

Abstract
Key points
Mitochondria are frequently implicated in the ageing of skeletal muscle, although the role of denervation in modulating mitochondrial function in ageing muscle is unknown.
We show that increased sensitivity to apoptosis initiation occurs prior to evidence of persistent denervation and is thus a primary mitochondrial defect in ageing muscle worthy of therapeutic targeting.
However, at more advanced age, mitochondrial function changes are markedly impacted by persistent sporadic myofibre denervation, suggesting the mitochondrion may be a less viable therapeutic target.
Abstract
Experimental denervation modulates mitochondrial function, where changes in both reactive oxygen species (ROS) and sensitivity to permeability transition are implicated in the resultant muscle atrophy. Notably, although denervation occurs sporadically in ageing muscle, its impact on ageing muscle mitochondria is unknown. Because this information has important therapeutic implications concerning targeting the mitochondrion in ageing muscle, we examined mitochondrial function in skeletal muscle from four groups of humans, comprising two active (mean ± SD age: 23.7 ± 2.7 years and 71.2 ± 4.9 years) and two inactive groups (64.8 ± 3.1 years and 82.5 ± 4.8 years), and compared this with a murine model of sporadic denervation. We tested the hypothesis that, although some alterations of mitochondrial function in aged muscle are attributable to a primary organelle defect, mitochondrial dysfunction would be impacted by persistent denervation in advanced age. Both ageing in humans and sporadic denervation in mice increased mitochondrial sensitivity to permeability transition (humans, P = 0.004; mice, P = 0.01). To determine the contribution of sporadic denervation to mitochondrial function, we pharmacologically inhibited the denervation‐induced ROS response. This reduced ROS emission by 60% (P = 0.02) in sporadically denervated mouse muscle, which is similar to that seen in humans older than 75 years (–66%, P = 0.02) but not those younger than 75 years. We conclude that an increased sensitivity to permeability transition is a primary mitochondrial defect in ageing muscle. However, at more advanced age, when muscle atrophy becomes more clinically severe, mitochondrial function changes are markedly impacted by persistent sporadic denervation, making the mitochondrion a less viable therapeutic target.
Keywords: aging, denervation, mitochondria, muscle atrophy, neuromuscular junction, sarcopenia
Key points
Mitochondria are frequently implicated in the ageing of skeletal muscle, although the role of denervation in modulating mitochondrial function in ageing muscle is unknown.
We show that increased sensitivity to apoptosis initiation occurs prior to evidence of persistent denervation and is thus a primary mitochondrial defect in ageing muscle worthy of therapeutic targeting.
However, at more advanced age, mitochondrial function changes are markedly impacted by persistent sporadic myofibre denervation, suggesting the mitochondrion may be a less viable therapeutic target.
Abbreviations
- AChR
ACh receptor
- ACR
acceptor control ratio
- AA
antimycin a
- AACOCF3
arachidonyl trifluoromethyl ketone
- CRC
calcium retention capacity
- cPLA2
cytoplasmic phospholipase A2
- DTT
dithiothreitol
- mPTP
mitochondrial permeability transition pore
- MHC
myosin heavy chain
- MuSK
muscle specific kinase
- NMJ
neuromuscular junction
- OA
old active
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- succ
succinate
- TMPD
N,N,N‧,N'‐tetramethyl‐p‐phenylenediamine dihydrochloride
- VOI
very old inactive
- VDAC
voltage‐dependent anion channel
maximal oxygen uptake
- WT
wild‐type
- YA
young active
Introduction
Mitochondria are frequently implicated in the age‐related atrophy of skeletal muscle (Hepple, 2014). Despite this, the evidence remains equivocal, particularly in humans where some indices of mitochondrial function are maintained in physically active subjects ≤75 years of age (Kent‐Braun & Ng, 2000; Hutter et al. 2007), even when significant whole muscle atrophy is documented (Gouspillou et al. 2014). Furthermore, the degree to which any mitochondrial alterations in ageing muscle represent a primary organelle defect worthy of therapeutic targeting vs. a consequence of other changes occurring with ageing is an important consideration when advancing to effective treatments. For example, persistent sporadic denervation, which results in the accumulation of severely atrophied angular fibres in muscle at very advanced age (>75 years in humans: Scelsi et al. 1980; or a similar relative age in rodent models: Rowan et al. 2011; Rowan et al. 2012) is a probable contributor to mitochondrial dysfunction in muscle at very advanced age based upon the effects of experimental denervation on mitochondrial reactive oxygen species (ROS) emission (Muller et al. 2007) and mitochondrial sensitization to permeability transition (Csukly et al. 2006). This is particularly important because an increase in mitochondrial ROS (Mansouri et al. 2006; Chabi et al. 2008; Picard et al. 2011) and increased sensitivity to mitochondrial permeability transition (Chabi et al. 2008; Picard et al. 2011; Gouspillou et al. 2014) are often seen in ageing muscle. Indeed, because the changes in mitochondrial function with denervation are involved in mediating the atrophy of denervated muscle (Bhattacharya et al. 2009; Romanello et al. 2010), we propose that attempting to ‘correct’ mitochondrial changes that are driven by denervated muscle fibres in ageing muscle could exacerbate matters by preventing denervation‐related atrophy, thereby placing a greater burden on the remaining innervated fibres.
