

Abstract
Key points
Biotin, a vitamin whose main role is as a coenzyme for carboxylases, accumulates at unusually large amounts within cells of the carotid body (CB).
In biotin‐deficient rats biotin rapidly disappears from the blood; however, it remains at relatively high levels in CB glomus cells. The CB contains high levels of mRNA for SLC5a6, a biotin transporter, and SLC19a3, a thiamine transporter regulated by biotin.
Animals with biotin deficiency exhibit pronounced metabolic lactic acidosis. Remarkably, glomus cells from these animals have normal electrical and neurochemical properties. However, they show a marked decrease in the size of quantal dopaminergic secretory events.
Inhibitors of the vesicular monoamine transporter 2 (VMAT2) mimic the effect of biotin deficiency. In biotin‐deficient animals, VMAT2 protein expression decreases in parallel with biotin depletion in CB cells.
These data suggest that dopamine transport and/or storage in small secretory granules in glomus cells depend on biotin.
Abstract
Biotin is a water‐soluble vitamin required for the function of carboxylases as well as for the regulation of gene expression. Here, we report that biotin accumulates in unusually large amounts in cells of arterial chemoreceptors, carotid body (CB) and adrenal medulla (AM). We show in a biotin‐deficient rat model that the vitamin rapidly disappears from the blood and other tissues (including the AM), while remaining at relatively high levels in the CB. We have also observed that, in comparison with other peripheral neural tissues, CB cells contain high levels of SLC5a6, a biotin transporter, and SLC19a3, a thiamine transporter regulated by biotin. Biotin‐deficient rats show a syndrome characterized by marked weight loss, metabolic lactic acidosis, aciduria and accelerated breathing with normal responsiveness to hypoxia. Remarkably, CB cells from biotin‐deficient animals have normal electrophysiological and neurochemical (ATP levels and catecholamine synthesis) properties; however, they exhibit a marked decrease in the size of quantal catecholaminergic secretory events, which is not seen in AM cells. A similar differential secretory dysfunction is observed in CB cells treated with tetrabenazine, a selective inhibitor of the vesicular monoamine transporter 2 (VMAT2). VMAT2 is highly expressed in glomus cells (in comparison with VMAT1), and in biotin‐deficient animals VMAT2 protein expression decreases in parallel with the decrease of biotin accumulated in CB cells. These data suggest that biotin has an essential role in the homeostasis of dopaminergic transmission modulating the transport and/or storage of transmitters within small secretory granules in glomus cells.
Keywords: arterial chemoreceptors, biotin, carotid body
Key points
Biotin, a vitamin whose main role is as a coenzyme for carboxylases, accumulates at unusually large amounts within cells of the carotid body (CB).
In biotin‐deficient rats biotin rapidly disappears from the blood; however, it remains at relatively high levels in CB glomus cells. The CB contains high levels of mRNA for SLC5a6, a biotin transporter, and SLC19a3, a thiamine transporter regulated by biotin.
Animals with biotin deficiency exhibit pronounced metabolic lactic acidosis. Remarkably, glomus cells from these animals have normal electrical and neurochemical properties. However, they show a marked decrease in the size of quantal dopaminergic secretory events.
Inhibitors of the vesicular monoamine transporter 2 (VMAT2) mimic the effect of biotin deficiency. In biotin‐deficient animals, VMAT2 protein expression decreases in parallel with biotin depletion in CB cells.
These data suggest that dopamine transport and/or storage in small secretory granules in glomus cells depend on biotin.
Abbreviations
- A
adrenaline
- ACC
acetyl CoA carboxylase
- AM
adrenal medulla
- BBGD
biotin‐responsive basal ganglia disease
- BD
biotin deficient
- CB
carotid body
- DA
dopamine
- DAB
diaminobenzidine
- MCC
3‐methylcrotonyl‐CoA carboxylase
- MCI
mitochondrial complex I
- NA
noradrenaline
- PC
pyruvate carboxylase
- PCC
propionyl‐CoA carboxylase
- PFA
paraformaldehyde
- Ro‐GFP
redox‐sensitive green fluorescent protein
- SCG
superior cervical ganglion
- SLC
solute carrier transporter
- TBZ
tetrabenazine
- TH
tyrosine hydroxylase
- TCA
tricarboxylic acid
- VE
minute ventilation
- VMAT
vesicular monoamine transporter
- VT
tidal volume
Introduction
Biotin is a water‐soluble vitamin that is essential as a coenzyme for the correct functioning of carboxylases, which catalyse fundamental steps in the metabolism of glucose, amino acids and fatty acids. In mammals there are five biotin‐dependent carboxylases: pyruvate carboxylase (PC), propionyl‐CoA carboxylase (PCC), 3‐methylcrotonyl‐CoA carboxylase (MCC) and two isoforms of acetyl CoA carboxylase (ACC‐1 and ACC‐2). PC, PCC and MCC are located in mitochondria where they catalyse anaplerotic reactions to replenish tricarboxylic acid (TCA) intermediates (oxalacetate, succinyl‐CoA and acetyl‐CoA, respectively). ACC‐1 (located in the cytosol) is necessary for fatty acid synthesis, whereas ACC‐2 seems to regulate mitochondrial fatty acid transport. The important role of biotin in cell intermediary metabolism is well established (see Mock, 1996; Zempleni et al. 2009), and it is also known that biotin or carboxylase deficiency can produce severe metabolic alterations in humans (Nyhan, 1988; Wolf, 2001; Pacheco‐Alvarez et al. 2002). Given its direct interaction with histones and other molecules, other emerging functions of biotin are also being studied (see Zempleni, 2005). However, whether biotin plays specific roles in the homeostasis of specialized tissues or organs is yet to be elucidated.
We observed that biotin, which is normally present in very small amounts in most tissues, is concentrated at unusually high levels in the carotid body (CB), an arterial chemoreceptor organ that plays a major role in the regulation of respiration. The CB contains O2‐sensitive glomus cells, which in response to hypoxia (and other stimuli such as hypercapnia, acidosis or hypoglucaemia) rapidly release transmitters to activate nerve fibres impinging upon the brain stem respiratory centre to elicit an adaptive hyperventilatory reflex response (for a recent review see López‐Barneo et al. 2016). While elucidation of the molecular mechanisms underlying the chemoreceptive properties of glomus cells is still in progress (for updated reviews see Peers, 2015; López‐Barneo et al. 2016), recent data strongly suggest that mitochondrial complex I (MCI) plays an essential role in acute O2 sensing (Ortega‐Sáenz et al. 2003; García‐Fernández et al. 2007 a; Fernández‐Agüera et al. 2015). Interestingly, glomus cells survive genetic mutations that abolish MCI function and contain high levels of succinate in comparison with other neural cells (Fernández‐Agüera et al. 2015). In contrast, they are severely damaged by genetic disruption of succinate dehydrogenase activity (Diaz‐Castro et al. 2012; Platero‐Luengo et al. 2014). Therefore, it seems that glomus cells possess special metabolic properties that allow them to optimally accomplish their chemoreceptor functions.
To elucidate the role of biotin in arterial chemoreceptors we have studied the properties of CB glomus cells in biotin‐deficient rats. We show that biotin is selectively concentrated in glomus cells as distinct from other cells of the sympathoadrenal lineage [superior cervical ganglion (SCG) neurons or chromaffin cells in the adrenal medulla (AM)]. In animals fed on a biotin‐free diet, biotin is rapidly depleted from the body; however, it is retained at relatively high level in the CB. Glomus cells from these biotin‐deficient rats, with a significant amount of stored biotin, have normal electrophysiology and dopamine content and show responsiveness to hypoxia. However, they exhibit a marked decrease in the size of quantal exocytotic events, which is compatible with alterations in the transport and/or condensation of dopamine into small secretory vesicles. These data demonstrate that biotin is necessary for the homeostasis of dopaminergic transmission. This new role of the vitamin could help to better understand its physiological and pharmacological properties.
Methods
Ethical approval
All procedures were approved by the Institutional Committee of the University of Seville for Animal Care and Use. Handling of the animals was conducted in accordance with the European Community Council directive of 24 November 1986 (86/609/EEC) for the Care and Use of Laboratory Animals. The experiments comply with the principles of animal research established by the Journal of Physiology (Grundy, 2015).
Animal care and generation of biotin‐deficient rats
For the experiments 2‐month‐old Wistar rats were supplied by Charles River Laboratories (Saint‐Germain‐Nuelles, France). Biotin deficiency was induced by feeding animals with a commercially available biotin‐deficient diet containing 30% egg white as a source of avidin (TD.81079, Harlan, Indianapolis, IN, USA). To set up the duration of the treatment a first series of experiments was done using 12 rats. A control group (three rats) and other three groups (three rats per group) were fed with the biotin‐deficient diet for 15, 30 and 60 days. These rats were killed by intraperitoneal sodium thiopental overdose and tissues were used to determine endogenous levels of biotin. After 2 months, animals developed the typical phenotypic features of biotin deficiency including hair loss, dry scaly skin, cracking in the corners of the mouth and loss of appetite (Whitehead, 1985; Velázquez‐Arellano et al. 2008). The rest of the experiments were performed on 4‐ to 5‐month‐old Wistar rats subjected for 10–12 weeks to an avidin‐enriched diet. For all experiments, rats of the same sex, age and weight were used as control. Control and biotin‐deficient (BD) animals were killed by intraperitoneal sodium thiopental overdose (120–150 mg kg−1).
Measurement of biotin levels in plasma
Biotin levels in plasma of control and BD animals were measured by ELISA (Biotin ELISA kit, Immunodiagnostic, Tyne & Wear, UK) following the manufacturer's instructions. The test is a competitive enzyme‐linked assay based on the sequential incubation of samples with streptavidin–enzyme conjugate and with biotin–albumin conjugate in microtitre plate wells. Blood samples were obtained from aorta or heart ventricles after animal death.
Detection of biotin expression by biotin–avidin amplification
Collected tissues were fixed in 4% paraformaldehyde (PFA) at 4°C for 2 h, washed in PBS, embedded in gelatin and cut into 50 μm thick slices in a vibratome (Leica VT1200S, Wetzlar, Germany). Slices were incubated for 5 min in H2O2 (0.03%) to quench endogenous peroxidase activity. Following permeabilization (PBS‐0.3% TritonX100) the slices from control and BD animals were incubated for 40 min in the biotin–avidin amplification kit reagent (Vectastain Elite ABC kit, Vector Labs, Burlingame, CA, USA). Sections were subsequently washed in PBS to remove excess solution and revealed with 3,3‐diaminobenzidine (DAB) substrate (Dako, Carpinteria, CA, USA). To avoid differences in the development time, samples from control and deficient rats were incubated and developed in the same well.
