

Abstract
Both the incidence of invasive fungal infections and rates of multidrug resistance associated with fungal pathogen Candida glabrata have increased in recent years. In this perspective, we will discuss the mechanisms underlying the capacity of C. glabrata to rapidly develop resistance to multiple drug classes, including triazoles and echinocandins. We will focus on the extensive genetic diversity among clinical isolates of C. glabrata, which likely enables this yeast to survive multiple stressors, such as immune pressure and antifungal exposure. In particular, over half of C. glabrata clinical strains collected from U.S. and non-U.S. sites have mutations in the DNA mismatch repair gene MSH2, leading to a mutator phenotype and increased frequencies of drug-resistant mutants in vitro. Furthermore, recent studies and data presented here document extensive chromosomal rearrangements among C. glabrata strains, resulting in a large number of distinct karyotypes within a single species. By analyzing clonal, serial isolates derived from individual patients treated with antifungal drugs, we were able to document chromosomal changes occurring in C. glabrata in vivo during the course of antifungal treatment. Interestingly, we also show that both MSH2 genotypes and chromosomal patterns cluster consistently into specific strain types, indicating that C. glabrata has a complex population structure where genomic variants arise, perhaps during the process of adaptation to environmental changes, and persist over time.
Keywords: antifungal resistance, Candida glabrata, DNA repair, MSH2, echinocandins, triazoles, karyotyping, MLST genotyping
Epidemiology of fungal infections and treatment options
Fungi infect billions of people around the world each year. Although most fungal infections remain superficial, millions develop invasive mycoses, which have alarmingly high mortality rates ranging from 30 to 90%, even with antifungal treatment (Pfaller and Diekema, 2007; Nivoix et al., 2008; Wingard et al., 2008; Roilides et al., 2009). According to the CDC and WHO, the number of deaths caused by fungal infections is comparable to the numbers attributed to the world's deadliest diseases, including malaria and tuberculosis. The majority of invasive fungal infections (IFIs) and deaths occur in immunocompromised individuals and are caused by species of Candida, Aspergillus, or Cryptococcus (Pfaller and Diekema, 2007). While Aspergillus and Cryptococcus are acquired from the environment, Candida is a human commensal and most infections arise endogenously. Candida species account for most mucosal and invasive fungal infections worldwide (Pfaller et al., 2006) and are associated with significant healthcare costs (Zaoutis et al., 2005; Pfaller and Diekema, 2010). Approximately 50% of candidemia cases in the U.S. are caused by C. albicans (Hajjeh et al., 2004; Azie et al., 2012). However, over the past 20 years there has been a shift toward non-albicans Candida species (Pfaller and Diekema, 2004; Diekema et al., 2012; Lockhart et al., 2012), and the identity of leading non-albicans species causing disease varies upon geographical location. In the U.S., C. glabrata accounts for ~25% of infections, followed by C. parapsilosis (~15%) and C. tropicalis (~10%) (Pfaller et al., 2011a,b; Azie et al., 2012; Lockhart et al., 2012). Decreases in C. albicans infections have been complemented by increases in C. glabrata infections in most U.S. cities (Lockhart et al., 2012). The reason for the steep increase in reported C. glabrata infections is not known, although advances in diagnostic methods, increases in the elderly population, geography, and widespread fluconazole use (see below) have been proposed to play roles (Pfaller et al., 2006, 2009; Diekema et al., 2012).
Patients undergoing procedures such as stem cell or organ transplantation, surgery, or cancer treatment are at high risk for developing life-threatening IFIs. Consequently, such patients are commonly placed on antifungal prophylaxis with either triazole or echinocandin drugs. The triazole class of antifungal drugs targets the biosynthesis of ergosterol, which is a critical component of fungal cell membranes, while the echinocandins block the biosynthesis of beta-1,3-glucan, a fundamental structural component of the cell wall. Both drug classes are recommended first-line therapy for a variety of IFIs caused by Candida species (Pappas et al., 2016). A consequence of the widespread use of triazole antifungals (e.g., fluconazole) for prophylaxis or therapy is the selection of Candida species that readily develop resistance, such as C. glabrata (Lortholary et al., 2011). Approximately 20–30% of C. glabrata strains, but less than 5% of C. albicans strains, exhibit fluconazole resistance in the U.S. (Castanheira et al., 2014). C. glabrata readily develops cross-resistance to azoles, including fluconazole, itraconazole, voriconazole and posaconazole, which is often associated with upregulation of ATP-binding cassette (ABC) transporters, such as CDR1 and SNQ2, that effectively pump the drugs out of the yeast cell (Sanglard et al., 1999, 2001; Izumikawa et al., 2003; Torelli et al., 2008). Upregulation of these drug efflux pumps is usually accompanied by a gain-of-function mutation in the transcription factor PDR1 (Vermitsky and Edlind, 2004; Tsai et al., 2006; Vermitsky et al., 2006), prompting the recent development of a novel inhibitor that interferes with Pdr1 binding to the Mediator complex preventing transcription initiation (Nishikawa et al., 2016). Specific pdr1 mutations in C. glabrata can also lead to a gain of fitness through enhanced adhesion and virulence (Ferrari et al., 2011; Vale-Silva et al., 2013, 2016). Additional mechanisms of triazole resistance, such as mutation of the drug target (ERG11; ergosterol biosynthesis) or mutation of genes (e.g., ERG3) that prevent the production of toxic intermediates caused by triazole action, have been identified in other Candida species (Cowen et al., 2014), yet rarely in C. glabrata (Hull et al., 2012).