On the basis of these issues, in the present study we addressed the impact of sporadic denervation on mitochondrial content and multiple mitochondrial functions in human limb muscle across a range of ages and physical activity levels: (i) Recreationally active men: young active (YA), mean ± SD age: 23.7 ± 2.7 years; old active (OA) 71.2 ± 4.9 years and (ii) Physically inactive men: old inactive (OI) 64.8 ± 3.1 years; very old inactive (VOI) 82.5 ± 4.8 years. To provide information on the effect of sporadic denervation on mitochondrial content and function independent of ageing, we examined a genetic model of sporadic denervation secondary to neuromuscular junction instability, neurotrypsin over‐expression in mice (Sarco mice: Vrijbloed et al. 2011). We hypothesized that some alterations of mitochondrial function at very advanced age would mirror those seen in Sarco mice and would be attenuated by pharmacologically blocking the denervation‐induced ROS signal (Bhattacharya et al. 2009), suggesting these alterations are secondary to sporadic denervation rather than primary organelle defects.
Methods
Ethical approval
Approval for human investigations was provided by the Faculty of Medicine IRB McGill University, the Montreal Chest Institute Research Ethics Board and the Comité Institutionnel d’Éthique de la Recherche avec des Êtres Humains de l'Université du Québec a‧ Montréal. All investigations were performed after written informed consent was provided and in accordance with the 1964 Declaration of Helsinki. Animal procedures were conducted with approval from the Animal Care Committee at McGill University (protocol 2012‐7189) and adhered to the Canadian Council on Animal Care guidelines (http://www.ccac.ca).
Experimental design
Human participants were recruited from the local Montreal area through the local press, and community organizations. Exclusion criteria included cardiac, motor or psychological pathology, as well as Coumadin (Bristol‐Myers Squibb Company, Princeton, NJ, USA) (warfarin) medication. Four groups of men were investigated: a young active group (YA, n = 11), an old active group (OA, n = 10), an old inactive group (OI, n = 8) and a very old inactive group (VOI, n = 8), based on age and self‐reported physical activity. The YA and OA groups have previously been reported in another study (Gouspillou et al. 2014), although the data obtained in the present study differ somewhat as a result of the de novo mitochondrial protein assessments on all samples that were completed for the current analyses (see below).
Neurotrypsin over‐expressing C57BL/6 mice [Sarco mice; (B6.C3‐Tg(PRSS12) 491 Zbz)] were provided by Neurotune (Schlieren, Switzerland). This mouse is a model of sporadic muscle fibre denervation caused by reduced activation of muscle specific kinase (MuSK), which in turn causes instability of the acetylcholine receptor cluster on the postsynaptic side of the neuromuscular junction (Butikofer et al. 2011). The mice were subsequently bred and maintained at the McGill University Health Centre vivarium under a 12:12 h light/dark cycle and had access to food and water ad libitum. Upon establishment of the colony, only heterozygous neurotrypsin over‐expressing mice were backcrossed with wild‐type (WT) C57BL/6CRL mice. Based upon previous work showing the soleus to be highly impacted by neurotrypsin over‐expression (Butikofer et al. 2011), we chose this muscle for our experiments. Furthermore, the fibre type distribution of the mouse soleus is quite similar to human vastus lateralis, which should facilitate translation of our results.