Immunocytochemistry and morphological studies
Animals were killed by sodium thiopental overdose (120–150 mg kg−1; i.p.), perfused with PBS and 4% paraformaldehyde in PBS, and the tissues (carotid bifurcations and adrenal glands) were dissected, washed with PBS, fixed at 4°C in 4% PFA and cryopreserved for 12 h in a 30% sucrose solution. The different tissues were embedded in Tissue‐Tek OCT (Sakura) and snap‐frozen by quenching in liquid nitrogen. Slices (thickness 10 μm) were cut with a cryostat and stained with the anti‐tyrosine hydroxylase (TH) polyclonal antibody (rabbit anti‐TH, Novus Biologicals, Littleton, CO, USA; 1:5000). The signal was detected using a fluorescent secondary antibody (anti‐rabbit IgG Alexa Fluor 594, 1:500; Molecular Probes, Carlsbad, CA, USA). To co‐localize biotin and TH, following the immunohistochemical labelling of TH+ cells, the slices were incubated for 30 min with Fluorescent Avidin DN (1:100, Vector Labs). Subsequently, a mixture of Fluorescent Avidin Biotin DN (1:100), pre‐incubated for 30 min was added and the mix incubated for 1 h. Finally the nuclei were stained with DAPI (1:1000; Sigma, St Louis, MO, USA). To detect vesicular monoamine transporter (VMAT) 1 and 2, the following antibodies were used: rabbit anti‐VMAT1 and VMAT2 (Phoenix Pharmaceuticals, Inc., Burlingame, CA, USA; 1:1000); secondary antibody goat anti‐rabbit IgG Alexa Fluor 594 (Molecular Probes, Invitrogen, 1:500). Images were acquired under a Leica TCS‐SP2 AOBS laser scanning fluorescence confocal microscope or an Olympus Bx‐61 microscope (Tokyo, Japan). Biotin levels were quantified on CB and AM micrographs using Image J software (NIH, Bethesda, MD, USA) and staining intensity was expressed as arbitrary units. A similar quantification was performed for CB slices stained for VMAT2, biotin and DAPI.
To study the location of biotin within glomus cells we performed an adenoviral infection of a primary culture of CB glomus cells with redox‐sensitive green fluorescent protein (Ro‐GFP) directed to the mitochondrial matrix (see Fernández‐Agüera et al. 2015). After 2 days of incubation cells were fixed and labelled with a fluorescent antibody against biotin.
Lactate plasma levels and urine pH measurements
Lactate plasma concentration was determined using the Lactate Plus Meter, a hand‐held testing device from Nova Biomedical (Runcorn, UK). To calibrate the device two control solutions of 1–1.6 and 4–5.4 mM lactate from the supplier were used. To test the lactate concentration in tail blood following a lancet puncture, the first blood drop was discarded and the second was assayed. The measurement was repeated twice per control and BD animals before the animals were killed (10–12 weeks after onset of treatment). Urine samples (approximately 200 μl) were collected in the morning and pH was determined by a standard pH meter.
ATP measurements
ATP content was measured in AM, SCG and CB as described previously (Díaz‐Castro et al 2012). In brief, tissues were homogenized in homogenization solution (100 mM Tris, 4 mM EDTA, pH 7.75), boiled for 3 min, chilled for 2 min, and then centrifuged for 1 min at 1000 g. The ATP concentration of the supernatants was assessed using the ATP Bioluminescence Assay CLS II kit (Roche Applied Science, Madison, WI, USA) following the manufacturer's instructions. For adrenal medulla and superior cervical ganglion tissue, ATP levels were normalized to protein content measured by the Bradford assay. Owing to the small size of the CB, ATP levels were normalized with the absorbance at 280 nm.
Amperometric recording of catecholamine secretion in slices
Animals were killed by intraperitoneal administration of a lethal dose of sodium thiopental (120–150 mg kg−1, i.p.). CBs and adrenal glands were dissected and placed on ice‐cooled and O2‐saturated modified Tyrode solution (mM: 148 NaCl, 2 KCl, 3 MgCl2, 10 Hepes, 10 glucose, pH 7.4). CB and adrenal gland slices were prepared after immersion in low melting point agarose as described previously (Pardal & López‐Barneo, 2002; García‐Fernández et al. 2007). CB slices (150 μm thick) were placed on 35 mm Petri dishes with Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum with or without biotin (SH 30068.03n; Thermo Scientific, Waltham, MA, USA), 1% penicillin/streptomycin, 1% l‐glutamine and 84 μU of insulin ml−1) and maintained at 37°C in a 5% CO2 incubator for 24 h before use. Adrenal gland slices (200 μm thick) were incubated for 15 min in recording solution bubbled with 95% O2 and 5% CO2 at 37°C before use. For the experiments, single slices were transferred to the chamber (volume ∼0.2 ml) placed on the stage of an upright microscope (Axioscope, Zeiss, Göttingen, Germany) and continuously perfused by gravity (flow 1–2 ml min−1) with a solution containing (mM): 117 NaCl, 4.5 KCl, 23 NaHCO3, 1 MgCl2, 2.5 CaCl2, 5 glucose and 5 sucrose. The ‘normoxic’ solution was bubbled with a gas mixture of 5% CO2, 20% O2 and 75 % N2 ( ∼150 mmHg). The ‘hypoxic’ solution was bubbled with 5% CO2 and 95 % N2 ( ∼10–20 mmHg). In the high K+ solutions, KCl replaced NaCl equimolarly. The osmolality of solutions was ∼300 mosmol kg−1 and pH was 7.4. All experiments were carried out at a chamber temperature of ∼36°C. Secretory events were recorded with a 10 μm carbon‐fibre electrode positioned under visual control. Amperometric currents were recorded with an EPC‐8 patch‐clamp amplifier (HEKA Electronics, Lambrecht/Pfaltz, Germany), filtered at 100 Hz and digitized at 250 Hz for storage in a computer. Data acquisition and analysis were carried out with an ITC‐16 interface (Instrutech Corporation, New York, USA) and PULSE/PULSEFIT software (HEKA Electronics). The secretion rate (fC min−1) was calculated as the amount of charge transferred to the recording electrode during a given period of time.
Patch clamp recordings in dispersed CB glomus cells
Dissected rat CBs were incubated at 37°C for 20 min in an enzyme solution (1 ml PBS with 0.6 mg collagenase type II, 0.3 mg trypsin, 10 μl elastase I and 10 μl CaCl2 from a 5 mM stock solution), and cells were mechanically dispersed and plated on glass cover slips treated with poly‐l‐lysine and kept in DMEM with 10% fetal bovine serum with or without biotin (SH 30068.03n; Thermo Scientific), 1% penicillin/streptomycin, 1% l‐glutamine and 84 μU insulin ml–1 at 37°C in a 5% CO2 incubator before use. Macroscopic ionic currents were recorded by using the whole‐cell configuration of the patch clamp technique as adapted to our laboratory (Levitsky & López‐Barneo, 2009; Ortega‐Sáenz et al. 2010). Patch electrodes (1.8–2.2 MΩ) were pulled from capillary glass tubes (Kimax; 1.5–1.6 mm OD, Kimble Products), fire polished on a microforge MF‐830 (Narishige, Tokyo, Japan) and coated with silicone elastomer (Sylgard 184; Dow Corning, Midland, MI, USA) to decrease capacitance. Voltage‐clamp recordings were obtained with an EPC‐8 patch clamp amplifier (Heka Elektronik) using standard voltage‐clamp protocols. Unless otherwise noted, the holding potential used was −80 mV. All experiments were conducted at room temperature (22–25°C). Experiments designed to estimate the cell's resting potential and input resistance were made using perforated patches with amphotericin B in the pipette solution. This solution also contained, in mM: 70 K2SO4, 30 KCl, 2 MgCl2, 1 EGTA, 10 Hepes, pH 7.2. The standard bath solution contained, in mM: 140 NaCl, 2.5 KCl, 10 Hepes, 10 glucose, 2.5 CaCl2, 4 MgCl2, pH 7.4. Macroscopic Ca2+ and Na+ currents were recorded in dialysed glomus cells. The solutions used for the recording of whole‐cell Na+ and Ca2+ currents contained (in mM): External (140 NaCl, 9 BaCl2, 1 CaCl2, 10 Hepes and 10 glucose; pH 7.4 and osmolality 300 mosmol kg−1); Internal (110 CsCl, 30 CsF, 10 EGTA, 10 Hepes and 4 ATP‐Mg; pH 7.2 and osmolality 290 mosmol kg−1). Cell capacitance was estimated from the time integral of capacitive current transients recorded by application of 2 ms depolarizing pulses of 20 mV from the holding potential of –80 mV. To calculate cell size, we assumed a constant membrane‐specific capacity of 1 μF cm−2.
Cytosolic Ca2+ measurements
For cytosolic Ca2+ measurements rat glomus cells were incubated in DMEM containing 4 μm Fura 2‐AM (Thermo Scientific, F1225) for 30 min at 37°C in a 5% CO2 incubator. For the experiments, a coverslip with loaded cells was placed in a recording chamber mounted on the stage of an inverted microscope (Eclipse Ti, Nikon) equipped with epifluorescence and photometry (Fernández‐Agüera et al. 2015). Alternating excitation wavelengths of 340 and 380 nm were used, and background fluorescence was subtracted before obtaining the F340/F380 ratio. Cytosolic [Ca2+] was digitized at a sampling interval of 1 s. All the experiments were performed at room temperature (∼25°C).
Measurement of catecholamine content
AMs were harvested and frozen in liquid nitrogen for posterior analysis of catecholamine content. Tissues were sonicated in 200 μl of a chilled monoamine stabilization solution containing 0.1 m HClO4, 0.02% EDTA and 1% ethanol. At this point the cellular extracts were centrifuged at 16,000 g for 10 min at 4°C. The resulting supernatant was filtered through a 30,000 MW exclusion membrane using Centrifugal Filter Devices (Millipore, Billerica, MA, USA) by centrifugation at 16,000 g for 30 min at 4°C. Filtered samples were applied to HPLC. Samples were analysed with an ALEXYS 100 (Antec Leyden, Zoeterwoude, The Netherlands) system equipped with a reversed‐phase C‐18 column (3 μm particle size, 150 × 2.1 mm dimension), a glassy carbon electrode and an in situ ISAAC reference electrode (all from Antec Leyden). To perform the catecholamine analysis of CBs, animals were perfused with PBS (to remove blood catecholamines present in this organ due to its high irrigation) and dissected CBs were lightly dissociated followed by manual pipetting in monoamine stabilization solution and then centrifugation at 16,000 g for 10 min. The concentrations of catecholamines for all tissues were expressed as nanograms per milligram of protein. Pelleted proteins were resuspended in 0.1 m NaOH for protein quantification by the Bradford assay.
RNA isolation and real‐time quantitative PCR
Total RNAs were isolated from CB, SCG and AM tissue with the RNeasy micro kit (Qiagen, Valencia, CA, USA) as previously described (Gao et al. 2013). Due to the small size of the CB, cRNA was amplified using an Ambion WT expression kit (Life Technologies, Carlsbad, CA, USA). Five hundred nanograms or less of total RNA (or amplified cRNA in the case of CB) was copied to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) in a final volume of 20 μl. Real‐time quantitative PCRs were performed in a 7500 Fast Real Time PCR System (Life Technologies). PCRs were performed in duplicate in a total volume of 20 μl containing 1–2.5 μl of cDNA solution and 1 μl of Taqman probe of the specific gene (Life Technologies). Gapdh was also estimated in each sample to normalize the amount of total RNA (or cRNA) used in order to perform relative quantifications.