In some settings, such as those involving hematologic malignancies, C. glabrata is isolated more often than C. albicans, making it the leading cause of IFIs (Hachem et al., 2008). Therefore, current treatment recommendations advise administration of an echinocandin to patients with known C. glabrata infections (Pappas et al., 2016). While the mechanism of echinocandin resistance is consistent across Candida species, the rates of resistance have increased the greatest (from 3 to 12%) in C. glabrata (Alexander et al., 2013) for reasons that are still largely unclear. Echinocandin resistance develops upon mutation of the catalytic subunits (Fks1/Fks2) that make up the echinocandin target enzyme, beta-1,3-glucan synthase. Mutations are generally found in the “hot spot” regions of either FKS gene and result in cross-resistance to all echinocandins (caspofungin, micafungin, and anidulafungin) (Perlin, 2015). Triazole-resistant clinical isolates (Pfaller et al., 2005; Messer et al., 2006) and laboratory strains (Niimi et al., 2006) that demonstrate increased drug efflux pump expression remain susceptible to the echinocandins in laboratory liquid assays. One study from 2003 (Schuetzer-Muehlbauer et al., 2003) did report reduced caspofungin susceptibility upon overexpression of the ABC efflux pump CDR2 in C. albicans, although this occurred under specific testing conditions and the other echinocandins (anidulafungin and micafungin) were not yet available for testing. Overall, the echinocandins do not appear to be substrates for drug efflux pumps conferring azole resistance. Echinocandins are also effective against some Candida biofilms, which are normally refractory to triazole treatment (Kuhn et al., 2002; Katragkou et al., 2008). A third class of antifungals, the polyenes, directly targets ergosterol in the cell membrane disrupting cellular permeability; however, treatment of invasive Candida infections with the polyene amphotericin B is only recommended as a second-line agent due to toxicity issues (Pappas et al., 2016).
The adaptive response
One factor that can influence the survival or persistence of fungi between the initial drug exposure and acquisition of resistance-conferring mutations is the adaptive response. Upon exposure to environmental stresses, such as temperature, ionic, oxidative, and osmolarity changes, and/or drug pressure, fungi activate stress responses to mitigate the harmful effects and avoid death. Of particular importance upon echinocandin exposure is the cell wall integrity (CWI) pathway, which regulates glucan synthesis through Rho1 and cell wall repair (Levin, 2005). Following cell wall stress, Rho1 activation leads to activation of protein kinase C (PKC1) and upregulation of the FKS genes. Targeting the CWI pathway through deletion of PKC1 or activated MAP kinases (e.g., CgSLT2) induces echinocandin hypersensitivity; while conversely, an increased activation of the pathway, such as through over-expression of CgSLT2, reduces susceptibility (increases tolerance) of C. glabrata to echinocandins (Cota et al., 2008; Miyazaki et al., 2010a; Schwarzmuller et al., 2014). Induction of the CWI pathway in Candida species (Stevens et al., 2006; Walker et al., 2013), including C. glabrata (Cota et al., 2008), has been linked to increased production of other cell wall components, such as chitin, potentially compensating for the loss of beta-1,3-glucans. Genetic (Ueno et al., 2011; Schwarzmuller et al., 2014) or chemical (Miyazaki et al., 2010a) targeting of chitin synthase has been shown to increase C. glabrata susceptibility to echinocandins in vitro. Regulators of the CWI pathway, such as Hsp90 and calcineurin, can similarly influence the susceptibility of C. glabrata to echinocandin and triazole drugs (Singh et al., 2009; Miyazaki et al., 2010b). Other epigenetic mechanisms, such as chromatin modification, may impact drug activity; for example, histone deacetylase inhibitors act synergistically with antifungals (Garnaud et al., 2016). In addition to genetic and epigenetic changes, alterations in chromosomal structure may also influence survival in vivo and antifungal susceptibility; this will be discussed in greater depth in subsequent sections of this perspective. Tolerance mechanisms may “prime” cells, particularly C. glabrata, to better survive adverse conditions in vivo and, in turn, better equip fungi to survive antifungal pressure. Inversely, abrogating the adaptive response of C. glabrata may increase the killing (fungicidal index) efficacy of antifungals. Thus, a better understanding of how fungi control these adaptive mechanisms, particularity in vivo, and how fitness may be affected is warranted.
In addition to individual cell responses to drugs, which are determined largely by cell genotype, population responses may also play a role in drug tolerance. Although acquisition of a pdr1 mutation is required for stable triazole resistance, studies have demonstrated the ability of C. glabrata subpopulations originating from a clonal population to develop heteroresistance to fluconazole through transient upregulation of efflux pumps (Claudino et al., 2009; Ben-Ami et al., 2016). Development of tolerance to fluconazole treatment at the population level may be one consequence of heteroresistance, which in turn, may allow the yeast to adapt and “buy time” until beneficial, stable mutations develop. As stated earlier, specific pdr1 mutations can also enhance the fitness of C. glabrata and increase virulence through adhesion upregulation (Vale-Silva et al., 2016). C. glabrata heteroresistance was not observed in response to the echinocandins, which may reflect its fungicidal mechanism of action (at least in vitro) (Ben-Ami et al., 2016) or potential associated fitness costs. However, yeast are exposed to additional pressures in vivo that may induce some kind of transient heterotolerance to the echinocandins that is not necessarily measurable in vitro.
The emergence of multidrug resistance in C. glabrata
While the individual mechanisms of C. glabrata triazole and echinocandin resistance have been extensively examined, the sudden emergence of strains that are resistant to both drug classes is surprising and alarming. Resistance to multiple antifungal classes, or multidrug resistance (MDR), has been reported most frequently in this fungus (Pfaller et al., 2012). A CDC/SENTRY Antimicrobial Surveillance Program study found that while none of the fluconazole-resistant C. glabrata isolates from 2001 to 2004 demonstrated echinocandin resistance, 11% of fluconazole-resistant isolates from 2006 to 2010 were also resistant to an echinocandin (Pfaller et al., 2012). Another study, performed at Duke Hospital in 2010, found that 14% of fluconazole-resistant isolates also exhibited resistance to at least one echinocandin and that 3.5% of all C. glabrata isolates were MDR (Alexander et al., 2013). Additionally, a survey of C. glabrata isolates obtained from a cancer hospital between 2005 and 2013 reported nearly 7% as MDR (Farmakiotis et al., 2014). As a consequence of current guidelines for prophylaxis and treatment, the majority of MDR C. glabrata strains have acquired resistance to triazole and echinocandin class antifungals, with rare isolates demonstrating resistance to amphotericin B (Farmakiotis et al., 2014; Cho et al., 2015). Collectively, the emergence of MDR in C. glabrata, although still uncommon, is a cause for grave concern given the fact that echinocandin and triazole use has increased significantly over the past 10 years (Pfaller et al., 2012), and the echinocandins are currently the preferred treatment for C. glabrata invasive infections. A better understanding of the mechanisms responsible for the emergence or perpetuation of MDR will help assess factors that can both prevent and overcome resistance.
