that are present in the bud via its central domain. An additional MEN-activating function of Lte1 is mediated by its N- and C-terminal GEF domains, which, we propose, directly activate the MEN GTPase Tem1. We conclude that control of the MEN by spindle position is exerted by both negative and positive regulatory elements that control the pathway’s GTPase activity.
INTRODUCTION
Polarized cell division is a defining characteristic of development and one mechanism by which cells produce progeny with distinct cell fates (Siller and Doe, 2009). Two well-known examples of asymmetric cell division are the meiotic divisions of the mammalian oocyte and the mitotic divisions of Drosophila germline stem cells. Because these asymmetric cell divisions rely on the unequal distribution of fate determinants within the cell, it is critical that the mitotic spindle and hence the plane of cell division are correctly placed with respect to these spatially restricted developmental cues. Evidence suggests that feedback mechanisms that sense spindle position are in place to ensure that this occurs. Drosophila germline stem cells, for example, delay the cell cycle if the spindle is not properly aligned along the axis of cell division (Cheng et al., 2008).
In budding yeast, the site of cytokinesis is determined well before cells enter mitosis. Division by budding also means that the connection between mother cell and bud is small and that the nucleus must squeeze through it in order to be partitioned into the bud. Cell proliferation by budding thus requires mechanisms that ensure that anaphase spindle disassembly and cytokinesis occur only after delivery of a DNA complement into the bud.
Yeast cells couple spindle disassembly and cytokinesis (collectively called exit from mitosis) to nuclear position through a mechanism known as the spindle position checkpoint (SPoC). If the spindle becomes misaligned and anaphase spindle elongation occurs in the mother cell compartment, the SPoC prevents exit from mitosis until the nucleus realigns along the mother–bud axis. In the absence of a functional SPoC, cells that do not deliver a DNA complement into the bud during anaphase inevitably become binucleate and anucleate (Figure 1A).
FIGURE 1:
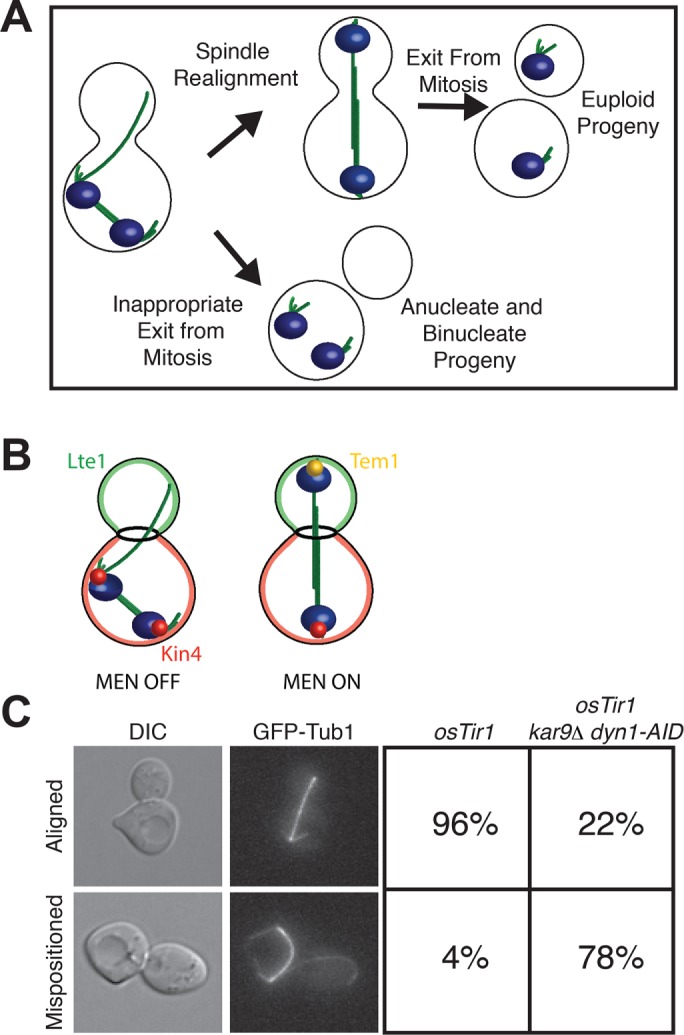
Characterization of cells with mispositioned spindles. (A) The function of the spindle position checkpoint. If the spindle becomes mispositioned in the mother cell compartment, most cells realign their spindles before exiting mitosis so that both daughter cells are euploid. A small fraction of the cells that misposition their spindles inappropriately exit from mitosis in the mother cell compartment and go on to produce one anucleate cell and one binucleate cell. The SPoC prevents cells from becoming anucleate/binucleate by arresting cells in late anaphase until the spindle has realigned along the mother–bud axis. The DNA is blue, and the spindle is green. (B) Regulation of exit from mitosis as described by the zone model. There are two zones: a mitotic exit–inhibitory zone in the mother cell compartment, and a mitotic exit–promoting zone in the bud cell compartment. If the anaphase spindle is mispositioned in the mother cell compartment, Kin4 (red) prevents Tem1 (yellow) from becoming enriched at SPBs, and the MEN cannot become active. It is only once one SPB escapes the inhibitory zone in the mother cell compartment and moves into the bud compartment where the positive regulator of the MEN Lte1 (green) resides that Tem1 can be enriched on SPBs and the MEN is activated. (C) Wild-type (A35699) or dyn1-AID kar9Δ (A35707) cells were grown in yeast extract/peptone/dextrose (YEPD) medium and arrested in G1 with 10 μg/ml α-factor. Cells were released into the cell cycle in YEPD medium and then monitored by live-cell microscopy in a Y04C flow cell. Depletion of dyn1-AID was induced in the flow cell with 100 μM auxin in YEPD medium. Cell cycle stage was assessed by spindle morphology using a GFP-tagged α-tubulin protein. Left, representative images of cells with either an aligned spindle (top) or mispositioned spindle (bottom); right, fraction of cells with aligned and mispositioned spindles in each strain (n = 100).
Spindle mispositioning prevents exit from mitosis by inhibiting the activation of a conserved Ras-like signal transduction cascade known as the mitotic exit network (MEN, also known as the Hippo pathway in mammals). Most MEN components localize to spindle pole bodies (SPBs; yeast centrosomes), and their SPB localization is critical for their function in regulating exit from mitosis (Valerio-Santiago and Monje-Casas, 2011). The target of this regulation is the GTPase Tem1; in its GTP-bound state, Tem1 recruits the PAK kinase Cdc15 to SPBs (Visintin and Amon, 2001; Rock and Amon, 2011; Scarfone et al., 2015). Cdc15, in turn, phosphorylates the SPB-localized MEN-scaffold protein Nud1 to create a binding site required for the recruitment and activation of the Dbf2-Mob1 kinase complex (Rock et al., 2013). In the final step of MEN signaling, activated Dbf2-Mob1, together with the polo kinase Cdc5, promotes the release of the protein phosphatase Cdc14 from its inhibitor in the nucleolus (Mohl et al., 2009; Manzoni et al., 2010). The released phosphatase then triggers exit from mitosis by reversing mitotic phosphorylation (Jaspersen et al., 1998; Visintin et al., 1998).
Control of the MEN by spindle position is mediated by the asymmetric localization of both positive and negative regulators of the MEN (Chan and Amon, 2010). The protein kinase Kin4 is an inhibitor of the MEN; it prevents the inactivation of the bipartite Tem1 GTPase-activating protein (GAP) complex Bub2/Bfa1 at SPBs (D’Aquino et al., 2005; Pereira and Schiebel, 2005). Its localization is restricted to the mother cell cortex and SPBs in the mother cell compartment. Thus, if anaphase occurs in the mother cell compartment, the MEN is inhibited by Kin4 because both SPBs are in the mother cell compartment (Figure 1B). Lte1 is an activator of the MEN that localizes to the bud compartment. Lte1 prevents Kin4 from associating with SPBs, rendering the Bub2-Bfa1 GAP inactive (Bertazzi et al., 2011; Falk et al., 2011). Thus, when one SPB moves out of the mother cell into the bud, where Lte1 antagonizes low levels of Kin4 that leak into the bud, MEN activation occurs.
Although the factors that exert spindle position control of the MEN have been identified, we have only a partial understanding of how they function to bring about this regulation. Here we use a new system to induce spindle misalignment that allowed us to assess SPoC activity in various checkpoint mutants. This analysis led to the remarkable finding that all SPoC mutants retain some SPoC competence. SPoC mutants transiently delay anaphase in response to spindle mispositioning before inappropriately exiting from mitosis. In contrast, in cells lacking the MEN GAP Bub2-Bfa1, exit from mitosis occurs with the same kinetics irrespective of whether the spindle is correctly or incorrectly aligned. We further show that the residual SPoC activity observed in SPoC mutants is due to the lack of a MEN-activating signal mediated by LTE1. This finding indicates that Lte1 has functions in promoting exit from mitosis in addition to antagonizing Kin4. Lte1 has homology to guanine nucleotide exchange factors (GEFs), and it has long been hypothesized that it could function as a GEF for Tem1 (Bardin et al., 2000; Seshan and Amon, 2005). Indeed, we find that the GEF domains of Lte1 are required for the protein’s mitotic exit–promoting function. We conclude that spindle position control of the MEN is exerted by both negative and positive regulatory mechanisms that control Tem1’s GTPase activity.