Plethysmography
To assess ventilatory function, awake and unrestricted rats were placed inside the plethysmography chambers (EMKA Technologies, Paris, France) as previously described (Macías et al. 2014). Chambers were constantly perfused with air containing either 21% O2 (normoxia) or 9% O2 (hypoxia). The experimental protocol consisted of two cycles of normoxia–hypoxia–normoxia with at least 5 min of stabilized O2 level in each condition. O2 tension and respiratory parameters were continuously monitored and recorded in each experiment.
Statistical analysis
Unless otherwise specified, data are expressed as mean ± standard error of the mean (SEM) with the number (n) of experiments indicated. Statistical analysis was performed by unpaired Student's t test. A value of P < 0.05 was considered to indicate statistical significant. One‐way ANOVA was used to compare means of three or more samples.
Results
Differential accumulation of biotin in peripheral nervous system tissues
We studied the level of biotin accumulated in several tissues, particularly in peripheral neural organs. Slices of the carotid artery bifurcation, shown in Fig. 1 A, illustrate the high level of biotin in the CB and the undetectable expression of the coenzyme in the SCG and the carotid arteries. AM cells also expressed biotin (Fig. 1 B) although at much lower levels than CB cells (see below). Interestingly, with the histochemical method used to detect biotin (ABC kit) we did not see any appreciable signal in liver or brain (cerebral cortex and striatum) tissue, even though these tissues are known to have high carboxylase activity (Mock, 1996; Pacheco‐Alvarez et al. 2004). Given the differential accumulation of biotin in the SCG, AM and CB, we examined the levels of expression in these tissues of SLC5a6, a sodium‐dependent multivitamin transporter member of the solute carrier transporter (SLC) family that mediates the uptake of biotin in numerous mammalian cell types (Spector & Mock, 1987; Grassl, 1992; Baur & Baumgärtner, 2000; for review see Said, 1999). SLC5a6 mRNA was markedly up‐regulated in the CB in comparison with the other tissues analysed (Fig. 1 C). Interestingly, similar qualitative differences were observed in the expression of SLC19a3, a thiamine transporter that appears to be regulated by biotin (Fig. 1 D) (Ganapathy et al. 2004; Vlasova et al. 2005; Zeng et al. 2005; Subramanian et al. 2006). Immunocytochemical studies showed the selective expression of biotin in TH‐positive CB glomus cells (Fig. 1 E, F) and by sequential scan detection the co‐localization of the coenzyme with mito‐Ro‐GFP, a genetic mitochondrial marker (Fig. 1 G). These data demonstrate the high level of biotin expression in mitochondria of sympathoadrenal paraganglia, particularly in the CB.
Figure 1. Accumulation of biotin in peripheral neural tissues.
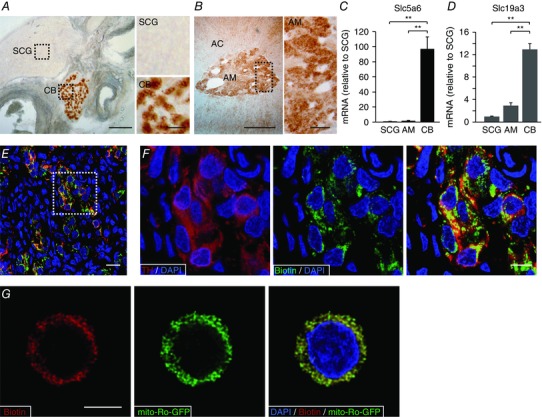
A and B, left: detection of biotin levels in slices of carotid artery bifurcation and adrenal gland. Scale bars 0.5 mm. Right: magnification of superior cervical ganglia (SCG), carotid body (CB) and adrenal medulla (AM) parenchyma indicated by the squares in the corresponding panels on left. Scale bars = 0.1 mm. C and D, Slc5a6 and SLC19a3 mRNA levels in CB and AM compared to SCG (mean ± SEM, n = 5 per group). **Statistical significance P < 0.01. E, fluorescence immunohistochemical detection of tyrosine hydroxylase (red) and endogenous biotin (green) in carotid body slices from control rats. Scale bar = 20 μm. F, magnification of the area inside the square in E to illustrate the co‐localization of biotin and TH. Scale bar = 10 μm. G, confocal image showing the immunofluorescent co‐localization of biotin with mito‐ro‐GFP expressed in a dispersed glomus cell by adenoviral infection. The mito‐ro‐GFP directs the expression of GFP to the mitochondrial matrix. Scale bar = 5 μm.
To investigate the functional role of biotin in CB and AM cells, we induced biotin deficiency in animals fed an avidin‐enriched diet (Whitehead, 1985; Velázquez‐Arellano et al. 2008). Figure 2 A–C shows a quantitative analysis of biotin levels in the CB (30,683 ± 5205 arbitrary units; n = 7 rats) and the AM (14,778 ± 3084 arbitrary units; n = 5 rats) and the time course of biotin reduction in the two tissue types from animals fed a biotin‐free diet for 60 days. In these experiments, CB (Fig. 2 A) and AM (Fig. 2 B) slices were processed in parallel and therefore the variations in the intensity of biotin staining indicate the different biotin levels present in CB and AM cells. The data clearly show the different biotin expression levels in CB and AM and that, whereas AM cells lose most of their biotin content in the first 2 weeks, CB cells maintain relatively high levels of the coenzyme after 2 months of biotin‐free treatment. At this stage, plasma biotin levels were negligible, decreasing from normal values of 21,569 ± 2840 to 37 ± 9 ng l−1 (n = 10) (Fig. 2 D). Weight gain was markedly diminished in animals fed an avidin‐enriched diet (Fig. 2 E) and they also showed signs of cachexia, which are characteristic of biotin deficiency (Whitehead, 1985). When maintained on a biotin‐free diet for longer time periods (∼10–12 weeks), rats died before biotin had completely disappeared from CB cells. At this time point, animals showed a pronounced lactic acidosis (Fig. 2 F) and aciduria (Fig. 2 G).
Figure 2. Biotin depletion in peripheral neural tissues during biotin deficiency.
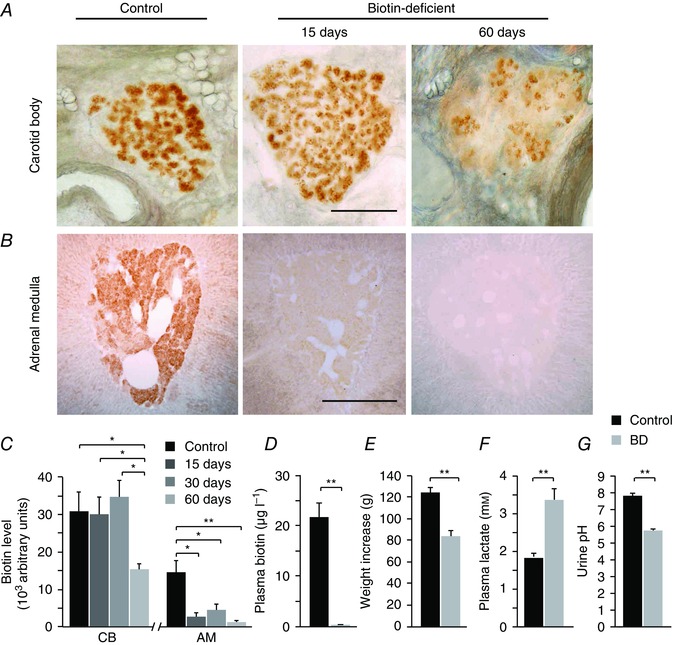
A and B, biotin levels in slices of the carotid artery bifurcation and adrenal gland in control animals (left), and after 15 (middle) or 60 (right) days in avidin‐enriched diet to generate biotin deficiency. Scale bar = 0.5 mm. Note the rapid disappearance of biotin from the adrenal medulla and the incomplete depletion of biotin from carotid body glomus cells. C, quantification of biotin content (arbitrary units) in CB and AM slices from control animals and after 15, 30 and 60 days in a biotin‐deficient diet [CB control, 30,683 ± 5205 arbitrary units (au), n = 7 rats, 25 slices; CB 15 days, 30,077 ± 4705 au, n = 3 rats, 5 slices; CB 30 days, 34,580 ± 4596 au, n = 3 rats, 7 slices; CB 60 days, 15,400 ± 1415 au, n = 3 rats, 5 slices; AM control, 14,778 ± 3084 au, n = 5 rats, 12 slices; AM 15 days, 2841 ± 1188 au, n = 3 rats, 4 slices; AM 30 days, 4629 ± 1595 au, n = 3 rats, 7 slices; AM 60 days, 1511 ± 421 au, n = 3 rats, 6 slices]. ANOVA CB control vs. CB 60 days P < 0.01; CB 15 days vs. CB 60 days P < 0.05; CB 30 days vs. CB 60 days P < 0.05; AM control vs. AM 15 days P < 0.01; AM control vs. AM 30 days P < 0.01; AM control vs. AM 60 days P < 0.01. D, quantification of plasma biotin levels by ELISA in control and biotin‐deficient (BD, 60 day diet) rats (control, 21.57 ± 2.84 μg l−1, n = 10; BD rats, 0.037 ± 0.01 μg l−1, n = 10, P < 0.001). E, body weight increase during the 2 month biotin‐free diet (between 2 and 4 months of age) in control (124 ± 5, n = 26) and BD (82 ± 5 g, n = 27) rats. Control vs. BD P < 0.01. F, plasma lactate concentration (mm): Control, 1.84 ± 0.09, n = 16 measurements in 6 rats; BD, 3.36 ± 0.3, n = 15 measurements in 6 rats; P < 0.01. G, urine pH in control (7.76 ± 0.13, n = 29 measurements in 6 animals) and BD (5.70 ± 0.14, n = 18 measurements in 6 animals) conditions at the end of treatment. BD P < 0.01.
Biotin‐deficient rats exhibit selective CB secretory dysfunction
To study whether biotin deficiency alters CB function we used amperometry to monitor catecholamine secretion from glomus cells in slices in response to hypoxia ( ∼15 mmHg), a characteristic physiological property of CB glomus cells (Fig. 3 A) (see López‐Barneo et al. 2016). Biotin deficiency resulted in a marked reduction of the secretory response induced by hypoxia (Fig. 3 A, B), with the secretion rate (the amount of transmitter released per minute, 1.56 ± 0.34 pC min−1, n = 13 rats) decreasing to less than 40% of that in control slices (3.92 ± 0.44 pC min−1, n = 23 rats; Fig. 3 C) in spite of the number of secretory events (spikes) remaining the same (Fig. 3 D). These data suggest that the small hypoxia‐induced secretory response observed in the absence of biotin was predominantly due to a generalized decrease in the size of quantal exocytotic events rather than to a marked loss of responsiveness to lowered O2 tension (see below). Indeed, a similar decrease in secretion rate and the size of secretory events with respect to controls was also observed in biotin‐deficient glomus cells activated by depolarization with high (40 mM) extracellular K+ (Fig. 3 E–H). The average charge of quantal events recoded during exposure to hypoxia decreased from 56.4 ± 3.7 fC (n = 970 spikes from 11 experiments) in controls to 24.3 ± 15 fC (n = 1088 spikes from 15 experiments) in biotin‐deficient animals. Frequency distribution histograms clearly show that glomus cells from biotin‐deficient rats exhibit a marked increase in the number of events with low dopamine content and a decrease in the number of large events compared with control animals (Fig. 3 I).