Many groups, including ours, have attributed the observed increases in drug resistance rates to the haploid genome of C. glabrata. While this is most likely a factor, our recent studies have suggested that other prominent biological factors may contribute more profoundly to resistance emergence. Here, we discuss genetic mechanisms, both at the chromosomal and nucleotide levels, which can increase the mutability and/or genetic diversity of C. glabrata. We propose that these mechanisms allow strains of C. glabrata to create genetically diverse populations within the human host and improve the yeast's chances of surviving multiple stressors, including antifungal treatment. We show that nucleotide polymorphisms and chromosomal rearrangements vary widely in C. glabrata yet group reliably into strain types. In this perspective, we will both summarize previously published studies and present new data that support these claims.
Genetic diversity and the mutator phenotype
All cells contain multiple mechanisms dedicated to repairing DNA damage that can result from either exogenous (e.g., chemicals or radiation) or endogenous (e.g., reactive oxygen radicals or DNA polymerase errors) sources. In eukaryotes these mechanisms include DNA-damage checkpoints and several highly conserved DNA repair systems, including double-strand break repair (DSBR), base-excision repair (BER), nucleotide-excision repair (NER), and mismatch repair (MMR). Defects in these mechanisms are often associated with increased mutation rates or genome rearrangements, and in humans they are associated with a predisposition to specific cancers (Hakem, 2008). Thus, defects in DNA repair may also underlie the emergence of drug-resistant variants in fungal pathogens such as C. glabrata. DNA repair has been extensively studied in the model organism Saccharomyces cerevisiae. Yet, its role in fungal pathogens, especially in connection to emergence of antifungal resistance, has only begun to be addressed.
One study investigated the involvement of DNA repair mechanisms in drug resistance in C. albicans and demonstrated that mutants of MMR and DSBR, but not BER or NER, lead to increased appearance of fluconazole-resistant colonies (Legrand et al., 2007, 2008). Recently, we examined the role of DNA repair in C. glabrata (Healey et al., 2016). We discovered that over half (55%) of all C. glabrata isolates (susceptible and resistant) recovered from patients contain mutations within the MMR gene MSH2. Strains with specific msh2 mutations exhibit a higher frequency of emergence of resistance in vitro, and deletion of MSH2 causes elevated propensity to breakthrough antifungal treatment in a mouse model of gastrointestinal (GI) colonization. Furthermore, analysis of serial clinical isolates demonstrated that msh2 mutations pre-date the emergence of antifungal resistance. Interestingly, MSH2 genotypes appear to be geographically dependent. The overwhelming majority of strains (75 of 76) that exhibit msh2-E231G/L269F or msh2-P208S/N890I were recovered at U.S. based medical centers. Strains containing either msh2-V239L/A942T or msh2-V239L were identified in both U.S. and non-U.S. (Switzerland, Qatar, and India) centers, although these genotypes were predominant (95 of 125) among the msh2 mutant alleles identified from the non-U.S. centers. These geographical differences are most likely a result of population and/or treatment differences (see “Strain type” section below). Our study suggests that msh2 mutation is a driver of the acquisition of antifungal resistance in C. glabrata. Whether other fungi exhibit similar mechanisms of genetic diversity through MMR remains to be addressed.
Through in vivo competition assays utilizing both GI colonization and bloodstream infection mouse models, we have demonstrated that complete loss of MMR function (msh2Δ) impacts fitness of C. glabrata in the host (Healey et al., 2016). However, strains exhibiting one of the aforementioned msh2 mutations lead to only partial loss-of-function (data not shown) and may not suffer from the extremely high mutation rate and fitness defect observed with msh2Δ. Additional mechanisms of DNA repair, including other MMR genes, and other cellular systems responsible for genome integrity maintenance, such as DNA-damage checkpoints, may also influence the mutagenic potential of C. glabrata clinical strains.
Genetic diversity of chromosome structure
Normally, all chromosomes in a cell are present at the same copy number; however, deviations from this norm, aka aneuploid isolates, have been reported in fungal pathogens (Bennett, 2010; Gerstein et al., 2015). A number of studies have suggested that variations in chromosome organization and copy number can be advantageous to fungal cells under certain conditions and may provide a route for rapid adaptation to stress, such as the host immune response or antifungal drugs. In C. albicans, chromosomal rearrangements (copy number variation, loss of heterozygosity, translocations, chromosome truncations) occur in response to stresses such as heat shock, host-pathogen interactions, and the presence of antifungal drugs (Selmecki et al., 2006; Bouchonville et al., 2009; Forche et al., 2009). As a result, the karyotypes (the number and size of chromosomes) between different isolates may vary drastically (Merz et al., 1988; Rustchenko-Bulgac, 1991; Pfaller et al., 1994). Even greater karyotypic variability has been observed in clinical isolates of C. glabrata. Karyotyping using pulsed-field gel electrophoresis (PFGE), which separates whole chromosomes, revealed that C. glabrata genome organization varies significantly among different isolates (Muller et al., 2009; Polakova et al., 2009). Similar to what has been described in C. albicans, the most frequent genome rearrangements identified were translocations of chromosomal arms and inter-chromosomal duplications, but formation of novel chromosomes was also reported (Muller et al., 2009; Polakova et al., 2009).