RESULTS
A system to assess spindle position checkpoint strength in SPoC mutants
In budding yeast, two partially redundant pathways position the mitotic spindle (reviewed in McNally, 2013). The first pathway is active throughout the cell cycle and depends on Kar9, an adaptor that links the plus-end microtubule–binding protein Bim1 (yeast EB1) to the type V myosin motor Myo2. This pathway functions to position the preanaphase spindle at the bud neck (Kopecká and Gabriel, 1998; Miller and Rose, 1998; Miller et al., 1999; Beach et al., 2000). The second spindle-positioning pathway is active in anaphase and depends on the minus-end–directed motor dynein (Muhua et al., 1994; Yeh et al., 1995; Heil-Chapdelaine et al., 2000; Farkasovsky and Küntzel, 2001). When either pathway is inactive, a fraction of cells initially misposition their spindles but then quickly realign them (Falk et al., 2016). In contrast, cells lacking both pathways are inviable because they cannot move the nucleus into the bud during anaphase (Miller and Rose, 1998).
The fact that MEN activity is controlled by spindle position was first recognized well more than a decade ago when it was shown that elimination of the Tem1 GAP or overexpression of the MEN activator Lte1 allowed cells with mispositioned spindles to inappropriately exit from mitosis (Bardin et al., 2000; Pereira et al., 2000). Many subsequent studies have identified the factors that inhibit the MEN in response to spindle mispositioning (D’Aquino et al., 2005; Pereira and Schiebel, 2005; Chan and Amon, 2009, 2010; Caydasi et al., 2010; Bertazzi et al., 2011; Falk et al., 2011). However, a detailed analysis of these mutants was difficult because we lacked a system that allowed us to generate synchronized populations of cells harboring mispositioned anaphase spindles. Deletion of either the KAR9 or the DYN1 pathway leads to only transient spindle mispositioning that is quickly corrected. Deletion of both genes causes high levels of spindle mispositioning but is lethal, and good conditional alleles for either gene were not available. To address this experimental limitation, we developed a system that allowed us to conditionally inactivate both spindle-positioning pathways. We generated cells that lacked KAR9 and harbored a depletion allele of DYN1 (dyn1-AID). To deplete cells of dynein, we used the indole-3-acetic acid (IAA; auxin) depletion system (Nishimura et al., 2009). IAA promotes the degradation of proteins containing an AID degron sequence by targeting them for ubiquitinylation by the SCF-Tir1 ubiquitin ligase (Gray et al., 2001; Dharmasiri et al., 2005; Kepinski and Leyser, 2005; Teale et al., 2006). The vast majority of kar9Δ cells depleted for dynein misposition their spindle upon entry into anaphase (Figure 1C). Thus this system allowed us to examine carefully the consequences of spindle mispositioning in SPoC mutants.
SPoC mutants vary in their checkpoint competency
Many genes have been identified whose inactivation leads to inappropriate mitotic exit in cells with mispositioned spindles. One way to measure the degree of checkpoint deficiency is to induce spindle mispositioning and then determine the percentage of multinucleate cells. Using the kar9Δ dyn1-AID system, we found that most SPoC mutants exhibited varying degrees of checkpoint competency. We arrested cells in the G1 stage of the cell cycle with α-factor pheromone and released them into the cell cycle in the presence of IAA to deplete dynein. This analysis showed that >50% of bub2∆ cells exited mitosis inappropriately and formed multinucleated cells (Figure 2A). Cells lacking KIN4 or RTS1 produce fewer multinucleate cells, indicating that SPOC activity is partially retained. In contrast, swe1∆ or bmh1Δ mutants, which were previously reported to harbor mild checkpoint defects (Caydasi et al., 2014; Moore et al., 2010), formed few if any multinucleate cells in our experimental setup (Figure 2A).
FIGURE 2:
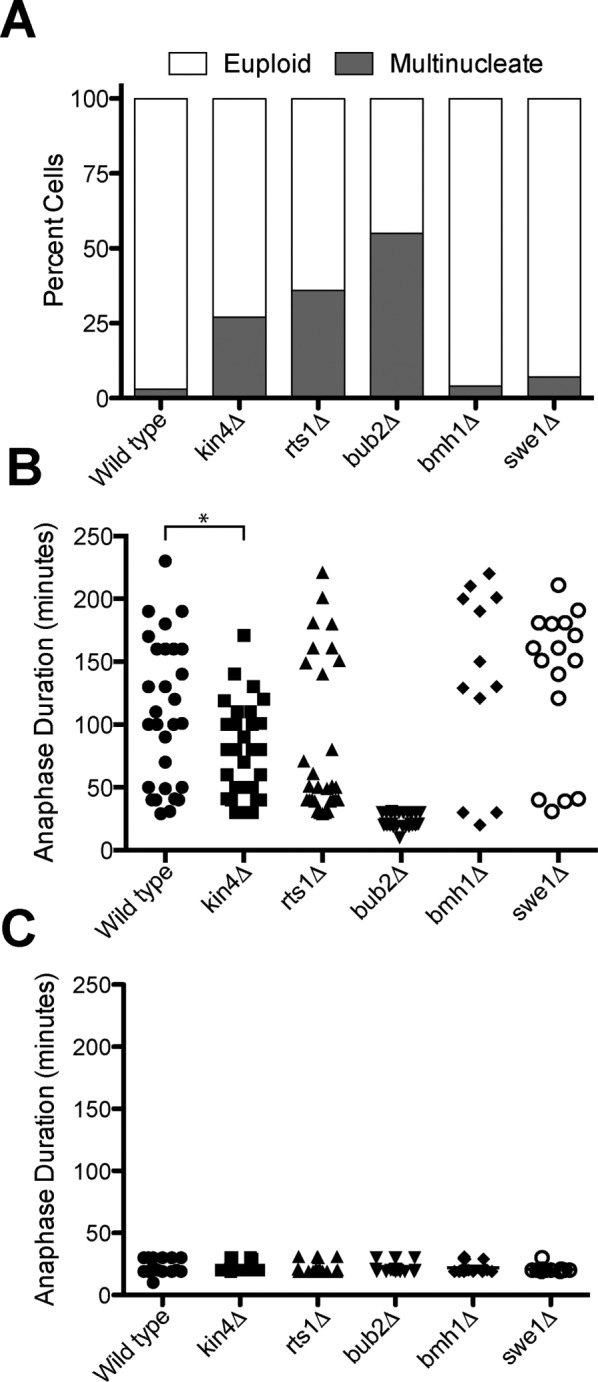
SPoC mutants retain some checkpoint activity. dyn1-AID kar9Δ (A35707), dyn1-AID kar9Δ kin4Δ (A35603), dyn1-AID kar9Δ rts1Δ (A37483), dyn1-AID kar9Δ bmh1Δ (A36544), dyn1-AID kar9Δ swe1Δ (A35146), and dyn1-AID kar9Δ bub2Δ (A36082) cells carrying GFP-tagged α-tubulin were grown in YEPD medium and arrested in the G1 phase of the cell cycle with 10 μg/ml α-factor pheromone. Cells were released into the cell cycle in YEPD medium and then monitored by live-cell microscopy in a Y04C flow cell. Depletion of dyn1-AID was induced in the flow cell with 100 μM auxin in synthetic complete medium. (A) Fraction of cells that become multinucleate. Number of nuclei was determined upon completion of the first cell cycle after factor release (n = 100). Cells that inappropriately exited from mitosis in the mother cell compartment and hence harbored two nuclei were scored as “multinucleate.” Cells that either arrested permanently with mispositioned spindles in the mother cell compartment or exited mitosis with aligned spindles were scores as “euploid.” (B, C) Anaphase duration was determined in cells that exit from mitosis with mispositioned (B) or correctly positioned (C) spindles. Sample sizes for B: dyn1-AID kar9Δ, 29; dyn1-AID kar9Δ kin4, 31; dyn1-AID kar9Δ rts1Δ, 30; dyn1-AID kar9Δ bmh1Δ, 12; dyn1-AID kar9Δ swe1Δ, 16; and dyn1-AID kar9Δ bub2Δ, 30. The p value in B is <0.05 (two-sided t test). Sample sizes for C: dyn1-AID kar9Δ, 18; dyn1-AID kar9Δ kin4, 20; dyn1-AID kar9Δ rts1Δ, 17; dyn1-AID kar9Δ bmh1Δ, 20; dyn1-AID kar9Δ swe1Δ, 20; and dyn1-AID kar9Δ bub2Δ, 19.
To examine specifically the fate of cells that inappropriately exit from mitosis when their spindle is mispositioned, we measured anaphase duration in these cells. In otherwise wild-type kar9Δ dyn1-AID cells, only a very small percentage of cells (∼4–11%; Adames et al., 2001; D’Aquino et al., 2005; Pereira and Schiebel, 2005; Falk et al., 2016) will inappropriately exit from mitosis when spindle mispositioning occurs. Even these cells, however, arrested in anaphase for prolonged periods of time before inappropriate exit from mitosis occurred compared with cells that underwent anaphase with correctly positioned spindles (compare Figure 2, B and C). In contrast, in cells lacking BUB2, anaphase took approximately the same amount of time in cells with mispositioned spindles as in cells with correctly positioned spindles (aligned 21.95 ± 4.33 vs. mispositioned 23.8 ± 5.6 min; Figure 2, B and C). In contrast, in the other checkpoint mutants, anaphase duration of cells that inappropriately exited from mitosis with mispositioned spindle was significantly longer than anaphase length of cells that harbored an aligned spindle (Figure 2, B and C). Perhaps the most surprising finding was that many cells with mispositioned spindles that lacked KIN4 were also extremely delayed in anaphase before inappropriately exiting mitosis (Figure 2, B and C). Our findings indicate that all SPoC mutants retain some SPoC activity. In contrast, hyperactivation of the MEN by eliminating the GAP makes cells completely insensitive to spindle position.