Figure 3. Selective carotid body dopamine secretory dysfunction in biotin‐deficient animals.
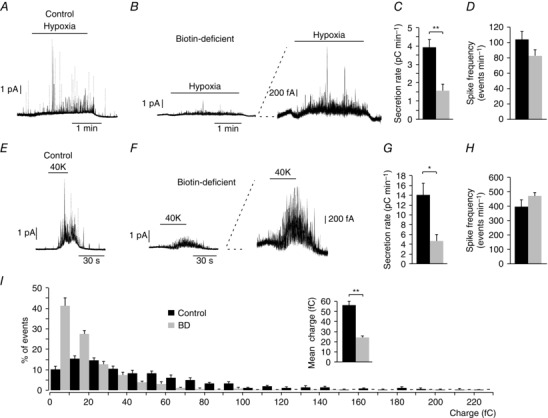
A and B, amperometric recordings showing glomus cell secretory responses to hypoxia in control and biotin‐deficient animals. Note in the trace at higher gain (B, right) that biotin‐deficient cells still retain their ability to respond to a decrease in O2 tension. C, average secretion rate induced by hypoxia in control (black, 3.92 ± 0.44 pC min−1, n = 23) and biotin‐deficient (BD) (grey, 1.56 ± 0.34 pC min−1, n = 13) animals. Control vs. BD hypoxia P < 0.01. D, spike frequency, measured in events per minute, elicited by hypoxia in control (black, 103.61 ± 10.51 events min−1, n = 13) and BD (grey, 82.34 ± 7.77 events min−1, n = 25) rats. Control vs. BD P > 0.05. E and F, secretory response to depolarization with 40 mM K+ in glomus cells from control and biotin‐deficient animals, respectively. Note in the trace at higher gain (F, right) that biotin‐deficient cells retain their ability to respond to high potassium. G, average secretion rate induced by high potassium in control (black, 14.1 ± 2.2 pC min−1, n = 20) and biotin‐deficient rats (grey, 4.6 ± 1.3 pC min−1, n = 19). Control vs. BD P < 0.05. H, quantification of the spike frequency induced in glomus cells by 40 mM K+ in control (black, 398.00 ± 45.42 events min−1, n = 9) and biotin‐deficient (grey, 470.76 ± 28.28 events min−1, n = 13) rats. Control vs. BD P > 0.05. I, frequency–charge distribution of individual exocytotic events elicited in response to hypoxia from glomus cells in carotid body slices prepared from control and biotin‐deficient rats (control, black, n = 970 spikes, 11 recordings; BD, grey, n = 1088 spikes, 15 recordings). The inset in I shows mean vesicle charge obtained from both groups of animals. Control, 56.4 ± 3.7 fc min−1, n = 970 spikes, 11 recordings; BD, 24.3 ± 1.5 fc min−1, n = 1088 spikes, 15 recordings. Control vs. BD P < 0.01.
Given the secretory alterations observed in glomus cells with decreased biotin content, we investigated whether a similar phenotype could be seen in AM chromaffin cells, which are also O2 sensitive (Thompson et al. 1997; Rychkov et al. 1998; García‐Fernández et al. 2007) and, as shown above, contain relatively high levels of biotin. Figure 4 A–C illustrates secretory responses to hypoxia in adult chromaffin cells, which have a secretion rate > 10 times higher than that in CB cells (compare values in Fig. 3 C and Fig. 4 C). However, neither the secretion rate nor the number of events induced by hypoxia and high K+ in adult AM cells was altered by biotin deficiency. As the quantal size (number of catecholamine molecules) of secretory events from chromaffin cells is on average ∼20 times larger than in glomus cells (compare bar charts of single events charges in Fig. 3 I and Fig. 4 E), we analysed in detail the frequency distribution of events with a size < 0.25 pC, which is within the range of those recorded in glomus cells. The small exocytotic events of chromaffin cells were also unaffected by the absence of biotin (Fig. 4 F), thus suggesting that the effect of the coenzyme is specific to the small secretory granules in glomus cells.
Figure 4. Secretory activity of chromaffin cells in adrenal medulla slices from control and biotin‐deficient animals.
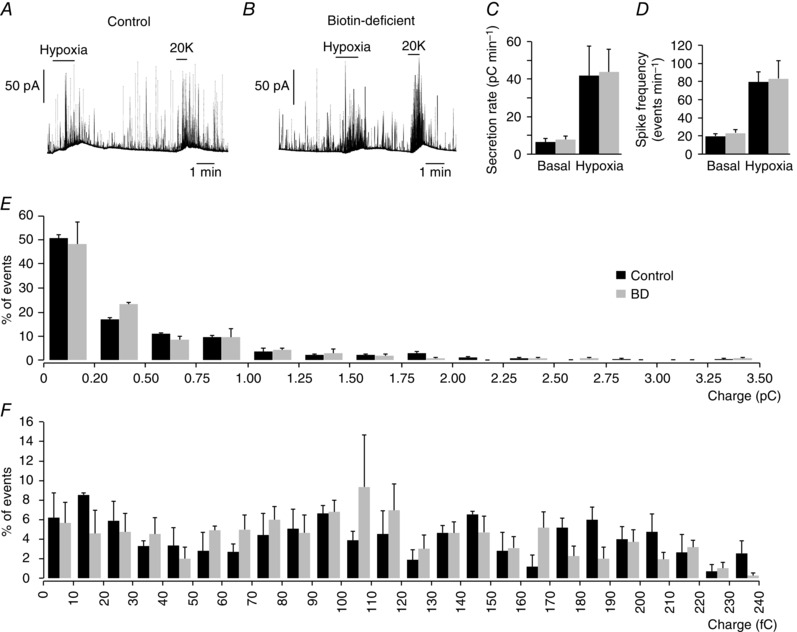
A and B, amperometric recording of secretory responses of chromaffin cells to hypoxia and to depolarization with 20 mM K+ in control and biotin‐deficient animals. C, secretion rate in pC min−1 recorded under basal and hypoxic conditions from control (black, 41.45 ± 15.56 pC min−1, n = 5) and biotin‐deficient (grey, 43.74 ± 12.40 pC min−1, n = 5) animals. D, spike frequency obtained under basal and hypoxic stimulation of control (black, 80 ± 11 spikes min–1, n = 5) and biotin‐deficient (grey, 83 ± 19 pC min−1, n = 5) animals. E, frequency–charge distribution of individual exocytotic events recorded from chromaffin cells of control and biotin‐deficient rats in response to hypoxia (control, black, n = 311 spikes, 3 recordings; biotin‐deficient, grey, n = 445 spikes, 4 recordings). F, frequency–charge distribution of small vesicles (from 0 to 240 fC) recorded from chromaffin cells of control and biotin‐deficient rats (control, black, n = 152 spikes, 3 recordings; biotin‐deficient, grey, n = 221 spikes, 4 recordings). No statistically significant differences were observed in C–F.
Biotin‐deficient cells have normal electrophysiological and neurochemical properties
To shed further light on the biotin‐dependent secretory phenotype in glomus cells, we investigated whether biotin deficiency alters the function of membrane ion channels that regulate transmembrane Ca2+ influx required for exocytosis and/or the metabolic machinery necessary for catecholamine synthesis in cells. Remarkably, the electrophysiological parameters (capacitance and input resistance) were similar in control and biotin‐deficient cells (Table 1), as were the amplitude (current density) and peak I–V relationships for voltage‐gated K+, Na+ and Ca2+ channels (Fig. 5 A–E, Table 1). Even the fast and slowly deactivating components of the Ca2+ current characteristic of glomus cells (Ortega‐Sáenz et al. 2010) were also practically unchanged in cells from biotin‐deficient samples (Fig. 5 F, G, Table 1). The resting membrane potential of biotin‐deficient cells tended to be slightly (∼5 mV) more depolarized than controls, although these differences were not statistically significant (Table 1).
Table 1.
Electrophysiological parameters of control and biotin‐deficient glomus cells
| Control | Biotin‐deficient | P (t test) | Voltage‐clamp configuration | |
|---|---|---|---|---|
| Input resistant (GΩ) | 6.8 ± 0.5 (n = 30) | 5.8 ± 0.5 (n = 21) | 0.19 | Perforated patch |
| V m resting (mV) | −53.6 ± 2.1 (n = 32) | −49.8 ± 1.8 (n = 24) | 0.18 | Perforated patch |
| Cell capacitance (pF) | 4.8 ± 0.3 (n = 50) | 4.5 ± 0.4 (n = 37) | 0.54 | Whole cell. Dialysed cells |
| Peak K+ current (V m = +50 mV (pA) | 411.9 ± 87.9 (n = 23) | 397.5 ± 55.3 (n = 20) | 0.89 | Perforated patch |
| Peak Na+ current (V m = +10 mV (pA) | −61.3 ± 17.0 (n = 12) | −62.8 ± 12.0 (n = 23) | 0.94 | Whole cell. Dialysed cells |
| Peak Ca2+ current (V m = 0 mV (pA) | −7.1 ± 0.8 (n = 40) | −6.5 ± 0.7 (n = 33) | 0.63 | Whole cell. Dialysed cells |
| Fast deactivating Ca2+current density (pA pF–1) | −34 ± 4 (n = 50) | −39 ± 4 (n = 36) | 0.41 | Whole cell. Dialysed cells |
| Slow deactivating Ca2+ current density (pA pF–1) | −7 ± 1 (n = 48) | −5 ± 1 (n = 33) | 0.24 | Whole cell. Dialysed cells |
| Tau fast deactivating Ca2+ current (μs) | 105 ± 6 (n = 50) | 97 ± 5 (n = 37) | 0.32 | Whole cell. Dialysed cells |
| Tau slow deactivating Ca2+ current (ms) | 1.0 ± 0.1 (n = 48) | 1.28 ± 0.14 (n = 34) | 0.09 | Whole cell. Dialysed cells |
Figure 5. Ionic currents of control and biotin‐deficient dispersed glomus cells.