Since genome plasticity has been connected with antifungal drug resistance in C. glabrata (Shin et al., 2007; Polakova et al., 2009), we analyzed the karyotypes of serial clinical C. glabrata isolates from our laboratory's strain collection. All strains were subjected to multi-locus sequence typing (MLST) analysis (Dodgson et al., 2003) to verify that each pair originated from the same lineage; indeed, both strains from each pair were of the same strain type (Figure 1A). MICs to fluconazole and the echinocandins were determined by the CLSI broth microdilution method (CLSI, 2012) and FKS1, FKS2, PDR1, and MSH2 genes were sequenced for all isolates (Figure 1A). In each case, a bloodstream isolate was obtained before and after echinocandin therapy, and in each case the first isolate was echinocandin sensitive, while the second isolate had acquired echinocandin resistance and an fks mutation. One set of isolates exhibited resistance to fluconazole and contained a pdr1 mutation. To determine whether these changes were also accompanied by large-scale chromosomal rearrangements, we performed PFGE. Karyotypes of the six isolates presented unique chromosomal profiles (Figure 1B). While strains isolated from the same patient were more similar to each other than strains isolated from different patients, even sequential isolates obtained from the same individual had distinct chromosomal bands, particularly among the larger chromosomes (Figure 1B). Thus, changes in chromosomal architecture occurred during microevolution of C. glabrata within the host, possibly in response to echinocandin treatment. Thus, genome rearrangements may be one mechanism used by C. glabrata to diversify the phenotypic properties of the population and promote adaptation under specific stress conditions.
Figure 1.

Electrophoretic karyotypes of three pairs of serial C. glabrata clinical isolates. (A) The minimum inhibitory concentrations (MIC), sequence type (ST), and genome mutations of two bloodstream isolates (A and B) from three individual patients (1, 2, and 3) are shown. (B) Karyotypes of the same isolates obtained with PFGE analysis. The karyotype of the C. glabrata reference strain CBS138/ATCC2001 is also shown. M: CHEF DNA chromosomal size marker (S. cerevisiae strain YNN295) (BIO-RAD). ANF, anidulafungin; MCF, micafungin; CSF, caspofungin; FLZ, fluconazole. *Gain of function mutations.
C. glabrata population structure: strain types are characterized by specific nucleotide polymorphisms and genome rearrangements
As described above, C. glabrata clinical isolates have highly variable chromosomal profiles. It has been reported that this fungus also shows genetic variability at the level of genomic nucleotide sequence (Lin et al., 2007). A MLST scheme based on six different loci has thus far identified over 80 different C. glabrata sequence types (STs, or clades) (Dodgson et al., 2003) and our preliminary analysis indicates that this number is likely much higher. For instance, MLST typing of 48 clinical strains identified 6 strains containing novel MLST alleles or novel configurations of previously reported alleles (data not shown). We also found that different msh2 alleles (see above and Figure 1A) are characteristic of distinct STs/clades, indicating that different STs may have different propensity toward mutability and acquiring drug resistant gene variants. This conclusion is significant because the distribution of C. glabrata STs varies both by geography and over time (Dodgson et al., 2003; Lott et al., 2010). For instance, Figure 2A shows that C. glabrata ST distribution in Atlanta area hospitals changed significantly between 1992 and 2008, the time period that includes the introduction of echinocandins (Lott et al., 2010). One noteworthy change is the increased prevalence of ST16, which carries an msh2 mutation and may therefore acquire drug resistance at elevated rates.
Figure 2.
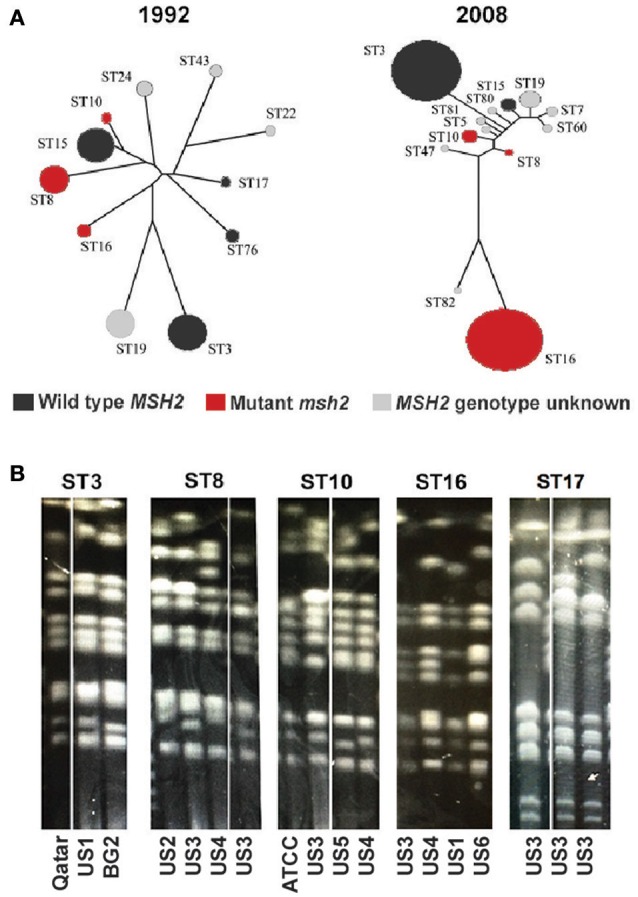
Temporal changes and chromosomal patterns associated with C. glabrata strain type. (A) The population structure of C. glabrata isolates obtained from candidemia patients in Atlanta hospitals during the indicated years. Each circle represents a particular ST and the msh2 profile of each ST is indicated by color (adapted from Lott et al., 2010). (B) Strains belonging to the same ST have similar chromosome patterns as determined by pulse field gel electrophoresis (PFGE). Chromosome configurations for strains of five different STs are shown and the origins of every clinical isolate are indicated below. Laboratory strains (BG2 and ATCC) or isolates recovered from individual patients at six separate U.S. medical centers (US1-6) or a medical center in Qatar were analyzed.