Modulating FEAR network activity differentiates between SPoC and MEN GAP mutants
The MEN is not the only pathway known to regulate Cdc14 localization and activity. The Cdc14 early anaphase release (FEAR) network is responsible for the initial release of Cdc14 from the nucleolus at the metaphase-to-anaphase transition (Stegmeier and Amon, 2004; reviewed in Rock and Amon, 2009). This release is transient and not essential for exit from mitosis but facilitates rDNA compaction, spindle midzone assembly, and priming of the MEN for its subsequent activation later in anaphase (Stegmeier and Amon, 2004; Rock and Amon, 2009). Of importance, previous studies also demonstrated that FEAR network function affects the strength of the spindle position checkpoint. The checkpoint arrest is inherently leaky in wild-type cells, with ∼4–11% of cells with mispositioned spindles exiting mitosis inappropriately (Adames et al., 2001; D’Aquino et al., 2005; Pereira and Schiebel, 2005). In contrast, in FEAR network mutants, SPoC arrest is complete. FEAR network mutants never exit from mitosis when their spindles are mispositioned (Scarfone et al., 2015; Falk et al., 2016; Gryaznova et al., 2016). This observation prompted us to investigate whether FEAR network activity would differentially affect the response of SPoC mutants and MEN GAP mutants to spindle mispositioning. For this analysis, we focused on bub2∆ and kin4∆ mutants because they are the best-characterized SPoC mutants and both have a severe SPoC defect. To inactivate the FEAR network, we deleted SPO12.
Remarkably, our double-mutant analysis showed that whereas spo12∆ bub2∆ mutants inappropriately exited mitosis when the spindle was mispositioned and produced multinucleate cells, spo12∆ kin4∆ mutants remained arrested in anaphase and did not produce multinucleate cells (Figure 3A). Consistent with this anaphase arrest, spo12∆ kin4∆ mutants with mispositioned spindles did not activate the MEN. Association of Dbf2–enhanced green fluorescent protein (eGFP) with both spindle pole bodies—a hallmark for MEN activation (Visintin and Amon, 2001)—occurred in kin4∆ cells with mispositioned spindles but did not take place in spo12∆ kin4∆ cells with mispositioned spindles (Figure 3B). We did note that a small fraction of spo12∆ single mutants exhibited weak Dbf2 localization on one SPB (Figure 3B). This association does not signify MEN activation and is likely due to hyperaccumulation of Dbf2 in cells that arrest/delay in mitosis, as occurs in the spo12∆ mutant (Visintin and Amon, 2001). We conclude that inactivation of the FEAR network suppresses the SPoC defect of cells lacking KIN4 but not of cells lacking BUB2.
FIGURE 3:
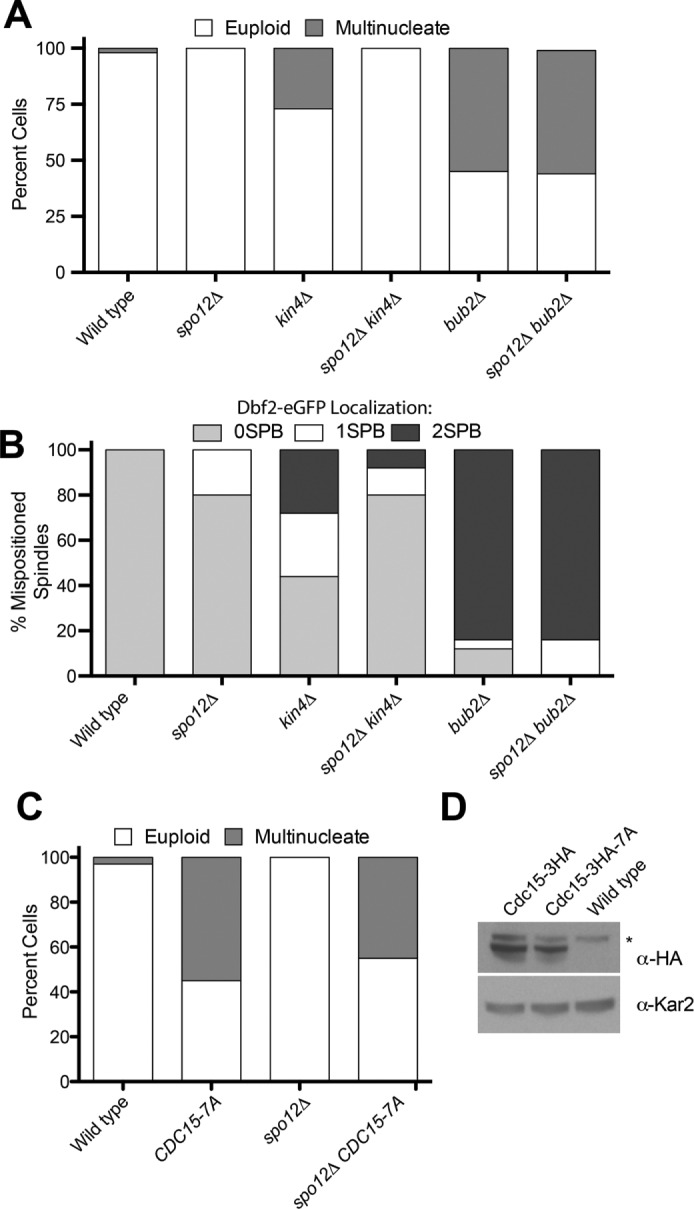
Inactivation of the FEAR network suppresses the SPoC defect of kin4Δ but not bub2Δ mutants. (A) Percentage multinucleate cells generated after one cell cycle. Cells were released from an α-factor pheromone–induced G1 arrest, and the percentage of multinucleate cells was assessed upon completion of the first cell cycle. Cells were scored as in Figure 2A. dyn1-AID kar9Δ (A35707), dyn1-AID kar9Δ spo12Δ (A35700), dyn1-AID kar9Δ kin4Δ (A35603), dyn1-AID kar9Δ spo12Δ kin4Δ (A36048), dyn1-AID kar9Δ bub2Δ (A36082), and dyn1-AID kar9Δ spo12Δ bub2Δ (A36965) cells harboring GFP-tagged α-tubulin were grown as described in Figure 2. Cells were released into the cell cycle in YEPD medium and then monitored by live-cell microscopy. Depletion of dyn1-AID was induced with 100 μM auxin in synthetic complete medium (n = 100). Note that dyn1-AID kar9Δ bubΔ (A36082) and dyn1-AID kar9Δ kin4Δ (A35603) are shown again from Figure 2A. (B) Dbf2-eGFP localization to SPBs in cells with mispositioned spindles in dyn1Δ (A36852), dyn1Δ spo12Δ (37134), dyn1Δ kin4Δ (A36782), dyn1Δ kin4Δ spo12Δ (A36851), dyn1Δ bub2Δ (A36850), and dyn1Δ spo12Δ bub2Δ (A36722) strains. Cells were incubated for 24 h at 14°C in YPD medium supplemented with 0.3 mM adenine hemisulfate, 0.4 mM l-tryptophan, 1 mM l-histidine, 0.2 mM uracil, and 1 mM l-leucine to induce spindle mispositioning. Cells were then fixed in 4% PFA and imaged. All strains expressed mCherry-tagged α-tubulin to mark the spindle. (C) Percentage of multinucleated and euploid cells was assessed in CDC15 (A38964), CDC15-7A (A38963), spo12Δ CDC15 (A38294), and spo12Δ CDC15-7A (A38290) strains that had mispositioned their spindles. Spindle mispositioning was induced as in Figure 2, and cells were analyzed as in Figure 2A. (D) Western blot analysis of whole-cell lysates to examine the expression of Cdc15-HA3 (A38294), Cdc15-HA3-7A (A38290), and the wild-type untagged control (A2587) strain. Asterisk indicates a cross-reacting band. Kar2 served as a loading control.
The FEAR network, while not essential for MEN activity, primes the pathway because Cdc14 released by the FEAR network dephosphorylates MEN components such as Cdc15 (Stegmeier et al., 2002). To determine whether the FEAR network promoted SPoC bypass by priming the MEN, we asked whether mimicking Cdc14-mediated dephosphorylation of Cdc15 allowed cells with mispositioned spindles to exit from mitosis. We replaced CDC15 with the CDC15-7A allele, in which phosphorylation sites targeted by Cdc14 are replaced by alanines (Jaspersen and Morgan, 2000; König et al., 2010). Remarkably, the CDC15-7A allele caused not only wild-type cells with mispositioned spindles to inappropriately exit from mitosis but also spo12∆ cells (Figure 3, C and D). We conclude that the FEAR network primes MEN activity, which counteracts SPoC-mediated inhibition of the MEN.
Loss of BUB2 but not KIN4 is sufficient to activate the MEN in metaphase
Further evidence that Bub2-Bfa1 and Kin4 exhibit differential inhibitory effects on the MEN came from our analysis of MEN regulation during metaphase. MEN activity is inhibited in cells arrested in metaphase (Visintin and Amon, 2001). Deletion of BUB2 causes activation of the MEN in metaphase, as judged by Dbf2-associated kinase activity (Figure 4; Visintin and Amon, 2001). We synchronized cells in G1 using pheromone and then released them into the cell cycle in the presence of the microtubule depolymerizing agent nocodazole. Nocodazole-treated cells arrest in metaphase due to activation of the spindle assembly checkpoint and Dbf2 kinase is not active (Figure 4, A and B). In contrast, Dbf2 kinase activity is greatly increased in cells lacking BUB2 (Figure 4). Deletion of KIN4, however, did not lead to activation of Dbf2 in metaphase-arrested cells. We conclude that inactivation of the MEN GAP causes complete deregulation of the MEN, whereas inactivation of the mother cell restricted MEN inhibitor Kin4 causes only partial activation of the MEN. This finding indicates that additional MEN repressors and/or activators control the activity of the signaling pathway in response to spindle mispositioning.