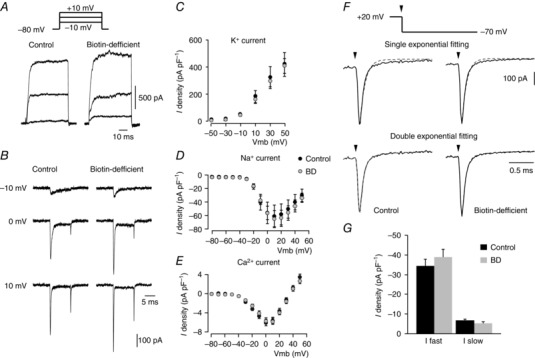
A, macroscopic outward potassium currents recorded in the perforated patch configuration of the patch‐clamp technique in glomus cells obtained from control (left) and biotin‐deficient (right) animals in response to depolarizing pulses from a holding potential of –80 mV to −10, 0 and +10 mV (a scheme of the pulse protocol is shown at the top). B, macroscopic inward sodium and calcium currents recorded with the whole‐cell configuration of the patch‐clamp technique in glomus cells from control (left) and biotin‐deficient (right) animals in response to depolarizing pulses from a holding potential of –80 mV to −10, 0 and +10 mV. C–E, peak current density versus voltage curves for K+ (n = 20–23 cells), Na+ (n = 12–23 cells) and Ca2+ (n = 33–40 cells) currents, respectively, obtained from dispersed glomus cells of control (black) and biotin‐deficient animals (grey) F, single (top) and double exponential (bottom) functions fitted to the Ca2+ tail current recorded on repolarization to –70 mV (vertical arrows) from a membrane potential of +20 mV in control (left) and biotin‐deficient (right) glomus cells. In the double exponential fit, the fast and slow time constant values were: control cells, 0.105 ± 0.006 (n = 50) and 0.999 ± 0.100 ms (n = 48); biotin‐deficient cells, 0.096 ± 0.005 (n = 37) and 1.280 ± 0.141 ms (n = 34) G, current density of the fast and slow components of the calcium tail currents recorded from glomus cells of control and biotin‐deficient rats (control cells: fast component, –34 ± 4 pA pF–1, n = 50; slow component, –7 ± 1 pA pF–1, n = 48; biotin‐deficient cells: fast component, –39 ± 4 pA pF–1, n = 36; slow component, –5 ± 1 pA pF–1, n = 33). No statistically significant differences were observed in C, D, E or G.
Given that biotin‐dependent carboxylases play a central role in several metabolic pathways, we tested whether energy or catecholamine metabolism where altered in biotin‐deficient sympathoadrenal cells. ATP levels in CB, AM and SCG were unaltered in avidin‐treated rats (Fig. 6 A). Similarly, mRNA levels of TH, the rate‐limiting enzyme in catecholamine biosynthesis, were also unchanged (or even slightly increased) in biotin‐deficient CB and AM cells (Fig. 6 B). Furthermore, HPLC analyses of CB and AM cell extracts demonstrated that catecholamine content was not decreased by biotin deficiency (Fig. 6 C, D); on the contrary, we even observed a statistically significant increase in the CB dopamine content in biotin‐deficient rats (Fig. 6 C). These data indicate that although the lack of biotin induces a marked secretory dysfunction in CB glomus cells, it affects neither glomus cell electrophysiology nor ATP or catecholamine synthesis. The high levels of dopamine observed in CBs from biotin‐deficient rats suggest that the deficiency of the coenzyme could selectively alter the process of dopamine transport into the small secretory vesicles present in glomus cells (see below).
Figure 6. Metabolic status and catecholamine synthesis in carotid body and adrenal medulla from control and biotin‐deficient animals.
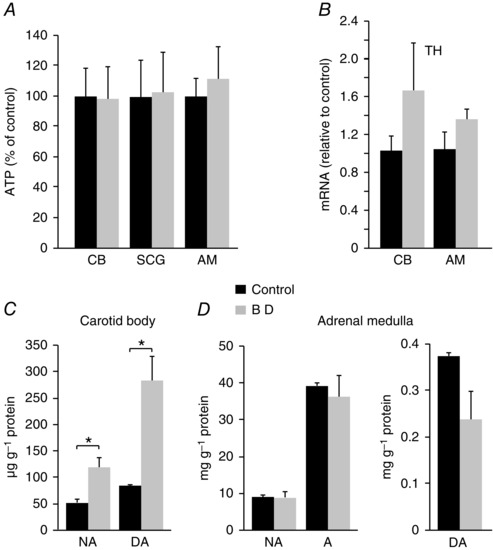
A, ATP levels in biotin‐deficient carotid body (CB), superior cervical ganglion (SCG) and adrenal medulla (AM) expressed relative to control. ATP values in nmol mg−1 protein: CB: Control (0.2 ± 0.04), BD (0.2 ± 0.04) n = 7, P > 0.05; SCG: Control (1.80 ± 0.45), BD (1.84 ± 0.47) n = 9, P > 0.05; AM: Control (81.8 ± 10.2), BD (91.4 ± 17.25), n = 7, P > 0.05. B, mRNA expression levels of tyrosine hydroxylase (TH) in the CB and AM of biotin‐deficient (grey) relative to control (black) animals (n = 5 per group). CB: Control (1.03 ± 0.15), BD (1.66 ± 0.51), P > 0.05; AM: Control (1.05 ± 0.16), BD (1.36 ± 0.1), P > 0.05. C. Noradrenaline (NA) and dopamine (DA) content, in μg g−1 of protein, measured by HPLC from carotid bodies of control and biotin‐deficient animals. NA: Control (52.21 ± 6.21), BD (119.34 ± 18.80), P < 0.05 (n = 3); DA: Control (84.04 ± 2.86), DA (283.47 ± 46.88), P < 0.05 (n = 3). D, noradrenaline (NA), adrenaline (A) (left) and dopamine (DA, right) content measured by HPLC, in mg g−1 of protein, from adrenal medullas of control and biotin‐deficient animals. NA: Control (8.85 ± 0.61), BD (8.62 ± 1.60), P > 0.05 (n = 4); A: Control (38.93 ± 0.98), BD (36.02 ± 5.83), P > 0.05 (n = 4); DA: Control (0.37 ± 0.01), BD (0.24 ± 0.06), P > 0.05 (n = 4).
Responsiveness of biotin‐deficient rats to hypoxia
Biotin or carboxylase deficiency and related disorders in humans produce a syndrome characterized by marked metabolic acidosis, ketonuria and accelerated breathing (Nyhan, 1988; Wolf, 2001; Mardach et al. 2002). These symptoms were also manifested in our animal model of biotin deficiency (see above Fig. 2 F, G). In addition, whole body plethysmography in unrestricted animals exposed to long‐term biotin deficiency (10–12 weeks) typically showed a marked (∼2–3‐fold) increase in basal respiratory frequency with respect to controls, which was also accompanied by a decrease in tidal volume (Fig. 7 A–C). Biotin‐deficient animals showed a clear trend to a higher minute volume than controls although this difference was not statistically significant (Fig. 7 C). Despite differences in basal respiration, control and biotin‐deficient animals showed an equivalent ventilatory response (increase in respiration rate) to hypoxia (Fig. 7 D, E). In agreement with these findings, we observed practically similar responses to high (30 mm) external K+ and hypoxia in single glomus cells loaded with Fura‐2 to measure cytosolic Ca2+ concentrations (López‐Barneo et al. 1999; Fernández‐Agüera et al. 2015) (Fig. 8). Taken together, these data indicate that the secretory dysfunction observed in the CB of biotin‐deficient animals is not due to alterations in cell electrophysiology, dopamine synthesis or cytosolic Ca2+ homeostasis, but rather they point to a primary defect in dopamine vesicular transport or storage in glomus cells.
Figure 7. Basal respiratory parameters and ventilatory response to hypoxia in control and biotin‐deficient animals.
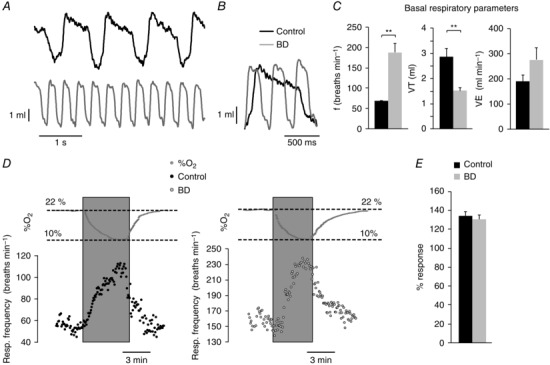
A, representative recordings of basal respiration in control and biotin‐deficient (BD) rats obtained by whole body plethysmography. B, superimposed traces from A to illustrate differences in respiratory frequency. C, average respiratory parameters from control (black, n = 18) and biotin‐deficient (grey, n = 14) animals during basal respiration. Respiratory frequency in breaths min−1 (left, control, 68.26 ± 1.85; BD, 187.76 ± 21.52; P < 0.001); tidal volume in ml (middle, control 2.85 ± 0.34; BD, 1.48 ± 0.17; P < 0.01); minute volume in ml min−1 (right, control, 192.63 ± 23.08; BD, 278.30 ± 47.05; P > 0.05). D, representative recordings of the ventilatory response to hypoxia (changes in %O2 are illustrated on top of the recordings) in control (left) and biotin‐deficient (right) rats. E, quantification of the increase in respiratory frequency in response to hypoxia of control (black, n = 13) and biotin‐deficient (grey, n = 8) animals. The effect of hypoxia is expressed as the percentage of frequency increase relative to the basal frequency in each group (control, 133.85 ± 4.31%; BD, 130.17 ± 4.50%; P > 0.05).
Figure 8. Responsiveness to hypoxia of control and biotin‐deficient glomus cells.
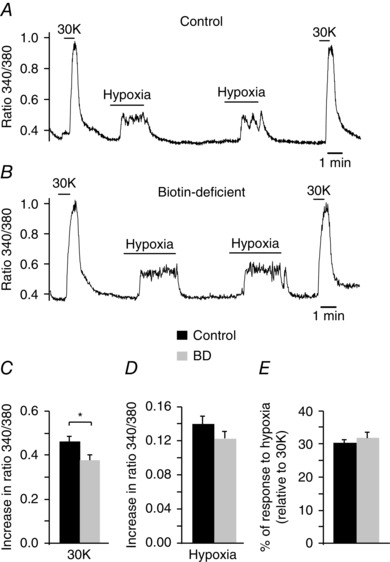
A and B, representative recordings of the ratiometric increase in cytosolic [Ca2+] elicited in Fura‐2 loaded control and biotin‐deficient dispersed glomus cells in response to hypoxia and 30 mM K+. C and D, quantification of cytosolic [Ca2+] increase elicited by 30 mM K+ and hypoxia in control (black, n = 53) and biotin‐deficient (BD, grey, n = 44) cells (30K, control: 0.46 ± 0.02; BD: 0.37 ± 0.02; P < 0.05; hypoxia, control: 0.14 ± 0.01; BD, 0.12 ± 0.0.1; P > 0.05) E, quantification of the hypoxic response (expressed as the percentage of the amplitude of the response to high potassium) in control (30 ± 1%) and BD (32 ± 2%) cells (P > 0.05).