The observation that different C. glabrata STs carry different msh2 alleles makes it highly likely that other important determinants of drug resistance or virulence also vary among STs. A straightforward way to identify such determinants is by genome sequencing. However, the great degree of genomic rearrangement among strains presents a barrier to typical shotgun sequencing approaches that use a reference strain as template. Indeed, while the genome of a reference C. glabrata laboratory strain (CBS138) has been sequenced, we were not able to use that strain's genomic sequence as a template for assembling the sequences of other strains, e.g., various clinical isolates, due to the high karyotypic variability among strains (Muller et al., 2009; Polakova et al., 2009; Bader et al., 2012; Figure 1B). Thus, it will be necessary to assemble the genomes of individual clinical C. glabrata isolates de novo using techniques such as optical restriction mapping and long read DNA sequencing (Neely et al., 2011; Rhoads and Au, 2015).
We also examined the chromosomal architecture of strains belonging to different STs. Because it was originally proposed that chromosomal rearrangements in C. glabrata arise as part of the virulence process or adaptation to drug pressure and emergence of resistance (Polakova et al., 2009), we expected that there would be no correspondence between ST identity and chromosomal configuration. However, we were surprised to discover similarities in chromosomal architecture among strains belonging to the same STs (Figure 2B). Together with data presented above (Figure 1B), this observation suggests that chromosomal variants of C. glabrata both exist as commensal populations, where different STs are characterized by specific chromosomal configurations, and arise de novo, perhaps in response to pressure from the host and antifungal drug treatment.
Conclusion
Accumulating evidence suggests that both increasing numbers of immunocompromised patients and the widespread use of triazoles, particularly fluconazole, have facilitated an increased prevalence of C. glabrata infections and more alarmingly, the emergence of multidrug resistant strains. Here we have summarized the current understanding of factors driving drug resistance in C. glabrata. Maximizing genetic diversity, a hallmark of C. glabrata, is most apparent by the high prevalence in the global population of mutator phenotypes mediated by mutations in MSH2. Additionally, new data is presented supporting the concept that C. glabrata has a surprisingly plastic and mutable genome. It appears that, in contrast to many organisms, C. glabrata can tolerate such a high degree of genome rearrangement and thrive under these circumstances, perhaps through acquisition of gain-of-fitness mutations. Continued tracking of C. glabrata strain types in medical centers around the globe will aid in the understanding of C. glabrata population structure and how pathogenesis and drug resistance are associated with different strains of C. glabrata.
Ethics statement
All isolates analyzed in new studies presented here are a part of the Perlin laboratory historic strain collection and were previously obtained from mycology laboratories without patient identifiers.
Author contributions
KH, CJ, ES, and DP wrote the paper. CJ, ES, and KH performed the experiments.
Funding
This research was supported by grants from the NIH (AI109025) and Astellas Pharma (Echinocandin Reference Center) to DP and by an Arnold O. Beckman postdoctoral fellowship from the Arnold and Mabel Beckman Foundation to KH.
Conflict of interest statement
DP has received research support and/or advised Merck, Astellas, Cidara, Synexis, Matinas, and Amplyx. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Alexander B. D., Johnson M. D., Pfeiffer C. D., Jiménez-Ortigosa C., Catania J., Booker R., et al. (2013). Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 56, 1724–1732. 10.1093/cid/cit136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azie N., Neofytos D., Pfaller M., Meier-Kriesche H. U., Quan S. P., Horn D. (2012). The PATH (Prospective Antifungal Therapy) Alliance(R) registry and invasive fungal infections: update 2012. Diagn. Microbiol. Infect. Dis. 73, 293–300. 10.1016/j.diagmicrobio.2012.06.012 [DOI] [PubMed] [Google Scholar]
- Bader O., Schwarz A., Kraneveld E. A., Tangwattanachuleeporn M., Schmidt P., Jacobsen M. D., et al. (2012). Gross karyotypic and phenotypic alterations among different progenies of the Candida glabrata CBS138/ATCC2001 reference strain. PLoS ONE 7:e52218. 10.1371/journal.pone.0052218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ami R., Zimmerman O., Finn T., Amit S., Novikov A., Wertheimer N., et al. (2016). Heteroresistance to fluconazole is a continuously distributed phenotype among Candida glabrata clinical strains associated with in vivo persistence. MBio 7:e00655-16. 10.1128/mBio.00655-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett R. J. (2010). Coming of age–sexual reproduction in Candida species. PLoS Pathog. 6:e1001155. 10.1371/journal.ppat.1001155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchonville K., Forche A., Tang K. E., Selmecki A., Berman J. (2009). Aneuploid chromosomes are highly unstable during DNA transformation of Candida albicans. Eukaryotic Cell 8, 1554–1566. 10.1128/EC.00209-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanheira M., Messer S. A., Jones R. N., Farrell D. J., Pfaller M. A. (2014). Activity of echinocandins and triazoles against a contemporary (2012) worldwide collection of yeast and moulds collected from invasive infections. Int. J. Antimicrob. Agents 44, 320–326. 10.1016/j.ijantimicag.2014.06.007 [DOI] [PubMed] [Google Scholar]
- Cho E. J., Shin J. H., Kim S. H., Kim H. K., Park J. S., Sung H., et al. (2015). Emergence of multiple resistance profiles involving azoles, echinocandins and amphotericin B in Candida glabrata isolates from a neutropenia patient with prolonged fungaemia. J. Antimicrob. Chemother. 70, 1268–1270. 10.1093/jac/dku518 [DOI] [PubMed] [Google Scholar]