FIGURE 4:

Cells lacking BUB2 but not KIN4 activate Dbf2 in metaphase. Wild-type (A1931), bub2Δ (A2472), and kin4Δ (A35432) cells containing a 3Myc-Dbf2 fusion were released from a pheromone-induced G1 arrest into medium containing nocodazole (15 μg/ml). After 80 min, α-factor pheromone was readded to prevent entry into the subsequent cell cycle. The amount of Dbf2-asssociated kinase activity (Dbf2 kinase) and immunoprecipitated 3MYC-Dbf2 (Dbf2-IP) was determined at the indicated times and then quantified (A, B). Synchrony of the release and progression through the cell cycle were determined by bud morphology (C).
Lte1 promotes exit from mitosis in a KIN4-dependent and -independent manner
Anaphase takes the same amount of time in bub2∆ cells whether or not the spindle is correctly positioned (∼22 min). The fact that anaphase duration is on average 76.2 ± 37.9 min in kin4∆ cells that exit from mitosis with incorrectly positioned spindles compared with 23.4 ± 4.77 min in kin4∆ cells with correctly position spindles (Figure 5A) indicates that there must be at least one additional regulator of the MEN. Such a regulator could be an activator of the MEN in the bud or a MEN inhibitor in the mother cell compartment. These two possibilities are of course not mutually exclusive.
FIGURE 5:

A Kin4-independent function for Lte1 in promoting exit from mitosis. (A) Rationale for why additional spatial regulators of the MEN must exist. Spindle position is not sensed by cells lacking BUB2, as indicated by the almost identical anaphase kinetics of cells that exit from mitosis with a mispositioned spindle and cells that exit from mitosis with an aligned spindle. In contrast, exit from mitosis is severely delayed in cells with a mispositioned spindle that lack KIN4 compared with kin4Δ cells with an aligned spindle. (B–G) Cells harboring GFP-tagged α-tubulin were arrested G1 with α-factor pheromone (10 μg/ml). Cells were then released into YEPD medium without α-factor but supplemented with 0.3 mM adenine hemisulfate, 0.4 mM l-tryptophan, 1 mM l-histidine, 0.2 mM uracil, and 1 mM l-leucine at 14°C. Fixed-cell fluorescence microscopy was performed, and cell cycle stage was assessed by spindle morphology. The following strains were analyzed. (B, C) Wild type (A33138), bub2Δ (A36608), kin4Δ (A38141), lte1Δ (A36718), and spo12Δ (A36605). (D, E) Wild type (A33138), kin4Δ spo12Δ (A36620), kin4Δ spo12Δ lte1Δ (A36607), spo12Δ bub2Δ (A36604), and bub2Δ spo12Δ lte1Δ (A36603). (F, G) Wild type (A33138), kin4Δ (A38141), lte1Δ (A36718), and kin4Δ lte1Δ (A38140). (B, D, F) Percentage of cells with metaphase spindles. (C, E, G) Percentage of anaphase spindles (n = 200).
One of the proteins that could regulate the MEN in parallel to Kin4 is Lte1. Cells lacking LTE1 arrest in anaphase at low temperature and exhibit synthetic lethality with deletions in genes encoding FEAR network components such as SPO12 (Shirayama et al., 1994; Stegmeier et al., 2002). Previous studies indicated that one function of Lte1 is to prevent small amounts of Kin4 that leak into the bud from associating with the MEN-bearing SPB in the bud (Bertazzi et al., 2011; Falk et al., 2011). To determine whether Lte1 has additional mitotic exit–promoting functions besides inhibiting Kin4 in the bud, we first asked whether cells lacking LTE1 were delayed in anaphase in the absence of KIN4. Cells were released from a pheromone-induced G1 arrest, and progression through mitosis at 14°C was analyzed. Consistent with previous data, we found that cells lacking LTE1 were severely delayed in exit from mitosis, as judged by the persistence of anaphase spindles (Figure 5C). Deleting KIN4 completely suppressed this anaphase delay (Figure 5G), which is consistent with previous findings showing that Lte1 antagonizes Kin4 function (Bertazzi et al., 2011; Falk et al., 2011). However, the analysis of cells in which mitotic exit solely depends on the MEN (in a FEAR network mutant) revealed a function of LTE1 besides inhibiting Kin4. kin4∆ spo12∆ lte1∆ cells were much more delayed in anaphase than kin4∆ spo12∆ cells (Figure 5E). In contrast, anaphase duration was nearly wild type in bub2∆ spo12∆ lte1∆ cells and identical to that of bub2∆ spo12∆ cells (Figure 5E). We conclude that by increasing the reliance of mitotic exit on the MEN (by inactivating the FEAR network), we were able to reveal a KIN4-independent function of LTE1 in promoting mitotic exit.
If Lte1 has a mitotic exit–promoting function besides inhibiting Kin4, targeting Lte1 to the mother cell should be more effective in triggering exit from mitosis in cells with mispositioned spindles than deleting KIN4. To test this hypothesis, we examined the consequences of expressing the LTE1-8N allele, which mislocalizes to the mother cell compartment (Geymonat et al., 2009), on anaphase duration in cells with mispositioned spindles. We found that cells harboring the LTE1-8N allele, like cells lacking BUB2, showed little or no anaphase delay when their spindles were mispositioned. Instead, such cells underwent exit from mitosis to produce multinucleate and anucleate cells (Figure 6, A and B). Like deletion of BUB2, LTE1-8N was able to trigger exit from mitosis in cells with mispositioned spindles even in the absence of FEAR network activity (Figure 6C). We conclude that LTE1 promotes exit from mitosis by activating the MEN in multiple ways. One of Lte1’s functions is to inhibit Kin4 (Bertazzi et al., 2011; Falk et al., 2011). The other function remains to be characterized.
FIGURE 6:
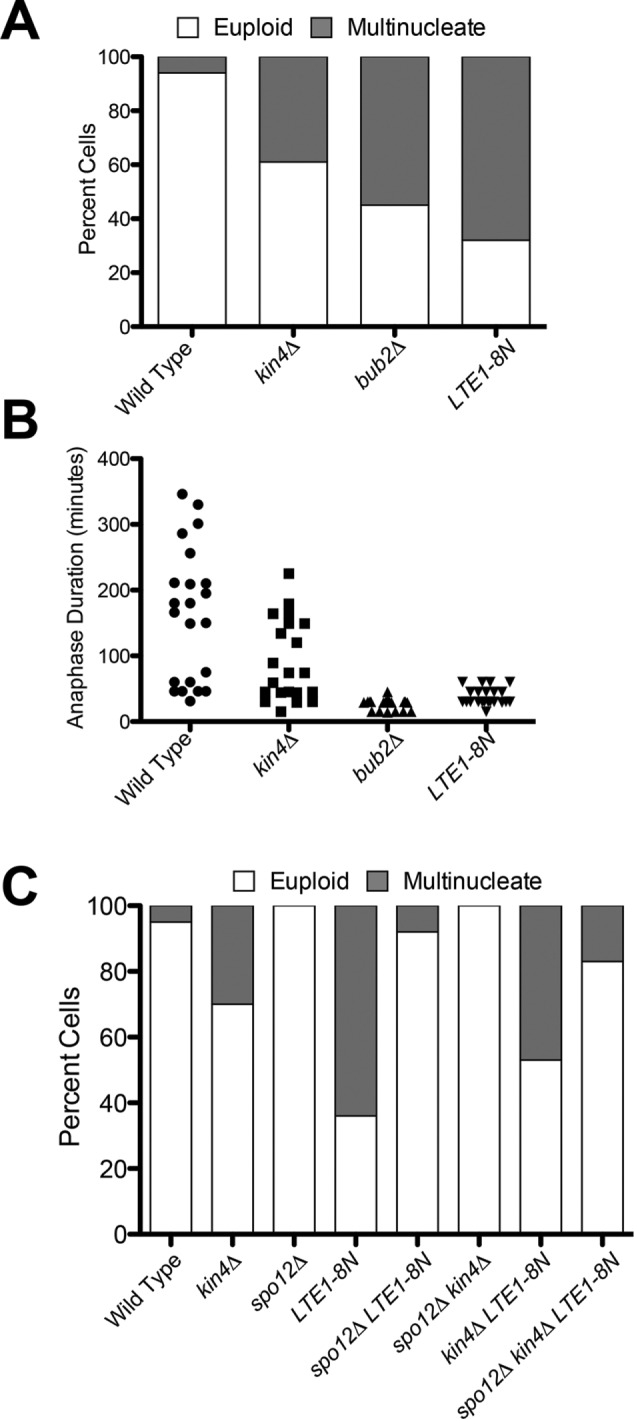
Mislocalizing Lte1 to the mother cell compartment bypasses the SPoC even in the absence of the FEAR. (A) The percentage of multinucleate cells was assessed in dyn1-AID kar9Δ (A35707), dyn1-AID kar9Δ kin4Δ (A35603), dyn1-AID kar9Δ bub2Δ (A36082), and dyn1-AID kar9Δ LTE1-8N (A36357) strains. Cells were cultured as in Figure 2. Spindle mispositioning was scored as in Figure 2A (n = 100). (B) Analysis of anaphase duration in cells that inappropriately exit from mitosis in the mother cell compartment. Cells from A were reanalyzed, and anaphase length was determined by spindle morphology (n = 22). (C) The percentage of multinucleate cells was assessed in dyn1-AID kar9Δ (A35707), dyn1-AID kar9Δ kin4Δ (A35603), dyn1-AID kar9Δ spo12 (A35700), dyn1-AID kar9Δ LTE1-8N (A36357), dyn1-AID kar9Δ LTE1-8N spo12Δ (A36358), dyn1-AID kar9Δ spo12Δ kin4Δ (A36048), dyn1-AID kar9Δ kin4Δ LTE1-8N (A36268), and dyn1-AID kar9Δ LTE1-8N kin4 Δ spo12Δ (A29611) strains. Cells were cultured as in Figure 2. Spindle mispositioning was scored as in Figure 2A (n = 100). Note that the anaphase duration of cells with aligned spindles in the LTE1-8N spo12Δ strain (A36358) was delayed in comparison to spo12Δ cells. This is most likely due to the fact that Lte1-8N is expressed off of its endogenous promoter and yet is distributed on both cortices. Therefore Lte1-8N is essentially depleted in the bud, and so, under sensitizing conditions (such as in the absence of FEAR network activity), cells are delayed in anaphase.