Biotin deficiency and homeostasis of the catecholamine transporter VMAT2
Catecholamine transport into vesicles is mainly mediated by VMAT1 and 2 (for a review see Eiden et al. 2004). Immunocytochemical analyses indicated that although VMAT1 is present in the CB glomeruli, VMAT2 is the most abundant transporter (Fig. 9 A). In contrast, VMAT1 is broadly expressed in the AM, while VMAT2 expression seems to be restricted to isolated groups of cells (Fig. 9 B). Although biotin deficiency did not alter VMAT1 and VMAT2 mRNA expression (Fig. 9 C), we studied the changes in VMAT2 protein levels, as it is known that biotin can affect post‐translational modification of proteins in addition to its metabolic role (Zempleni et al. 2009). As the small size of CB tissue precluded us from performing Western blots or other analyses for direct protein quantification, VMAT2 and biotin levels in CB tissue from control and biotin‐deficient animals were studied in parallel by immunocytochemistry (optical density quantification). VMAT2 appeared in biotin‐positive cells as well as in sympathetic nerve fibres innervating the CB. Interestingly, in avidin‐treated animals VMAT2 fluorescence in glomus cells decreased in parallel with the reduction of biotin levels, whereas the VMAT2 signal in nerve fibres remained unaltered (Fig. 9 D, E). These data indicate a selective down‐regulation of VMAT2 protein expression in biotin‐deficient glomus cells.
Figure 9. VMAT expression in carotid body and adrenal medulla: effect of biotin deficiency.
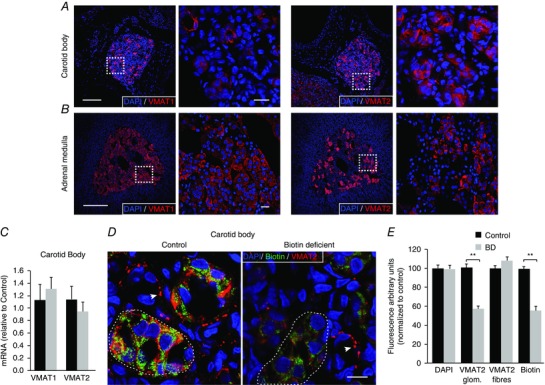
A and B, confocal images of carotid body and adrenal medulla slices illustrating the expression of VMAT1 (left) and VMAT2 (right). Scale bars = 0.5 mm. The squares in the low‐magnification images indicate the area expanded in the accompanying panels. Scale bars = 20 μm. Note that CB cells mainly express VMAT2. C, quantification of VMAT1 and VMAT2 mRNA expression in biotin‐deficient animals (grey) relative to control (black). VMAT1: Control (1.11 ± 0.28), biotin‐deficient (1.29 ± 0.24), P > 0.05; n = 4; VMAT2: Control (1.04 ± 0.18), biotin‐deficient (0.85 ± 0.13), P > 0.05, n = 5. D, confocal images illustrating biotin (green) and VMAT2 (red) co‐expression in CB slices of control and biotin‐deficient rats. Note the parallel decrease in both biotin and VMAT2 expression levels in the CB parenchyma (several CB glomeruli are encircled by the discontinuous line) in biotin‐deficient rats. In contrast, the intensity of VMAT2 immunostaining in fibres (white arrows) remained unaltered. Scale bar = 10 μm. E, quantification of changes in the level of intensity of VMAT2 (in glomeruli and fibres) and biotin labelling expressed as a percentage relative to control. DAPI has been quantified for reference (Control, 16 slices, 4 rats; BD, 20 slices, 5 rats). Decrease in fluorescence, expressed as a percentage relative to control: DAPI, 100.1 ± 2.4%, P > 0.05; VMAT2 glom., 58.2 ± 2.7%, P < 0.001; VMAT2 fibres, 108.5 ± 4.3%, P > 0.05; biotin, 56.4 ± 4.1%, P < 0.001.
To further support the potential modulation of VMAT2 protein expression by biotin, we incubated CB slices with tetrabenazine (TBZ), a selective inhibitor of VMAT2 (Ugolev et al. 2013). With this treatment we attempted to reproduce the phenotype observed in biotin‐deficient cells. As expected, overnight incubation of CB slices with TBZ (50 μm) produced an inhibition of the secretory response of CB glomus cells induced by either hypoxia or high extracellular K+ which was similar to that observed under biotin deficiency (Fig. 10 A–C). In contrast, in chromaffin cells subjected to the same treatment the secretory responses to several stimuli (high extracellular K+, hypoxia and hypercapnia) remained unaltered (Fig. 10 D–F). Furthermore, in CB cells treated with TBZ, the frequency distribution of quantal events was modified in a manner similar to that observed above in biotin‐deficient animals (Fig. 10 G). In agreement with the data shown in Fig. 4 (see above), the inhibition of VMAT2 did not produce any change in the distribution of quantal events in AM cells (Fig. 10 H).
Figure 10. Dysfunction of carotid body dopamine secretion after pharmacological inhibition of VMAT2.
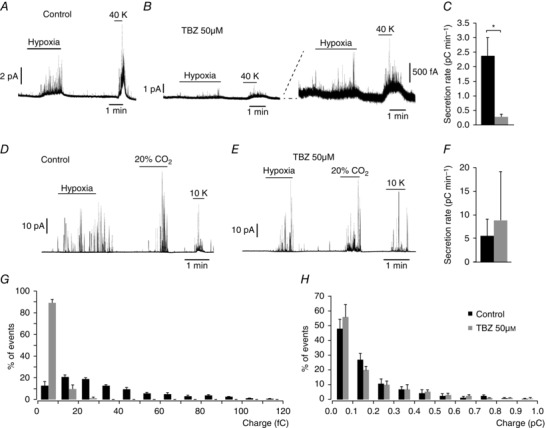
A and B, amperometric recordings showing secretory response to hypoxia and to depolarization with 40 mM K+ of glomus cell in slices incubated overnight in control medium and in the presence of 50 μM TBZ (specific VMAT2 inhibitor). Note in the scaled traces (in B, right) that TBZ treatment mimics the secretory dysfunction observed in biotin‐deficient glomus cells. C, average secretion rate, in pC min−1, induced by hypoxia in control (2376 ± 634 fC min−1, n = 5 recordings) and TBZ‐treated (274 ± 99 fC min−1, n = 6 recordings) glomus cells. Statistical significance P < 0.005. D and E, amperometric recordings showing AM chromaffin cell secretory response to hypoxia, hypercapnia and to depolarization with 10 mM K+ of control and TBZ‐treated cells. Note that in contrast to the carotid body, similar responses were recorded from both groups. F, average secretion rate induced by hypoxia control (black, 5243 ± 3763 fC min−1, n = 4 recordings) and TBZ‐treated cells (grey, 9134 ± 10,306 fC min−1, n = 2 recordings). Statistical significance P > 0.05. G and H, frequency–charge distribution of individual exocytotic events elicited in response to hypoxia and 20 mM K+ from glomus and chromaffin cells in carotid body and adrenal medulla slices, respectively, incubated in control medium (black, CC control, n = 772 spikes from 7 experiments; AM control, n = 384 spikes from 7 experiments) and in 50 μM TBZ (grey, CC TBZ n = 278 spikes from 5 experiments; AM TBZ, n = 314 spikes from 5 experiments).
Discussion
Accumulation of biotin in peripheral arterial chemoreceptors
Biotin is normally supplied by dietary intake, as it cannot be synthesized in mammalian cells. Although biotin can bind to histones and other proteins, it is found most abundantly attached to the ε‐amino group of a specific lysine residue in carboxylases (see for reviews Mock, 1996; Zempleni, 2009). Biotin is efficiently transported into cells with highly active gluconeogenesis and lipid synthesis (liver) or Kreb's cycle (brain), and it has been reported that in biotin‐deficient animals biotin deposited in liver is lost earlier than in brain tissue (Pacheco‐Alvarez et al. 2004). With our histochemical detection method, however, we did not see any measurable biotin signal in liver, brain, carotid arteries or SCG, suggesting that biotin levels in the CB (or even in the AM) are probably several orders of magnitude higher than in tissues (e.g. liver or brain) classically considered to have high biotin‐dependent carboxylase activity. As expected, CB cells showed high levels of SLC5a6 mRNA expression, the protein of which is a well‐known biotin transporter (see Said, 1999), and biotin was preferentially located in glomus cell mitochondria, where most carboxylases are expressed. Interestingly, the depletion of CB biotin was not complete after 2 months on the avidin‐enriched diet even though the animals presented signs of advanced biotin deficiency (cachexia and aciduria) and biotin had practically disappeared from the blood and other tissues examined. These data demonstrate the capacity of glomus cells to accumulate unusually high levels of biotin, a novel characteristic without precedent in any other cell type.
Secretory phenotype of CB cells in biotin‐deficient animals: modulation of dopamine vesicular content by biotin
We observed a clear and highly reproducible decrease in the size of quantal catecholaminergic secretory events restricted to CB glomus cells in biotin‐deficient animals (not seen in AM cells). However, it is remarkable that despite the severe systemic affectation evident in biotin‐deficient animals, the electrophysiological and neurochemical properties of CB cells as well as the secretory responses of AM chromaffin cells remained unaltered. ATP levels did not change in peripheral nervous tissues (AM, SCG and CB) from biotin‐deficient animals, which agrees with previous data in heart (Velázquez‐Arellanos et al 2008) and suggests that energy metabolism was not affected by the absence of biotin. Moreover, we found that the levels of TH mRNA and catecholamines in biotin‐deficient CBs were normal or, as in the case of dopamine, even increased. Based on these results, we hypothesized that the decrease in quantal dopaminergic content in biotin‐deficient glomus cells could be secondary to alterations in the biogenesis of secretory granules and/or accumulation of dopamine within the vesicles. The fact that the number of secretory events (spikes) induced by either hypoxia or high extracellular K+ did not change in the absence of biotin in AM or CB cells indicates that vesicle biogenesis was not profoundly altered. This was also supported by a preliminary electron microscope study in which we found that the number of small dense‐core vesicles, characteristic of CB chemoreceptors (see for a recent description Platero‐Luengo et al. 2014) appeared to be similar in control and biotin‐deficient glomus cells (data not shown). However, as this morphological study, which was designed to analyse changes (rarefaction) in the density of the vesicle matrix, did not produce robust and reproducible results it was not continued.