- Claudino A. L., Peixoto Junior R. F., Melhem M. S., Szeszs M. W., Lyon J. P., Chavasco J. K., et al. (2009). Mutants with heteroresistance to amphotericin B and fluconazole in Candida. Braz. J. Microbiol. 40, 943–951. 10.1590/S1517-83822009000400028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI (2012). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard 3rd Edn., Vol. 28 Wayne, PA: Clinical and Laboratory Standards Institute document M27-S4. [Google Scholar]
- Cota J. M., Grabinski J. L., Talbert R. L., Burgess D. S., Rogers P. D., Edlind T. D., et al. (2008). Increases in SLT2 expression and chitin content are associated with incomplete killing of Candida glabrata by caspofungin. Antimicrob. Agents Chemother. 52, 1144–1146. 10.1128/AAC.01542-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen L. E., Sanglard D., Howard S. J., Rogers P. D., Perlin D. S. (2014). Mechanisms of antifungal drug resistance. Cold Spring Harb. Perspect. Med. 5:a019752. 10.1101/cshperspect.a019752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekema D., Arbefeville S., Boyken L., Kroeger J., Pfaller M. (2012). The changing epidemiology of healthcare-associated candidemia over three decades. Diagn. Microbiol. Infect. Dis. 73, 45–48. 10.1016/j.diagmicrobio.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Dodgson A. R., Pujol C., Denning D. W., Soll D. R., Fox A. J. (2003). Multilocus sequence typing of Candida glabrata reveals geographically enriched clades. J. Clin. Microbiol. 41, 5709–5717. 10.1128/JCM.41.12.5709-5717.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmakiotis D., Tarrand J. J., Kontoyiannis D. P. (2014). Drug-resistant Candida glabrata infection in cancer patients. Emerging Infect. Dis. 20, 1833–1840. 10.3201/eid2011.140685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S., Sanguinetti M., Torelli R., Posteraro B., Sanglard D. (2011). Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS ONE 6:e17589. 10.1371/journal.pone.0017589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A., Magee P. T., Selmecki A., Berman J., May G. (2009). Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 182, 799–811. 10.1534/genetics.109.103325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnaud C., Champleboux M., Maubon D., Cornet M., Govin J. (2016). Histone deacetylases and their inhibition in Candida species. Front. Microbiol. 7:1238. 10.3389/fmicb.2016.01238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein A. C., Fu M. S., Mukaremera L., Li Z., Ormerod K. L., Fraser J. A., et al. (2015). Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. mBio 6:e01340–15. 10.1128/mBio.01340-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachem R., Hanna H., Kontoyiannis D., Jiang Y., Raad I. (2008). The changing epidemiology of invasive candidiasis: Candida glabrata and Candida krusei as the leading causes of candidemia in hematologic malignancy. Cancer 112, 2493–2499. 10.1002/cncr.23466 [DOI] [PubMed] [Google Scholar]
- Hajjeh R. A., Sofair A. N., Harrison L. H., Lyon G. M., Arthington-Skaggs B. A., Mirza S. A., et al. (2004). Incidence of bloodstream infections due to Candida species and in vitro susceptibilities of isolates collected from 1998 to 2000 in a population-based active surveillance program. J. Clin. Microbiol. 42, 1519–1527. 10.1128/JCM.42.4.1519-1527.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R. (2008). DNA-damage repair; the good, the bad, and the ugly. EMBO J. 27, 589–605. 10.1038/emboj.2008.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey K. R., Zhao Y., Perez W. B., Lockhart S. R., Sobel J. D., Farmakiotis D., et al. (2016). Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commun. 7:11128. 10.1038/ncomms11128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C. M., Parker J. E., Bader O., Weig M., Gross U., Warrilow A. G., et al. (2012). Facultative sterol uptake in an ergosterol-deficient clinical isolate of Candida glabrata harboring a missense mutation in ERG11 and exhibiting cross-resistance to azoles and amphotericin B. Antimicrob. Agents Chemother. 56, 4223–4232. 10.1128/AAC.06253-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumikawa K., Kakeya H., Tsai H. F., Grimberg B., Bennett J. E. (2003). Function of Candida glabrata ABC transporter gene, PDH1. Yeast 20, 249–261. 10.1002/yea.962 [DOI] [PubMed] [Google Scholar]
- Katragkou A., Chatzimoschou A., Simitsopoulou M., Dalakiouridou M., Diza-Mataftsi E., Tsantali C., et al. (2008). Differential activities of newer antifungal agents against Candida albicans and Candida parapsilosis biofilms. Antimicrob. Agents Chemother. 52, 357–360. 10.1128/AAC.00856-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn D. M., George T., Chandra J., Mukherjee P. K., Ghannoum M. A. (2002). Antifungal susceptibility of Candida biofilms: unique efficacy of amphotericin B lipid formulations and echinocandins. Antimicrob. Agents Chemother. 46, 1773–1780. 10.1128/AAC.46.6.1773-1780.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand M., Chan C. L., Jauert P. A., Kirkpatrick D. T. (2007). Role of DNA mismatch repair and double-strand break repair in genome stability and antifungal drug resistance in Candida albicans. Eukaryotic Cell. 6, 2194–2205. 10.1128/EC.00299-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand M., Chan C. L., Jauert P. A., Kirkpatrick D. T. (2008). Analysis of base excision and nucleotide excision repair in Candida albicans. Microbiology 154, 2446–2456. 10.1099/mic.0.2008/017616-0 [DOI] [PubMed] [Google Scholar]