Lte1’s GEF domains promote exit from mitosis
Lte1 harbors two putative GEF domains, but whether the protein functions as a Tem1 GEF is not known. In fact, several lines of evidence support the idea that Lte1 is not a GEF for Tem1. Purified Lte1 does not exhibit GEF activity toward Tem1 in vitro (Geymonat et al., 2002). Furthermore, the Lte1 GEF domains were shown to be primarily required to anchor Lte1 to the bud cell cortex via interactions with Ras (Yoshida et al., 2003). Finally, it was reported that Lte1’s two GEF domains are dispensable for promoting exit from mitosis (Yoshida et al., 2003). Despite these multiple reports arguing against a role for Lte1’s GEF domains in MEN regulation, we decided to reevaluate their role in exit from mitosis.
Lte1 inhibits Kin4 (Bertazzi et al., 2011; Falk et al., 2011), but whether Lte1’s GEF domains were important for Kin4 inhibition was not known. To address this question, we analyzed an allele of LTE1, lte1-ΔEcoRI, which lacks amino acids 327–894 but retains both the N- and C-terminal GEF domains (Geymonat et al., 2009). A previous study showed that this allele was not able to support Lte1’s mitotic exit–promoting functions, suggesting that the central 567 amino acids were essential for LTE1 function (Geymonat et al., 2009). Consistent with earlier reports, we found that lte1-ΔEcoRI-GFP was well expressed and localized to the bud cell cortex (Figure 7B; Geymonat et al., 2009). In fact, the lte1-ΔEcoRI-GFP mutant protein localized to the bud cell cortex even more robustly than wild-type Lte1 (A.S., unpublished observations).
FIGURE 7:
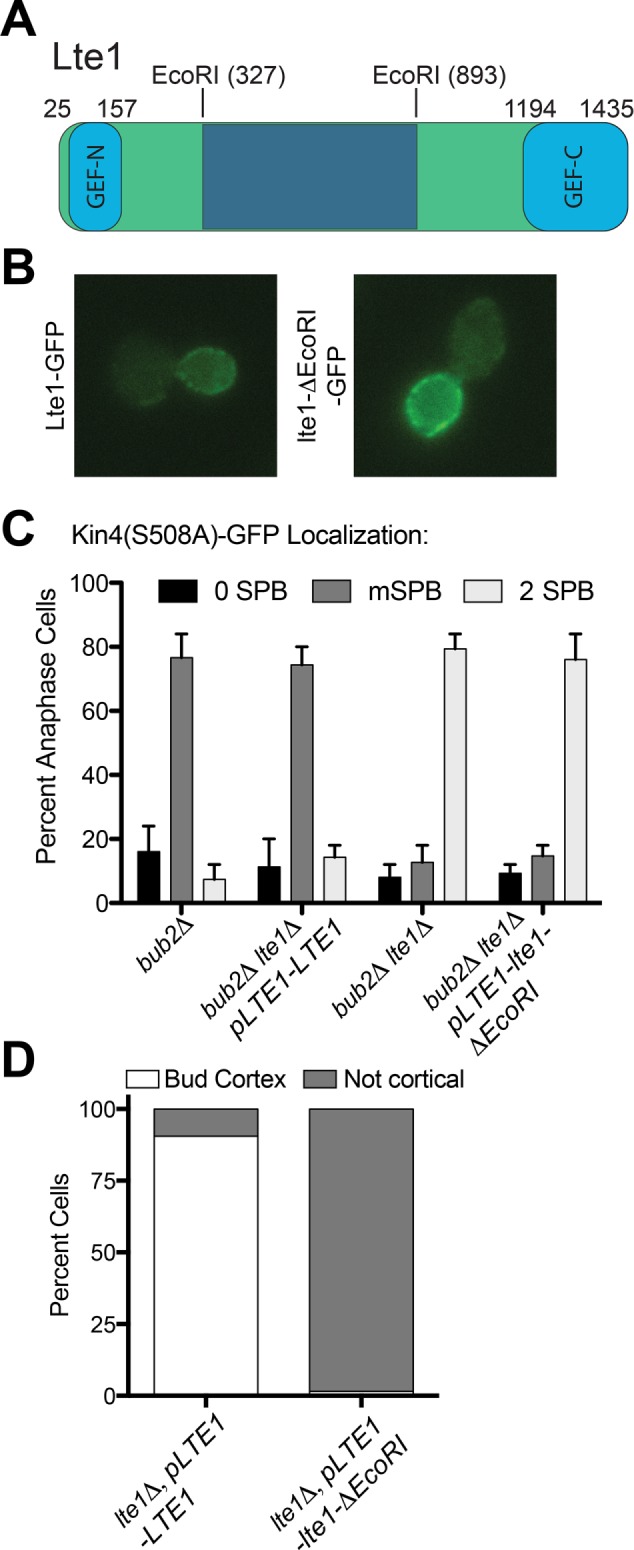
The central domain of Lte1 binds to and inhibits Kin4. (A) Domain structure of Lte1 and position of the EcoRI sites that depict the boundaries of the EcoRI deletion allele. (B) Localization of GFP-tagged Lte1 (AS184) and GFP-tagged lte1-ΔEcoRI (AS351). Log-phase cells were fixed in 4% PFA and then imaged. Image acquisition conditions were the same for both strains. (C) Cells expressing GFP-tagged Kin4-S508A and Tub1-mCherry were arrested in G1 with 10 μg/ml α-factor pheromone. G1-arrested cells were then released into pheromone-free YEPD medium. Kin4(S508A)-GFP localization to SPBs was examined in late anaphase cells with the following genetic backgrounds: bub2∆ (A26874), bub2∆, lte1∆ (A26170), bub2∆, lte1∆, pLTE1-LTE1 (A38199), and bub2∆, lte1∆, pLTE1-lte1DecoRI (A38120). n ≥ 45. Error bars represent the range. (D) Wild-type (AS365) and lte1ΔEcoRI (AS367) cells containing the kin4(1-341)-GFP truncation were grown to log phase, and Kin4(1-341)-GFP localization was examined.
To assess the importance of Lte1’s GEF domains, we first asked whether the lte1-ΔEcoRI allele was able to prevent Kin4 from localizing to SPBs. To address this question, we used an allele of KIN4 (KIN4-S508A) that localizes to both the mother and bud cell compartments and thus efficiently associates with the SPB in the daughter cell in the absence of LTE1 (Chan and Amon, 2010; Falk et al., 2011). Association of Kin4 with the daughter cell SPB (henceforth dSPB) prevents exit from mitosis, which made it necessary to conduct this analysis in cells lacking BUB2 (Chan and Amon, 2010; Falk et al., 2011). Consistent with previous observations, we found that Kin4-S508A-GFP localized to both the mother cell SPB (henceforth mSPB) and dSPB in cells lacking LTE1 (lte1Δ bub2Δ strain; 2 SPB category; Figure 7C). Cells expressing the lte1-ΔEcoRI allele exhibited a similar localization pattern (Figure 7C). This finding indicates that Lte1’s central domain is required for preventing Kin4 from associating with SPBs.
Not only does Lte1 prevent Kin4 from associating with SPBs, but the protein also binds to Kin4 (Bertazzi et al., 2011; Falk et al., 2011). This is most easily observed when analyzing the localization pattern of the kin4(1-341) truncation. This truncated protein exhibits the same localization pattern as Lte1, and its bud localization depends on LTE1 (Falk et al., 2011). The bud cortex localization of kin4(1-341) was abrogated in cells expressing the lte1-ΔEcoRI allele (Figure 7D). Consistent with the observation that the lte1-ΔEcoRI allele was defective in interacting with and inhibiting Kin4 was the observation that KIN4-S508A became very toxic to cells in which lte1-ΔEcoRI allele is the sole copy of LTE1 and caused cells to delay in anaphase (Figure 8, A and C). Together these results indicate that the central domain of Lte1 binds to and inhibits Kin4 thereby preventing it from associating with dSPBs.