Catecholamine transport into secretory vesicles is mediated by VMAT1 and VMAT2, which exhibit different levels of tissue‐specific expression (for reviews see Shuldiner et al. 1995; Eiden et al. 2004). Our immunocytochemical analyses showed that, as reported previously (Eiden et al. 2004), both transporters, but preferentially VMAT1, are expressed in chromaffin cells. In contrast, CB cells preferentially express VMAT2 (Koerner et al. 2004), which is the transporter normally present in neurons (Eiden et al. 2004; Schafer et al. 2013). While VMAT1 and VMAT2 mRNA expression in the whole CB seemed to be unaffected by biotin deficiency, we observed a decrease in VMAT2 protein levels in biotin‐deficient glomus cells. Although the small size of the CB precluded precise Western blot experiments from being performed (for which a much larger amount of tissue is needed) the quantification of confocal immunocytochemical images indicated a clear decrease in VMAT2 protein expression in parallel with the reduction of biotin levels in glomus cells. This finding, together with the fact that the biotin‐dependent secretory phenotype in glomus cells was mimicked by a selective VMAT2 inhibitor, strongly suggests that biotin is required for normal VMAT2 protein expression and function. Interestingly, VMAT2 detected by immunocytochemistry in small fibres (possibly varicosities of efferent autonomic fibres innervating the CB) (Nurse, 2014) seemed to be unaffected by the lack of biotin, thus suggesting that the modulatory role of the vitamin is selectively exerted on VMAT2 in dopaminergic vesicles. As the substantia nigra is among the brain nuclei exhibiting a high biotin content (McKay et al. 2004), it would be interesting to determine in future experiments if VMAT2 expressed in central dopaminergic neurons (soma and terminals) is also modulated by biotin (see below). Biotin could influence VMAT2 stability and turnover by post‐translational modification of the protein in several possible ways. VMAT2 could be directly biotinylated as there is considerable evidence indicating that, in addition to carboxylases, histones and other proteins can also bind biotin to modulate their function (Zempleni, 2005). Moreover, biotin is known to activate the guanyl cyclase/cyclic GMP/protein kinase G pathway (Stockert et al. 1992; Stocker & Ren, 1997), which could indirectly lead to VMAT2 modulation. Indeed, phosphorylation and nitration of VMAT2 residues have been reported (Watabe & Nakaki, 2008; Ramamoorthy et al. 2011). In addition to VMAT2 modulation, biotin could also regulate dopamine accumulation in the secretory granules. In this regard, it has been suggested that dopamine molecules in the CB granules could be packed in micelle‐like supramolecular structures formed by amphipathic N‐acyl dopamine, a condensation product of fatty acid chains and dopamine (Pokorski & Matysiak, 1998). This proposal, although not supported by experimental evidence, must be taken into consideration, as biotin is essential for the synthesis of fatty acids, which are known to influence brain dopaminergic transmission (Zimmer et al. 2002).
In parallel with the CB cellular phenotype, biotin‐deficient animals showed systemic alterations characterized by a marked increase in respiration rate, which is probably due to activation of the central and peripheral chemoreceptors by metabolic lactic acidosis. Hyperventilation could be also favoured by the reduction of CB dopamine release, since it is believed that this transmitter normally exerts an autocrine and/or paracrine inhibitory action on CB chemoreceptor cells (Benot & López‐Barneo, 1990; Bain et al. 2015). However, it is noteworthy that biotin‐deficient animals showed a practically normal hyperventilatory response to hypoxia, which suggests that acetylcholine and ATP, the excitatory transmitters at the afferent chemosensory synapse (see Iturriaga & Alcayaga, 2004; Shirahata et al. 2007; Nurse, 2014), are probably stored and released from secretory vesicles which, unlike the small dopaminergic granules, are unaffected by biotin deficiency. It could also be possible that acidosis greatly enhances the secretory response to hypoxia of glomus cells, a phenomenon that is not seen in vitro where external pH is controlled.
Medical implications
Frank biotin deficiency is a rare disease, although some cases of genetic deficiencies involving individual carboxylases, holocarboxylase synthetase, biotinidase or even biotin transporters have been reported (Nyhan, 1988; Wolf, 2001; Mardach et al. 2002; Pacheco‐Alvarez et al. 2002). Complete loss of function of these enzymes appears to be incompatible with life, as the genetic defects manifested in patients show only partial deficiency of biotin metabolism that can be compensated for by treatment with high doses of the vitamin. Marginal biotin deficiency can be encountered during pregnancy and in smokers or users of some anticonvulsant drugs (Zempleni & Mock, 1999; Mock et al. 2002, Zempleni et al. 2009). Severe biotin deficiency causes alterations to the skin and developmental delays in children as well as encephalopathy with ataxia, hypotonia and seizures. Although, to our knowledge, specific defects of the central and/or peripheral dopaminergic systems have not been reported in biotin‐deficient patients, high doses of biotin are known to improve the symptoms of biotin‐responsive basal ganglia disease (BBGD), a rare autosomal recessive neurological disorder caused by mutations in the gene encoding SLC19a3, a thiamine transporter (Ozand et al. 1998; Rajgopal et al. 2001; Zeng et al. 2005). BBGD typically presents with recurrent subacute encephalopathy leading to seizures, extrapyramidal motor deficits and eventually coma, which are accompanied by characteristic lesions in the basal ganglia (caudate and putamen) and other parts of the brain (Ozand et al. 1998; Tabarki et al. 2013). Most patients recover fully from the crisis after administration of high doses of biotin (or in some cases biotin plus thiamine). BBGD pathogenesis and the precise mechanisms by which the vitamin is effective in improving BBGD are unknown, as biotin does not seem to be a substrate of SLC19a3 (Subramanian et al. 2006; Tabarki et al. 2013). However, biotin can modulate SLC19a3 mRNA expression in human leukocytes, which has led to the suggestion that it could favour thiamine uptake by cells (Vlasova et al. 2005). This idea is supported by our findings in the CB where in parallel with high levels of biotin we observed a constitutive up‐regulation of SLC19a3 mRNA. Another possibility is that by up‐regulating VMAT2 protein expression biotin potentiates dopaminergic transmission and compensates for the lack of thiamin. Given the medical interest of BBGD and other pathologies affecting the nigrostriatal pathway (e.g. Parkinson's disease), future studies are warranted to test whether, as occurs in the dopaminergic glomus cells, biotin can also modulate dopamine vesicular transport/accumulation in central neurons.
Other possible actions of biotin in peripheral chemoreceptor cells
Taken together, the experimental facts described in this report suggest that in addition to the regulation of vesicular dopamine homeostasis, biotin could have other essential roles in CB chemoreceptor cells. However, it is highly likely that the signs of CB biotin deficiency were not fully manifested in the current study because, as indicated above, animals died or reached extreme cachexia before biotin was completely eliminated from glomus cells. Biotin deficiency has detrimental effects on TCA intermediates (e.g. oxaloacetate or succinate) that are necessary as substrates for mitochondrial oxidative phosphorylation as well as for the biosynthesis of essential TCA derivatives (e.g. heme) (Atamna et al. 2007). As a considerable body of knowledge supports the idea that mitochondria play a fundamental role in acute oxygen sensing (see López‐Barneo et al. 2016), it can be speculated that biotin is essential for the metabolic specialization of CB cells necessary to carry out their chemosensory functions. In this regard, it is noteworthy that the CB contains high levels of succinate (which could result from the activity of biotin‐dependent carboxylases) in comparison with the brain or other areas of the peripheral nervous system (Fernández‐Agüera et al. 2015). Indeed, we have proposed a model for CB glomus cell acute oxygen sensing based on MCI signalling to membrane channels, which could be favoured by the succinate‐dependent accumulation of reduced quinone during hypoxia (Fernández‐Agüera et al. 2015; López‐Barneo et al. 2016). In addition to glomus cell metabolic specializations necessary for responsiveness to acute hypoxia, biotin could also participate in CB growth during acclimatization to chronic hypoxia, a process that depends on the activation of a resident population of neural stem cells, which are instructed to proliferate and differentiate in response to low (Platero‐Luengo et al. 2014). It is conceivable that histone biotinylation, which is known to modulate gene expression (see Zempleni et al. 2009), favours activation of the gene programme required for maturation of CB progenitors into differentiated chemosensory glomus cells (Navarro‐Guerrero et al. 2016). In sum, additional roles of biotin in CB physiology might be unveiled in future studies on animal models with selective biotin elimination from glomus cells.
Additional information
Competing interests
The authors have no conflicts of interest to declare.
Author contributions
P.O.‐S., D.M., J.A.R‐G., P.G.‐R., K.L.L., V.B.‐H. and I.A.‐M. performed the experiments and participated in the interpretation of data and the design of figures. P.O.‐S. and J.L.‐B. designed and wrote the first draft of the paper. J.L.B. wrote the final draft of the paper and supervised the project. All authors read and approved the final manuscript.
Funding
This research was supported by the Botín Foundation, the Spanish Ministries of Health and of Economy and Competitiveness (SAF2012‐39343 and PIE13/0004) and the European Research Council (ERC:PRJ201502629).
Acknowledgements
We wish to thank Professor Juan J. Toledo‐Aral for bringing to our attention the high level of endogenous staining of the CB using the amplification ABC kit.