- Levin D. E. (2005). Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 69, 262–291. 10.1128/MMBR.69.2.262-291.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. Y., Chen Y. C., Lo H. J., Chen K. W., Li S. Y. (2007). Assessment of Candida glabrata strain relatedness by pulsed-field gel electrophoresis and multilocus sequence typing. J. Clin. Microbiol. 45, 2452–2459. 10.1128/JCM.00699-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart S. R., Iqbal N., Cleveland A. A., Farley M. M., Harrison L. H., Bolden C. B., et al. (2012). Species identification and antifungal susceptibility testing of Candida bloodstream isolates from population-based surveillance studies in two U.S. cities from 2008 to 2011. J. Clin. Microbiol. 50, 3435–3442. 10.1128/JCM.01283-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortholary O., Desnos-Ollivier M., Sitbon K., Fontanet A., Bretagne S., Dromer F., et al. (2011). Recent exposure to caspofungin or fluconazole influences the epidemiology of candidemia: a prospective multicenter study involving 2,441 patients. Antimicrob. Agents Chemother. 55, 532–538. 10.1128/AAC.01128-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott T. J., Frade J. P., Lockhart S. R. (2010). Multilocus sequence type analysis reveals both clonality and recombination in populations of Candida glabrata bloodstream isolates from U.S. surveillance studies. Eukaryot Cell 9, 619–625. 10.1128/EC.00002-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz W. G., Connelly C., Hieter P. (1988). Variation of electrophoretic karyotypes among clinical isolates of Candida albicans. J. Clin. Microbiol. 26, 842–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer S. A., Diekema D. J., Boyken L., Tendolkar S., Hollis R. J., Pfaller M. A. (2006). Activities of micafungin against 315 invasive clinical isolates of fluconazole-resistant Candida spp. J. Clin. Microbiol. 44, 324–326. 10.1128/JCM.44.2.324-326.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T., Inamine T., Yamauchi S., Nagayoshi Y., Saijo T., Izumikawa K., et al. (2010a). Role of the Slt2 mitogen-activated protein kinase pathway in cell wall integrity and virulence in Candida glabrata. FEMS Yeast Res. 10, 343–352. 10.1111/j.1567-1364.2010.00611.x [DOI] [PubMed] [Google Scholar]
- Miyazaki T., Yamauchi S., Inamine T., Nagayoshi Y., Saijo T., Izumikawa K., et al. (2010b). Roles of calcineurin and Crz1 in antifungal susceptibility and virulence of Candida glabrata. Antimicrob. Agents Chemother. 54, 1639–1643. 10.1128/AAC.01364-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H., Thierry A., Coppée J. Y., Gouyette C., Hennequin C., Sismeiro O., et al. (2009). Genomic polymorphism in the population of Candida glabrata: gene copy-number variation and chromosomal translocations. Fungal Genet. Biol. 46, 264–276. 10.1016/j.fgb.2008.11.006 [DOI] [PubMed] [Google Scholar]
- Neely R. K., Deen J., Hofkens J. (2011). Optical mapping of DNA: single-molecule-based methods for mapping genomes. Biopolymers 95, 298–311. 10.1002/bip.21579 [DOI] [PubMed] [Google Scholar]
- Niimi K., Maki K., Ikeda F., Holmes A. R., Lamping E., Niimi M., et al. (2006). Overexpression of Candida albicans CDR1, CDR2, or MDR1 does not produce significant changes in echinocandin susceptibility. Antimicrob. Agents Chemother. 50, 1148–1155. 10.1128/AAC.50.4.1148-1155.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa J. L., Boeszoermenyi A., Vale-Silva L. A., Torelli R., Posteraro B., Sohn Y. J., et al. (2016). Inhibiting fungal multidrug resistance by disrupting an activator-Mediator interaction. Nature 530, 485–489. 10.1038/nature16963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivoix Y., Velten M., Letscher-Bru V., Moghaddam A., Natarajan-Amé S., Fohrer C., et al. (2008). Factors associated with overall and attributable mortality in invasive aspergillosis. Clin. Infect. Dis. 47, 1176–1184. 10.1086/592255 [DOI] [PubMed] [Google Scholar]
- Pappas P. G., Kauffman C. A., Andes D. R., Clancy C. J., Marr K. A., Ostrosky-Zeichner L., et al. (2016). Clinical practice guideline for the management of candidiasis: 2016 update by the infectious diseases society of America. Clin. Infect. Dis. 62, e1–e50. 10.1093/cid/civ933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlin D. S. (2015). Mechanisms of echinocandin antifungal drug resistance. Ann. N.Y. Acad. Sci. 1354, 1–11. 10.1111/nyas.12831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A., Boyken L., Hollis R. J., Messer S. A., Tendolkar S., Diekema D. J. (2005). In vitro activities of anidulafungin against more than 2,500 clinical isolates of Candida spp., including 315 isolates resistant to fluconazole. J. Clin. Microbiol. 43, 5425–5427. 10.1128/JCM.43.11.5425-5427.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A., Castanheira M., Lockhart S. R., Ahlquist A. M., Messer S. A., Jones R. N. (2012). Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J. Clin. Microbiol. 50, 1199–1203. 10.1128/JCM.06112-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A., Diekema D. J. (2004). Twelve years of fluconazole in clinical practice: global trends in species distribution and fluconazole susceptibility of bloodstream isolates of Candida. Clin. Microbiol. Infect. 10(Suppl. 1), 11–23. 10.1111/j.1470-9465.2004.t01-1-00844.x [DOI] [PubMed] [Google Scholar]
- Pfaller M. A., Diekema D. J. (2007). Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20, 133–163. 10.1128/CMR.00029-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A., Diekema D. J. (2010). Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 36, 1–53. 10.3109/10408410903241444 [DOI] [PubMed] [Google Scholar]
- Pfaller M. A., Messer S. A., Hollis R. J., Boyken L., Tendolkar S., Kroeger J., et al. (2009). Variation in susceptibility of bloodstream isolates of Candida glabrata to fluconazole according to patient age and geographic location in the United States in 2001 to 2007. J. Clin. Microbiol. 47, 3185–3190. 10.1128/JCM.00946-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A., Messer S. A., Moet G. J., Jones R. N., Castanheira M. (2011b). Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in Intensive Care Unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008–2009). Int. J. Antimicrob. Agents. 38, 65–69. 10.1016/j.ijantimicag.2011.02.016 [DOI] [PubMed] [Google Scholar]