FIGURE 8:
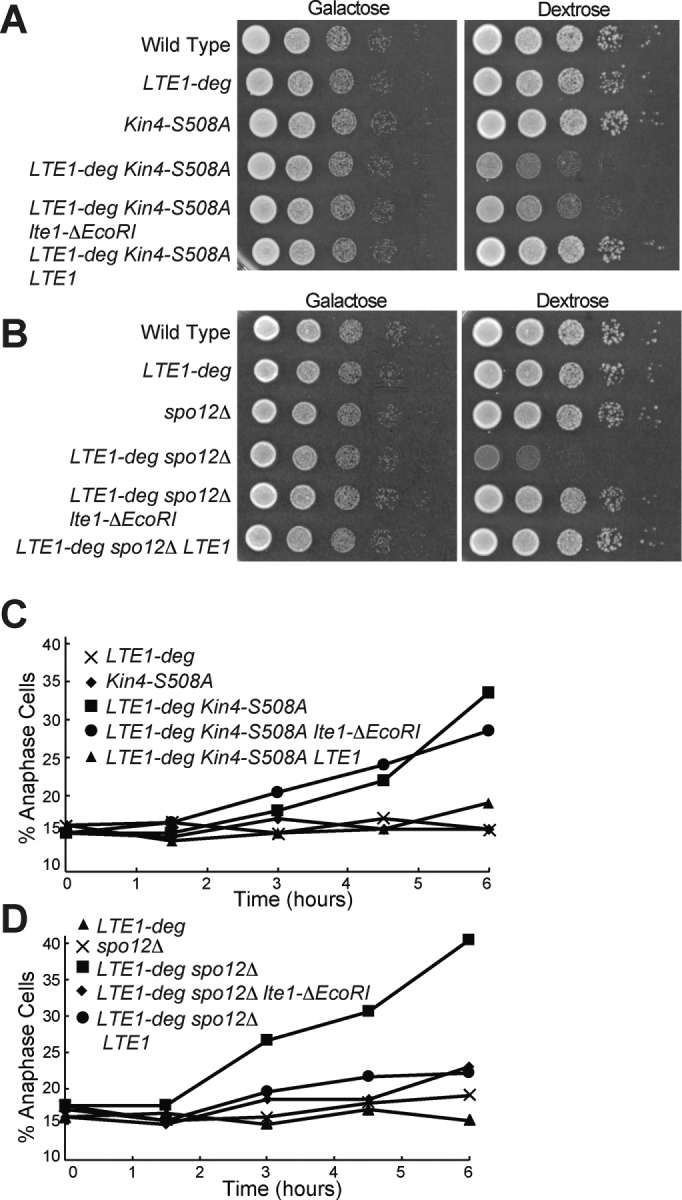
The GEF domains of Lte1 promote exit from mitosis. (A, B) Tenfold serial dilutions of overnight cultures of wild type (A2587), pGAL1-10-URL-3HA-LTE1 (A23686), KIN4-S508A (A21299), pGAL1-10-URL-3HA-LTE1 KIN4-S508A (A24084), pGAL1-10-URL-3HA-LTE1 KIN4-S508A pLTE1-lte1ΔEcoRI (AS336), pGAL1-10-URL-3HA-LTE1 KIN4-S508A pLTE1-LTE1 (A38247), spo12Δ (A4874), pGAL1-10-URL-3HA-LTE1 spo12Δ (A24543), pGAL1-10-URL-3HA-LTE1 spo12Δ pLTE1-lte1ΔEcoRI (AS338), and pGAL1-10-URL-3HA-LTE1 spo12Δ pLTE1-LTE1 (A38260) cells were spotted onto YEP plates containing either raffinose and galactose or glucose and incubated at 30°C. (C, D) pGAL1-10-URL-3HA-LTE1 (A23686), KIN4-S508A (A21299), pGAL1-10-URL-3HA-LTE1 KIN4-S508A (A24084), pGAL1-10-URL-3HA-LTE1 KIN4-S508A, pLTE1-lte1ΔEcoRI (AS336), pGAL1-10-URL-3HA-LTE1 KIN4-S508A pLTE1-LTE1 (A38247), spo12Δ (A4874), pGAL1-10-URL-3HA-LTE1 spo12Δ (A24543), pGAL1-10-URL-3HA-LTE1 spo12Δ pLTE1-lte1ΔEcoRI (AS338), and pGAL1-10-URL-3HA-LTE1 spo12Δ pLTE1-LTE1 (A38260) cells were grown overnight in YEP medium with raffinose and galactose to log phase and diluted into YEPD medium. Cells were grown for 6 h at 30°C, and samples were taken at the indicated times to determine the percentage of anaphase cells (n = 200). Note that the data for C and D are from the same experiment, and so the pGAL1-10-URL-3HA-LTE1 data from C are also shown in D.
We next asked whether the lte1-ΔEcoRI allele suppressed the synthetic lethality of spo12Δ lte1Δ double mutants. In this mutant, unlike in the KIN4-S508A mutant, little Kin4 is present in the daughter cell, making the central domain of Lte1 less important and thus allowing us to observe potential additional functions for the GEF domains of Lte1. Indeed, we found that the lte1-ΔEcoRI allele suppressed the lethality of spo12Δ lte1Δ double mutants to the same extent as wild-type LTE1 (Figure 8, B and D). Furthermore, an lte1-∆EcoRI lte1∆ spo12∆ strain is viable not only at 30°C but even at low temperatures (Supplemental Figure S1). These results indicate that under conditions in which Kin4 localization is largely restricted to the mother cell, Lte1’s GEF domains are sufficient to fulfill LTE1’s mitotic exit–promoting functions. This observation leads us to conclude that Lte1’s GEF domains are important for promoting MEN activity and hence exit from mitosis.
DISCUSSION
Spindle position controls the activity of the MEN. Previous studies suggested that this was solely mediated by a MEN inhibitor residing in the mother cell and an antagonist of this inhibitor in the bud. Here we show that a bud-localized activating signal also controls MEN activity. We find that cells lacking the mother cell localized SPoC component Kin4, unlike cells lacking MEN GAP function, retain partial SPoC competency. We further show that this is due to the fact that an activating bud-localized signal is necessary to fully activate the MEN. This MEN activator is LTE1, which we show not only prevents Kin4 from associating with the MEN-bearing SPB in the bud, but also activates the MEN in additional ways. On the basis of the observation that this MEN-activating function depends on the protein’s GEF activity, we propose that Lte1 has a dual function in activating the MEN. Via its central domain, the protein antagonizes Kin4. Through its N-terminal and C-terminal GEF domains, the protein directly activates the MEN GTPase Tem1.
Spindle position controls MEN activity through an inhibitor in the mother cell and an activator in the bud
We used a new spindle-mispositioning system to perform an in-depth analysis of factors that have been identified as components of the spindle position checkpoint machinery. By analyzing the length of anaphase in cells with mispositioned spindles, we discovered that cells lacking BUB2 are completely SPoC deficient. In contrast, cells with mispositioned spindles that lack KIN4 exhibit a transient delay in anaphase before exiting mitosis prematurely. Similarly, other SPoC mutants were also delayed in anaphase and produced even fewer multinucleate cells than kin4Δ cells.
The difference between MEN mutants that have lost all regulation (bub2Δ) and mutants that fail to regulate the MEN GAP in response to spindle mispositioning (kin4Δ) was striking in cells lacking FEAR network function. In this genetic background, the phenotype was binary; kin4Δ cells with mispositioned spindles arrested in anaphase, whereas bub2Δ cells with mispositioned spindles exited from mitosis as quickly as cells with correctly positioned spindles. Together these findings demonstrate that other factors restrain exit from mitosis in response to spindle mispositioning in kin4Δ cells. Residual SPoC activity in kin4Δ strains could mean that there is still some residual GAP activity that prevents full Tem1 activation. Alternatively, pathways acting in parallel to KIN4 could be required for the full activation of Tem1. Our analysis of LTE1 mutants suggests the latter.
Lte1 promotes exit from mitosis in multiple ways
Lte1 harbors homology to guanine nucleotide exchange factors (Shirayama et al., 1994). This observation previously led to the proposal that Lte1 may function as a GEF for Tem1 (Bardin et al., 2000). However, subsequent studies suggested that this was not the case. First, truncations of LTE1’s GEF domains were shown to not affect the gene’s ability to promote exit from mitosis when overexpressed (Yoshida et al., 2003). In contrast, deletion of LTE1’s central domain was shown to be essential for exit from mitosis (Yoshida et al., 2003). Finally, Lte1 purified from yeast cells did not exhibit GEF activity toward Tem1 in vitro (Geymonat et al., 2009). These observations, together with the finding that Lte1 prevented the small amounts of Kin4 that are present in the bud from associating with SPBs in the bud (Bertazzi et al., 2011; Falk et al., 2011), led to the conclusion that although Lte1 has homology to GEFs, it actually does not function as a MEN GEF. Instead, its GEF domains were proposed to anchor Lte1 at the bud cortex by binding to Ras (Yoshida et al., 2003; Seshan and Amon, 2005).
Our results show that Lte1 has functions besides inhibiting bud-localized Kin4 from associating with SPBs. kin4Δ spo12Δ lte1Δ cells exhibit a significantly longer anaphase delay than kin4Δ spo12Δ cells. In support of the idea that Lte1’s GEF domains provide this additional function, we find that a LTE1 mutant allele that contains only the protein’s GEF domains as the sole source of LTE1 in cells is fully capable of performing LTE1’s mitotic exit functions. The lte1-∆EcoRI mutant suppressed the lethality of a lte1Δ spo12Δ double mutant.
We find that whereas the GEF domains of LTE1 are essential for the protein’s mitotic exit–promoting functions under all growth conditions, the central domain of Lte1 becomes important for exit from mitosis only in cells in which large amounts of Kin4 localize to the bud, as is the case for cells with the KIN4-S508A allele. This finding suggests that Lte1’s central domain mediates Kin4 inhibition. Indeed, we find that this region of Lte1 is required for Kin4 binding. We therefore propose that Lte1 not only inhibits Kin4 but also functions as a GEF for Tem1. Indeed, the vast majority of GTPases have been shown to be regulated by GEFs, and so it is not inconceivable that Tem1 is also controlled by such a protein (Bos et al., 2007). However, whereas Lte1’s GEF domains are sufficient to activate the MEN, it is important to note that Lte1 has never been shown to have GEF activity in vitro. Therefore both an in-depth biochemical analysis of Lte1 and further examination of how Lte1’s GEF domains activate the MEN are critical to elucidate the function of Lte1.
Why is cell cycle arrest in response to spindle mispositioning not absolute?