References
- Atamna H, Newberry J, Erlitzki R, Schultz CS & Ames BN (2007). Biotin deficiency inhibits heme synthesis and impairs mitochondria in human lung fibroblasts. J Nutr 137, 25–30. [DOI] [PubMed] [Google Scholar]
- Bain AR, Dujic Z, Hoiland RL, Barak OF, Madden D, Drvis I, Stembridge M, MacLeod DB, MacLeod DM & Ainslie PN (2015). Peripheral chemoreflex inhibition with low‐dose dopamine: new insight into mechanisms of extreme apnea. Am J Physiol Regul Integr Comp Physiol 309, R1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur B & Baumgartner ER (2000). Biotin and biocytin uptake into cultured primary calf brain microvessel endothelial cells of the blood–brain barrier. Brain Res 858, 348–355. [DOI] [PubMed] [Google Scholar]
- Benot AR & Lopez‐Barneo J (1990). Feedback inhibition of Ca2+ currents by dopamine in glomus cells of the carotid body. Eur J Neurosci 2, 809–812. [DOI] [PubMed] [Google Scholar]
- Diaz‐Castro B, Pintado CO, Garcia‐Flores P, Lopez‐Barneo J & Piruat JI (2012). Differential impairment of catecholaminergic cell maturation and survival by genetic mitochondrial complex II dysfunction. Mol Cell Biol 32, 3347–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiden LE, Schafer MK, Weihe E & Schutz B (2004). The vesicular amine transporter family (SLC18): amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine. Pflugers Arch 447, 636–640. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Aguera MC, Gao L, Gonzalez‐Rodriguez P, Pintado CO, Arias‐Mayenco I, Garcia‐Flores P, Garcia‐Perganeda A, Pascual A, Ortega‐Saenz P & Lopez‐Barneo J (2015). Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab 22, 825–837. [DOI] [PubMed] [Google Scholar]
- Ganapathy V, Smith SB & Prasad PD (2004). SLC19: the folate/thiamine transporter family. Pflugers Arch 447, 641–646. [DOI] [PubMed] [Google Scholar]
- Gao L, Hidalgo‐Figueroa M, Escudero LM, Diaz‐Martin J, Lopez‐Barneo J & Pascual A (2013). Age‐mediated transcriptomic changes in adult mouse substantia nigra. PLoS One 8, e62456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Fernandez M, Ortega‐Saenz P, Castellano A & Lopez‐Barneo J (2007. a). Mechanisms of low‐glucose sensitivity in carotid body glomus cells. Diabetes 56, 2893–2900. [DOI] [PubMed] [Google Scholar]
- Garcia‐Fernandez M, Mejias R & Lopez‐Barneo J (2007. b). Developmental changes of chromaffin cell secretory response to hypoxia studied in thin adrenal slices. Pflugers Arch 454, 93–100. [DOI] [PubMed] [Google Scholar]
- Grassl SM (1992). Human placental brush‐border membrane Na+‐pantothenate cotransport. J Biol Chem 267, 22902–22906. [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturriaga R & Alcayaga J (2004). Neurotransmission in the carotid body: transmitters and modulators between glomus cells and petrosal ganglion nerve terminals. Brain Res Brain Res Rev 47, 46–53. [DOI] [PubMed] [Google Scholar]
- Koerner P, Hesslinger C, Schaefermeyer A, Prinz C & Gratzl M (2004). Evidence for histamine as a transmitter in rat carotid body sensor cells. J Neurochem 91, 493–500. [DOI] [PubMed] [Google Scholar]
- Levitsky KL & Lopez‐Barneo J (2009). Developmental change of T‐type Ca2+ channel expression and its role in rat chromaffin cell responsiveness to acute hypoxia. J Physiol 587, 1917–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Barneo J, Ortega‐Saenz P, Gonzalez‐Rodriguez P, Fernandez‐Aguera MC, Macias D, Pardal R & Gao L (2016). Oxygen‐sensing by arterial chemoreceptors: mechanisms and medical translation. Mol Aspects Med 47–48, 90–108. [DOI] [PubMed] [Google Scholar]
- Lopez‐Barneo J, Pardal R, Montoro RJ, Smani T, Garcia‐Hirschfeld J & Urena J (1999). K+ and Ca2+ channel activity and cytosolic [Ca2+] in oxygen‐sensing tissues. Respir Physiol 115, 215–227. [DOI] [PubMed] [Google Scholar]
- Macías D, Fernández‐Agüera MC, Bonilla‐Henao V & Loópez‐Barneo J (2014). Deletion of the von Hippel‐Lindau gene causes sympathoadrenal cell death and impairs chemoreceptor‐mediated adaptation to hypoxia. EMBO Mol Med 6, 1577–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardach R, Zempleni J, Wolf B, Cannon MJ, Jennings ML, Cress S, Boylan J, Roth S, Cederbaum S & Mock DM (2002). Biotin dependency due to a defect in biotin transport. J Clin Invest 109, 1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BE, Molineux ML & Turner RW (2004). Biotin is endogenously expressed in select regions of the rat central nervous system. J Comp Neurol 473, 86–96. [DOI] [PubMed] [Google Scholar]
- Mock DM (1996). Biotin. International Life Sciences Institute Press, Washington, DC. [Google Scholar]
- Mock DM, Quirk JG & Mock NI (2002). Marginal biotin deficiency during normal pregnancy. Am J Clin Nutr 75, 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro‐Guerrero E, Platero‐Luengo A, Linares‐Clemente P, Cases I, Lopez‐Barneo J & Pardal R (2016). Gene expression profiling supports the neural crest origin of adult rodent carotid body stem cells and identifies CD10 as a marker for mesectoderm‐committed progenitors. Stem Cells 34, 1647–1650. [DOI] [PubMed] [Google Scholar]
- Nurse CA (2014). Synaptic and paracrine mechanisms at carotid body arterial chemoreceptors. J Physiol 592, 3419–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyham W (1988). Multiple carboxylase deficiency. Int J Biochem 20, 363–370. [DOI] [PubMed] [Google Scholar]
- Ortega‐Saenz P, Levitsky KL, Marcos‐Almaraz MT, Bonilla‐Henao V, Pascual A & Lopez‐Barneo J (2010). Carotid body chemosensory responses in mice deficient of TASK channels. J Gen Physiol 135, 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega‐Saenz P, Pardal R, Garcia‐Fernandez M & Lopez‐Barneo J (2003). Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J Physiol 548, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozand PT, Gascon GG, Al Essa M, Joshi S, Al Jishi E, Bakheet S, Al Watban J, Al‐Kawi MZ & Dabbagh O (1998). Biotin‐responsive basal ganglia disease: a novel entity. Brain 121, 1267–1279. [DOI] [PubMed] [Google Scholar]
- Pacheco‐Alvarez D, Solorzano‐Vargas RS & Del Rio AL (2002). Biotin in metabolism and its relationship to human disease. Arch Med Res 33, 439–447. [DOI] [PubMed] [Google Scholar]
- Pacheco‐Alvarez D, Solorzano‐Vargas RS, Gravel RA, Cervantes‐Roldan R, Velazquez A & Leon‐Del‐Rio A (2004). Paradoxical regulation of biotin utilization in brain and liver and implications for inherited multiple carboxylase deficiency. J Biol Chem 279, 52312–52318. [DOI] [PubMed] [Google Scholar]
- Pardal R & Lopez‐Barneo J (2002). Carotid body thin slices: responses of glomus cells to hypoxia and K+‐channel blockers. Respir Physiol Neurobiol 132, 69–79. [DOI] [PubMed] [Google Scholar]
- Peers C (2015). Acute oxygen sensing–Inching ever closer to an elusive mechanism. Cell Metab 22, 753–754. [DOI] [PubMed] [Google Scholar]
- Platero‐Luengo A, Gonzalez‐Granero S, Duran R, Diaz‐Castro B, Piruat JI, Garcia‐Verdugo JM, Pardal R & Lopez‐Barneo J (2014). An O2‐sensitive glomus cell‐stem cell synapse induces carotid body growth in chronic hypoxia. Cell 156, 291–303. [DOI] [PubMed] [Google Scholar]
- Pokorski M & Matysiak Z (1998). Fatty acid acylation of dopamine in the carotid body. Med Hypotheses 50, 131–133. [DOI] [PubMed] [Google Scholar]
- Rajgopal A, Edmondnson A, Goldman ID & Zhao R (2001). SLC19A3 encodes a second thiamine transporter ThTr2. Biochim Biophys Acta 1537, 175–178. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Shippenberg TS & Jayanthi LD (2011). Regulation of monoamine transporters: role of transporter phosphorylation. Pharmacol Ther 129, 220–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov GY, Adams MB, McMillen IC & Roberts ML (1998). Oxygen‐sensing mechanisms are present in the chromaffin cells of the sheep adrenal medulla before birth. J Physiol 509, 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said HM (1999). Cellular uptake of biotin: mechanisms and regulation. J Nutr 129, 490S–493S. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Weihe E & Eiden LE (2013). Localization and expression of VMAT2 across mammalian species: a translational guide for its visualization and targeting in health and disease. Adv Pharmacol 68, 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner S, Shirvan A & Linial M (1995). Vesicular neurotransmitter transporters: from bacteria to humans. Physiol Rev 75, 369–392. [DOI] [PubMed] [Google Scholar]
- Shirahata M, Balbir A, Otsubo T & Fitzgerald RS (2007). Role of acetylcholine in neurotransmission of the carotid body. Respir Physiol Neurobiol 157, 93–105. [DOI] [PubMed] [Google Scholar]
- Spector R & Mock D (1987). Biotin transport through the blood‐brain barrier. J Neurochem 48, 400–404. [DOI] [PubMed] [Google Scholar]
- Stockert RJ, Paietta E, Racevskis J & Morell AG (1992). Posttranscriptional regulation of the asialoglycoprotein receptor by cGMP. J Biol Chem 267, 56–59. [PubMed] [Google Scholar]
- Stockert RJ & Ren Q (1997). Cytoplasmic protein mRNA interaction mediates cGMP‐modulated translational control of the asialoglycoprotein receptor. J Biol Chem 272, 9161–9165. [DOI] [PubMed] [Google Scholar]
- Subramanian VS, Marchant JS & Said HM (2006). Biotin‐responsive basal ganglia disease‐linked mutations inhibit thiamine transport via hTHTR2: biotin is not a substrate for hTHTR2. Am J Physiol Cell Physiol 291, C851–859. [DOI] [PubMed] [Google Scholar]
- Tabarki B, Al‐Shafi S, Al‐Shahwan S, Azmat Z, Al‐Hashem A, Al‐Adwani N, Biary N, Al‐Zawahmah M, Khan S & Zuccoli G (2013). Biotin‐responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology 80, 261–267. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson A & Nurse CA (1997). Developmental loss of hypoxic chemosensitivity in rat adrenomedullary chromaffin cells. J Physiol 498, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolev Y, Segal T, Yaffe D, Gros Y & Schuldiner S (2013). Identification of conformationally sensitive residues essential for inhibition of vesicular monoamine transport by the noncompetitive inhibitor tetrabenazine. J Biol Chem 288, 32160–32171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez‐Arellano A, Hernandez‐Esquivel Mde L, Sanchez RM, Ortega‐Cuellar D, Rodriguez‐Fuentes N, Cano S, Leon‐Del‐Rio A & Carvajal K (2008). Functional and metabolic implications of biotin deficiency for the rat heart. Mol Genet Metab 95, 213–219. [DOI] [PubMed] [Google Scholar]
- Vlasova TI, Stratton SL, Wells AM, Mock NI & Mock DM (2005). Biotin deficiency reduces expression of SLC19A3, a potential biotin transporter, in leukocytes from human blood. J Nutr 135, 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe M & Nakaki T (2008). Mitochondrial complex I inhibitor rotenone inhibits and redistributes vesicular monoamine transporter 2 via nitration in human dopaminergic SH‐SY5Y cells. Mol Pharmacol 74, 933–940. [DOI] [PubMed] [Google Scholar]
- Whitehead CC (1985). Assessment of biotin deficiency in animals. Ann N Y Acad Sci 447, 86–96. [DOI] [PubMed] [Google Scholar]
- Wolf B (2001). Disorders of biotin metabolism In: The Metabolic and Molecular Basis of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, eds. McGraw‐Hill, New York, NY. [Google Scholar]
- Zempleni J (2005). Uptake, localization, and noncarboxylase roles of biotin. Annu Rev Nutr 25, 175–196. [DOI] [PubMed] [Google Scholar]
- Zempleni J & Mock DM (1999). Biotin biochemistry and human requirements. J Nutr Biochem 10, 128–138. [DOI] [PubMed] [Google Scholar]
- Zempleni J, Wijeratne SSK & Hassan YI (2009). Biotin Biofactors 35, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng WQ, Al‐Yamani E, Acierno JS, Jr , Slaugenhaupt S, Gillis T, MacDonald ME, Ozand PT & Gusella JF (2005). Biotin‐responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. Am J Hum Genet 77, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer L, Vancassel S, Cantagrel S, Breton P, Delamanche S, Guilloteau D, Durand G & Chalon S (2002). The dopamine mesocorticolimbic pathway is affected by deficiency in n‐3 polyunsaturated fatty acids. Am J Clin Nutr 75, 662–667. [DOI] [PubMed] [Google Scholar]