- Pfaller M. A., Moet G. J., Messer S. A., Jones R. N., Castanheira M. (2011a). Candida bloodstream infections: comparison of species distributions and antifungal resistance patterns in community-onset and nosocomial isolates in the SENTRY Antimicrobial Surveillance Program, 2008–2009. Antimicrob. Agents Chemother. 55, 561–566. 10.1128/AAC.01079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M. A., Pappas P. G., Wingard J. R. (2006). Invasive fungal pathogens: current epidemiological trends. Clinical Infectious Diseases. 43, S3–S14. 10.1086/504490 [DOI] [Google Scholar]
- Pfaller M. A., Rhine-Chalberg J., Redding S. W., Smith J., Farinacci G., Fothergill A. W., et al. (1994). Variations in fluconazole susceptibility and electrophoretic karyotype among oral isolates of Candida albicans from patients with AIDS and oral candidiasis. J. Clin. Microbiol. 32, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poláková S., Blume C., Zárate J. A., Mentel M., Jørck-Ramberg D., Stenderup J., et al. (2009). Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc. Natl. Acad. Sci. U.S.A. 106, 2688–2693. 10.1073/pnas.0809793106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A., Au K. F. (2015). PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13, 278–289. 10.1016/j.gpb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roilides E., Zaoutis T. E., Walsh T. J. (2009). Invasive zygomycosis in neonates and children. Clin. Microbiol. Infect. 15(Suppl. 5), 50–54. 10.1111/j.1469-0691.2009.02981.x [DOI] [PubMed] [Google Scholar]
- Rustchenko-Bulgac E. P. (1991). Variations of Candida albicans electrophoretic karyotypes. J. Bacteriol. 173, 6586–6596. 10.1128/jb.173.20.6586-6596.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanglard D., Ischer F., Bille J. (2001). Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 45, 1174–1183. 10.1128/AAC.45.4.1174-1183.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanglard D., Ischer F., Calabrese D., Majcherczyk P. A., Bille J. (1999). The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43, 2753–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetzer-Muehlbauer M., Willinger B., Krapf G., Enzinger S., Presterl E., Kuchler K. (2003). The Candida albicans Cdr2p ATP-binding cassette (ABC) transporter confers resistance to caspofungin. Mol. Microbiol. 48, 225–235. 10.1046/j.1365-2958.2003.03430.x [DOI] [PubMed] [Google Scholar]
- Schwarzmüller T., Ma B., Hiller E., Istel F., Tscherner M., Brunke S., et al. (2014). Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 10:e1004211. 10.1371/journal.ppat.1004211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A., Forche A., Berman J. (2006). Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313, 367–370. 10.1126/science.1128242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J. H., Chae M. J., Song J. W., Jung S. I., Cho D., Kee S. J., et al. (2007). Changes in karyotype and azole susceptibility of sequential bloodstream isolates from patients with Candida glabrata candidemia. J. Clin. Microbiol. 45, 2385–2391. 10.1128/JCM.00381-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. D., Robbins N., Zaas A. K., Schell W. A., Perfect J. R., Cowen L. E. (2009). Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 5:e1000532. 10.1371/journal.ppat.1000532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens D. A., Ichinomiya M., Koshi Y., Horiuchi H. (2006). Escape of Candida from caspofungin inhibition at concentrations above the MIC (paradoxical effect) accomplished by increased cell wall chitin; evidence for beta-1,6-glucan synthesis inhibition by caspofungin. Antimicrob. Agents Chemother. 50, 3160–3161. 10.1128/AAC.00563-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torelli R., Posteraro B., Ferrari S., La Sorda M., Fadda G., Sanglard D., et al. (2008). The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol. Microbiol. 68, 186–201. 10.1111/j.1365-2958.2008.06143.x [DOI] [PubMed] [Google Scholar]
- Tsai H. F., Krol A. A., Sarti K. E., Bennett J. E. (2006). Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50, 1384–1392. 10.1128/AAC.50.4.1384-1392.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno K., Namiki Y., Mitani H., Yamaguchi M., Chibana H. (2011). Differential cell wall remodeling of two chitin synthase deletants Deltachs3A and Deltachs3B in the pathogenic yeast Candida glabrata. FEMS Yeast Res. 11, 398–407. 10.1111/j.1567-1364.2011.00728.x [DOI] [PubMed] [Google Scholar]
- Vale-Silva L. A., Moeckli B., Torelli R., Posteraro B., Sanguinetti M., Sanglard D. (2016). Upregulation of the adhesin gene EPA1 mediated by PDR1 in Candida glabrata Leads to enhanced host colonization. mSphere. 1:e00065–15. 10.1128/mSphere.00065-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale-Silva L., Ischer F., Leibundgut-Landmann S., Sanglard D. (2013). Gain-of-function mutations in PDR1, a regulator of antifungal drug resistance in Candida glabrata, control adherence to host cells. Infect. Immun. 81, 1709–1720. 10.1128/IAI.00074-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermitsky J. P., Earhart K. D., Smith W. L., Homayouni R., Edlind T. D., Rogers P. D. (2006). Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol. Microbiol. 61, 704–722. 10.1111/j.1365-2958.2006.05235.x [DOI] [PubMed] [Google Scholar]
- Vermitsky J. P., Edlind T. D. (2004). Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 48, 3773–3781. 10.1128/AAC.48.10.3773-3781.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. A., Gow N. A., Munro C. A. (2013). Elevated chitin content reduces the susceptibility of Candida species to caspofungin. Antimicrob. Agents Chemother. 57, 146–154. 10.1128/AAC.01486-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingard J. R., Ribaud P., Schlamm H. T., Herbrecht R. (2008). Changes in causes of death over time after treatment for invasive aspergillosis. Cancer 112, 2309–2312. 10.1002/cncr.23441 [DOI] [PubMed] [Google Scholar]
- Zaoutis T. E., Argon J., Chu J., Berlin J. A., Walsh T. J., Feudtner C. (2005). The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis. Clin. Infect. Dis. 41, 1232–1239. 10.1086/496922 [DOI] [PubMed] [Google Scholar]