Budding yeast cells determine the site of cytokinesis as soon as they enter the cell cycle and so must work to ensure that each daughter cell ends up with one nucleus after anaphase. Yeast cells have multiple redundant mechanisms to ensure that each daughter cell remains euploid. The first level of control comprises two redundant pathways that align the spindle along the mother–bud axis. However, microtubules are inherently cold sensitive, and so at low temperatures especially, this can lead to spindle mispositioning. When this occurs, an additional layer of control, the so-called spindle position checkpoint, becomes important. This regulatory mechanism buys the cell time during anaphase so that it can thread the anaphase spindle into the bud.
It has long been known that not all cells arrest in anaphase in response to spindle mispositioning. Approximately 4–11% of cells, after an initial short anaphase delay, exit from mitosis, producing binucleate and anucleate cells (Adames et al., 2001; Falk et al., 2016). This observation could have been dismissed as a leakiness of the SPoC, that is, as being the result of suboptimal regulatory mechanisms. However, the observation that the checkpoint arrest is irreversible in FEAR network mutants raises the possibility that there is perhaps purpose to this leakiness. We propose that incomplete penetrance of the checkpoint arrest provides a survival advantage. During conditions that elicit spindle mispositioning and hence prolonged checkpoint arrest, such as low temperatures, it could be beneficial to escape the cell cycle arrest and have another attempt at producing a euploid daughter cell in the next cell division.
Checkpoint leakiness is not unique to the SPoC: cell-to-cell variability in the robustness of the spindle assembly checkpoint and the DNA damage checkpoint has also been reported (Galgoczy and Toczyski, 2001; Musacchio and Salmon, 2007). As in the SPoC, cells that are held in a checkpoint arrest due to DNA damage or lack of spindle tension do not benefit from indefinitely maintaining this arrest. Instead, many cells adapt to the checkpoint stress and continue through the cell cycle still harboring DNA lesions, improperly segregated chromosomes, or the wrong number of nuclei. Understanding the mechanisms that allow some cells but not others to escape the SPoC-mediated anaphase arrest will be an important next step in understanding the MEN and checkpoint control of the cell cycle.
MATERIALS AND METHODS
Strains and plasmids
All yeast strains used in this study were derivatives of W303 (A2587). Strain genotypes are listed in Supplemental Table S1. Culturing conditions are described in the figure legends. The pFA6-3V5-IAA17-KanMx6, pGPD1-OsTIR1-LEU2, and pGPD1-osTIR::HIS3 auxin depletion plasmids were constructed by Leon Chan, Thomas Eng, and Vincent Guacci (D. Koshland and K. Weis labs at the University of California, Berkeley). The CDC15-HA3-LEU2 and CDC15-HA3-7A-LEU2 strains are described in Jaspersen et al. (1998). The GFP-TUB1 construct is described in Straight et al. (1997). The mCherry-TUB1 fusion is described in Khmelinskii et al. (2007).
To generate the pLTE1-LTE1 and pLTE1-lte1-∆EcoRI plasmids, plasmid pNH605-pMET25-LTE1 (p1946) was digested with EcoRI to generate pNH605-pMET25-lte1-∆EcoRI (plasmid 11). Plasmids 1946 and 11 were then digested with Psp0MI and Xho1 to replace the MET25 promoter with the LTE1 promoter to generate pNH605-pLTE1-LTE1I (plasmid 23) and pNH605-pLTE1-lte1-∆EcoRI (plasmid 10). pNH605-pLTE1-LTE1I and pNH605-pLTE1-lte1-∆EcoRI were digested with Pme1 for integration at the LEU2 locus. pNH605-pLTE1-LTE1I and pNH605-pLTE1-lte1-∆EcoRI were GFP-tagged by standard PCR-based techniques (Longtine et al., 1998).
Live-cell imaging
All time-lapse imaging was performed at 25°C using Y04C Cellasic microfluidic chambers (EMD Millipore Billerica, MA). Images were captured using a Zeiss Axio Observer.Z1 inverted microscope (Zeiss, Thornwood, NY) equipped with a Heliophor Pumped Phosphor Light Engine (89 North, Chroma Technology, Bellows Falls, VT) and a Hamamatsu ORCA-ER C4742-80 charge-coupled device camera (Hamamatsu. Middlesex, NJ). Images were acquired and processed with MetaMorph software (Molecular Devices, Sunnyvale, CA).
Kin4 images were acquired with a DeltaVision Elite microscope (GE Healthcare Bio-Sciences, Pittsburgh, PA). Images were taken with a 100× plan-Apo objective, an InsightSSI solid-state light source, and a CoolSNAP HQ2 camera.
Fixed-cell imaging
Cells for Figures 3B and 7B were fixed with 4% paraformaldehyde (PFA) for 3 min, washed with potassium phosphate/sorbitol buffer (1.2 M sorbitol, 0.1 M potassium phosphate, pH 7.5), and treated with 0.05% Triton X-100 for 5 min. Cells were again washed with potassium phosphate/sorbitol buffer, resuspended in potassium phosphate/sorbitol buffer with 4′,6-diamidino-2-phenylindole dihydrochloride, and imaged. Cells for Figure 5 were fixed with 4% PFA for 3 min, washed with potassium phosphate/sorbitol buffer, and then resuspended in potassium phosphate/sorbitol buffer. Imaging for Figure 3B was performed on a Zeiss Axioplan 2 (Zeiss, Thornwood, NY) with a Hamamatsu Orca-R2 (Hamamatsu, Middlesex, NJ) camera and a 63× objective. Images were acquired with Volocity software (PerkinElmer). Imaging for Figure 7B was performed on a DeltaVision Elite microscope (GE Healthcare Bio-Sciences). Images were taken with a 100× plan-Apo objective, an InsightSSI solid-state light source, and a CoolSNAP HQ2 camera. Indirect immunofluorescence for α-tubulin (Tub1) was performed as described previously (Kilmartin and Adams, 1984; Hochwagen et al., 2005).
Dbf2 kinase assay
The kinase activity of Dbf2 was assayed as previously described (Visintin and Amon 2001) with the following modifications. Immunoprecipitation of Dbf2-3Myc was performed on ∼5 mg of total protein with an anti-c-MYC antibody coupled to agarose beads (Sigma-Aldrich) for 90 min at 4°C with rotation. Kinase reactions were performed for 60 min with constant mixing using Histone H1 (Roche) as a substrate. Histone H1 phosphorylation was measured using a Typhoon Trio variable mode imager (GE Healthcare). Immunoblot of total Dbf2-Myc was measured using ECL Plus (GE Healthcare) and fluorescence imaging.
Western blot analysis
Cells were harvested by centrifugation, resuspended in 5% trichloroacetic acid, and incubated for at least 10 min on ice. They were then washed in acetone, and the cell pellet was left to dry. Cells were then lysed in sample buffer (10 mM Tris, pH 7.5, 1 mM EDTA [ethylenediaminetetraacetic acid], 2.5 mM dithiothreitol) using a bead mill and glass beads. SDS sample buffer was added to the lysate, which was then boiled for 5 min. Cdc15 was detected using a mouse anti-hemagglutinin antibody at a 1:500 dilution (HA.11; Covance). A rabbit anti-Kar2 antibody was used as a loading control and used at 1:200,000.
Supplementary Material
Acknowledgments
We are grateful to members of the Amon lab for critical reading of the manuscript. We also thank Matthew Powers for technical support. We thank Vincent Guacci, Leon Chan, and Thomas Eng for plasmids. The anti-Kar2 antibody was a gift from Mark Rose (Princeton University). This work was funded by National Institutes of Health Grant HD085866 to A.A. and supported in part by Koch Institute Support (Core) Grant P30-CA14051 from the National Cancer Institute. A.A. is an investigator of the Howard Hughes Medical Institute and of the Paul F. Glenn Foundation for Biomedical Research.
Abbreviations used:
- FEAR
Cdc14 early anaphase release network
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- MEN
mitotic exit network
- SPB
spindle pole body
- SPoC
spindle position checkpoint.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-08-0563) on October 26, 2016.
REFERENCES
- Adames NR, Oberle JR, Cooper JA. The surveillance mechanism of the spindle position checkpoint in yeast. J Cell Biol. 2001;153:159–168. doi: 10.1083/jcb.153.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin AJ, Visintin R, Amon A. A mechanism for coupling exit from mitosis to partitioning of the nucleus. Cell. 2000;102:21–31. doi: 10.1016/s0092-8674(00)00007-6. [DOI] [PubMed] [Google Scholar]
- Beach DL, Thibodeaux J, Maddox P, Yeh E, Bloom K. The role of the proteins Kar9 and Myo2 in orienting the mitotic spindle of budding yeast. Curr Biol. 2000;10:1497–1506. doi: 10.1016/s0960-9822(00)00837-x. [DOI] [PubMed] [Google Scholar]
- Bertazzi DT, Kurtulmus B, Pereira G. The cortical protein Lte1 promotes mitotic exit by inhibiting the spindle position checkpoint kinase Kin4. J Cell Biol. 2011;193:1033–1048. doi: 10.1083/jcb.201101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Caydasi AK, Kurtulmus B, Orrico MIL, Hofmann A, Ibrahim B, Pereira G. Elm1 kinase activates the spindle position checkpoint kinase Kin4. J Cell Biol. 2010;190:975–989. doi: 10.1083/jcb.201006151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Micoogullari Y, Kurtulmus B, Palani S, Pereira G. The 14-3-3 protein Bmh1 functions in the spindle position checkpoint by breaking Bfa1 asymmetry at yeast centrosomes. Mol Biol Cell. 2014;25:2143–2151. doi: 10.1091/mbc.E14-04-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LY, Amon A. The protein phosphatase 2A functions in the spindle position checkpoint by regulating the checkpoint kinase Kin4. Genes Dev. 2009;23:1639–1649. doi: 10.1101/gad.1804609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LY, Amon A. Spindle position is coordinated with cell-cycle progression through establishment of mitotic exit-activating and -inhibitory zones. Mol Cell. 2010;39:444–454. doi: 10.1016/j.molcel.2010.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Türkel N, Hemati N, Fuller MT, Hunt AJ, Yamashita YM. Centrosome misorientation reduces stem cell division during ageing. Nature. 2008;456:599–604. doi: 10.1038/nature07386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aquino KE, Monje-Casas F, Paulson J, Reiser V, Charles GM, Lai L, Shokat KM, Amon A. The protein kinase Kin4 inhibits exit from mitosis in response to spindle position defects. Mol Cell. 2005;19:223–234. doi: 10.1016/j.molcel.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Dharmasiri N, Dharmasiri S, Estelle M. The F-box protein TIR1 is an auxin receptor. Nature. 2005;435:441–445. doi: 10.1038/nature03543. [DOI] [PubMed] [Google Scholar]
- Falk JE, Chan LY, Amon A. Lte1 promotes mitotic exit by controlling the localization of the spindle position checkpoint kinase Kin4. Proc Natl Acad Sci USA. 2011;108:12584–12590. doi: 10.1073/pnas.1107784108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk JE, Tsuchiya D, Verdaasdonk J, Lacefield S, Bloom K, Amon A. Spatial signals link exit from mitosis to spindle position. Elife. 2016;5:e14036. doi: 10.7554/eLife.14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkasovsky M, Küntzel H. Cortical Num1p interacts with the dynein intermediate chain Pac11p and cytoplasmic microtubules in budding yeast. J Cell Biol. 2001;152:251–262. doi: 10.1083/jcb.152.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgoczy DJ, Toczyski DP. Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol Cell Biol. 2001;21:1710–1718. doi: 10.1128/MCB.21.5.1710-1718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geymonat M, Spanos A, de Bettignies G, Sedgwick SG. Lte1 contributes to Bfa1 localization rather than stimulating nucleotide exchange by Tem1. J Cell Biol. 2009;187:497–511. doi: 10.1083/jcb.200905114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geymonat M, Spanos A, Smith SJM, Wheatley E, Rittinger K, Johnston LH, Sedgwick SG. Control of mitotic exit in budding yeast. In vitro regulation of Tem1 GTPase by Bub2 and Bfa1. J Biol Chem. 2002;277:28439–28445. doi: 10.1074/jbc.M202540200. [DOI] [PubMed] [Google Scholar]
- Gray WM, Kepinski S, Rouse D, Leyser O, Estelle M. Auxin regulates SCF(TIR1)-dependent degradation of AUX/IAA proteins. Nature. 2001;414:271–276. doi: 10.1038/35104500. [DOI] [PubMed] [Google Scholar]
- Gryaznova Y, Koca Caydasi A, Malengo G, Sourjik V, Pereira G. A FRET-based study reveals site-specific regulation of spindle position checkpoint proteins at yeast centrosomes. Elife. 2016;5:159. doi: 10.7554/eLife.14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil-Chapdelaine RA, Oberle JR, Cooper JA. The cortical protein Num1p is essential for dynein-dependent interactions of microtubules with the cortex. J Cell Biol. 2000;151:1337–1344. doi: 10.1083/jcb.151.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochwagen A, Wrobel G, Cartron M, Demougin P, Niederhauser-Wiederkehr C, Boselli MG, Primig M, Amon A. Novel response to microtubule perturbation in meiosis. Mol Cell Biol. 2005;25:4767–4781. doi: 10.1128/MCB.25.11.4767-4781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Tinker-Kulberg RL, Morgan DO. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9:2803–2817. doi: 10.1091/mbc.9.10.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Morgan DO. Cdc14 activates cdc15 to promote mitotic exit in budding yeast. Curr Biol. 2000;10:615–618. doi: 10.1016/s0960-9822(00)00491-7. [DOI] [PubMed] [Google Scholar]
- Kepinski S, Leyser O. The Arabidopsis F-box protein TIR1 is an auxin receptor. Nature. 2005;435:446–451. doi: 10.1038/nature03542. [DOI] [PubMed] [Google Scholar]
- Khmelinskii A, Lawrence C, Roostalu J, Schiebel E. Cdc14-regulated midzone assembly controls anaphase B. J Cell Biol. 2007;177:981–993. doi: 10.1083/jcb.200702145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König C, Maekawa H, Schiebel E. Mutual regulation of cyclin-dependent kinase and the mitotic exit network. J Cell Biol. 2010;188:351–368. doi: 10.1083/jcb.200911128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecká M, Gabriel M. The aberrant positioning of nuclei and the microtubular cytoskeleton in Saccharomyces cerevisiae due to improper actin function. Microbiology. 1998;144:1783–1797. doi: 10.1099/00221287-144-7-1783. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Mckenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Manzoni R, Montani F, Visintin C, Caudron F, Ciliberto A, Visintin R. Oscillations in Cdc14 release and sequestration reveal a circuit underlying mitotic exit. J Cell Biol. 2010;190:209–222. doi: 10.1083/jcb.201002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally FJ. Mechanisms of spindle positioning. J Cell Biol. 2013;200:131–140. doi: 10.1083/jcb.201210007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RK, Matheos D, Rose MD. The cortical localization of the microtubule orientation protein, Kar9p, is dependent upon actin and proteins required for polarization. J Cell Biol. 1999;144:963–975. doi: 10.1083/jcb.144.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RK, Rose MD. Kar9p is a novel cortical protein required for cytoplasmic microtubule orientation in yeast. J Cell Biol. 1998;140:377–390. doi: 10.1083/jcb.140.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohl DA, Huddleston MJ, Collingwood TS, Annan RS, Deshaies RJ. Dbf2-Mob1 drives relocalization of protein phosphatase Cdc14 to the cytoplasm during exit from mitosis. J Cell Biol. 2009;184:527–539. doi: 10.1083/jcb.200812022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Chudalayandi P, Heil-Chapdelaine RA, Cooper JA. The spindle position checkpoint is coordinated by the Elm1 kinase. J Cell Biol. 2010;191:493–503. doi: 10.1083/jcb.201006092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhua L, Karpova TS, Cooper JA. A yeast actin-related protein homologous to that in vertebrate dynactin complex is important for spindle orientation and nuclear migration. Cell. 1994;78:669–679. doi: 10.1016/0092-8674(94)90531-2. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Pereira G, Höfken T, Grindlay J, Manson C, Schiebel E. The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol Cell. 2000;6:1–10. [PubMed] [Google Scholar]
- Pereira G, Schiebel E. Kin4 kinase delays mitotic exit in response to spindle alignment defects. Mol Cell. 2005;19:209–221. doi: 10.1016/j.molcel.2005.05.030. [DOI] [PubMed] [Google Scholar]
- Rock JM, Amon A. The FEAR network. Curr Biol. 2009;19:R1063–R1068. doi: 10.1016/j.cub.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JM, Amon A. Cdc15 integrates Tem1 GTPase-mediated spatial signals with Polo kinase-mediated temporal cues to activate mitotic exit. Genes Dev. 2011;25:1943–1954. doi: 10.1101/gad.17257711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JM, Lim D, Stach L, Ogrodowicz RW, Keck JM, Jones MH, Wong CCL, Yates JR, Winey M, Smerdon SJ, et al. Activation of the yeast Hippo pathway by phosphorylation-dependent assembly of signaling complexes. Science. 2013;340:871–875. doi: 10.1126/science.1235822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarfone I, Venturetti M, Hotz M, Lengefeld J, Barral Y, Piatti S. Asymmetry of the budding yeast Tem1 GTPase at spindle poles is required for spindle positioning but not for mitotic exit. PLoS Genet. 2015;11:e1004938. doi: 10.1371/journal.pgen.1004938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshan A, Amon A. Ras and the Rho effector Cla4 collaborate to target and anchor Lte1 at the bud cortex. Cell Cycle. 2005;4:940–946. doi: 10.4161/cc.4.7.1785. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Matsui Y, Tanaka K, Toh-E A. Isolation of a CDC25 family gene, MSI2/LTE1, as a multicopy suppressor of ira1. Yeast. 1994;10:451–461. doi: 10.1002/yea.320100404. [DOI] [PubMed] [Google Scholar]
- Siller KH, Doe CQ. Spindle orientation during asymmetric cell division. Nat Cell Biol. 2009;11:365–374. doi: 10.1038/ncb0409-365. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Amon A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet. 2004;38:203–232. doi: 10.1146/annurev.genet.38.072902.093051. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Visintin R, Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 2002;108:207–220. doi: 10.1016/s0092-8674(02)00618-9. [DOI] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW. Mitosis in living budding yeast: anaphase A but no metaphase plate. Science. 1997;277:574–578. doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- Teale WD, Paponov IA, Palme K. Auxin in action: signalling, transport and the control of plant growth and development. Nat Rev Mol Cell Biol. 2006;7:847–859. doi: 10.1038/nrm2020. [DOI] [PubMed] [Google Scholar]
- Valerio-Santiago M, Monje-Casas F. Tem1 localization to the spindle pole bodies is essential for mitotic exit and impairs spindle checkpoint function. J Cell Biol. 2011;192:599–614. doi: 10.1083/jcb.201007044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Amon A. Regulation of the mitotic exit protein kinases Cdc15 and Dbf2. Mol Biol Cell. 2001;12:2961–2974. doi: 10.1091/mbc.12.10.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2:709–718. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K. Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharomyces cerevisiae. J Cell Biol. 1995;130:687–700. doi: 10.1083/jcb.130.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Ichihashi R, Toh-e A. Ras recruits mitotic exit regulator Lte1 to the bud cortex in budding yeast. J Cell Biol. 2003;161:889–897. doi: 10.1083/jcb.200301128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.