The numbers of participants and animals were based upon sample size calculations made from observed variation of the mitochondrial function indices investigated (Picard et al. 2010; Gouspillou et al. 2014). The present study aimed to determine the extent to which mitochondrial functional changes with age represent a primary defect of the mitochondrion vs. a secondary (physiological) response to persistent sporadic denervation.
Sample collection
On the day of the experiment, 8‐month‐old male animals were killed with CO2 asphyxiation followed by cervical dislocation. Soleus muscle of Sarco mice was used in our mouse experiments because the fibre type distribution in mouse soleus corresponds relatively closely to human vastus lateralis muscle (i.e. ∼50/50 type I and type IIa fibres). Human muscle samples were obtained from the vastus lateralis using the Bergström needle biopsy technique (Bergstrom, 1975).
Both human and mouse samples required for histology were snap frozen in isopentane pre‐cooled in liquid nitrogen and stored at –80°C, whereas those for mitochondrial function investigations (70–100 mg from humans and 12–20 mg from mice) were placed in ice‐cold buffer A (2.77 mm CaK2EGTA, 7.23 mm K2EGTA, 6.56 mm MgCL2, 0.5 mm dithiothreitol (DTT), 50 mm KMES, 20 mm imidazol, 20 mm taurine, 5.3 mm Na2ATP, 15 mm phosphocreatine, pH7.3) for manual dissection under a stereomicroscope.
Myofibre size determination
Muscle sections were cut on a HM505E cryostat (Microm, Walldorf, Germany) at –24°C and mounted on frosted glass slides, which were then stored at –80°C. The myofibre cross‐sectional area was determined from images of muscle sections immunofluorescently labelled for the basal lamina and myosin heavy chain (MHC) isoforms. Slides were first removed from –80°C, allowed to defrost in slide boxes for 30 min and then air dried for 1 h. Sections were washed in PBS and then blocked using 10% normal goat serum in PBS. Sections were then incubated for 1 h at room temperature with rabbit polyclonal IgG anti‐laminin (L9393, dilution 1:750; Sigma, St Louis, MO, USA), and the antibodies: MHCI (BA‐F8 mouse monoclonal IgG2b, dilution 1:25), MHCIIa (Sc71, mouse monoclonal IgG1, dilution 1:200) and MHCIIx (6H1 mouse monoclonal IgGM, dilution 1:25) and the mouse sections were also incubated with MHCIIb (BF‐F3 mouse monoclonal IgM, dilution 1:200). All antibodies were purchased from Developmental Studies Hybridoma Bank (University of Iowa). Because of the presence of four MHC isoforms in mouse soleus, the labelling of mouse tissue was performed over two serial sections. After incubation, sections were washed (3 × 5 min in PBS) and then reacted against Alexa Fluor 488 IgG goat anti‐rabbit, Alexa Fluor 350 IgG2b goat anti‐mouse, Alexa Fluor 594 IgG1 goat anti‐mouse and Alexa Fluor 488 IgM goat anti‐mouse (Life Technologies, Grand Island, NY, USA). After a final washing step, slides were mounted using Prolong Gold Hard Set Mounting Medium (Invitrogen, Carlsbad, CA, USA). Images were captured using an Axio Imager M2 fluorescence microscope (Carl Zeiss, Oberkochen, Germany), and analysed using ImageJ (National Institutes of Health, Bethesda, MD, USA). For human samples a ‘grid’ plug‐in was used, followed by systematic sampling until at least 300 fibres had been analysed, whereas mice had all fibres from the soleus muscle cross‐section counted.
Whole muscle mount for neuromuscular junction (NMJ) fragmentation analysis
Neurotrypsin over‐expression in mice causes a marked disturbance of NMJ postsynaptic morphology and numerous denervation‐related ageing phenotypes in skeletal muscle (Butikofer et al. 2011). To confirm this observation in our colony of neurotrypsin over‐expressing (Sarco) mice, soleus muscles were harvested to examine the extent of NMJ fragmentation in WT and Sarco mice. Muscles were washed (3 × 5 min in PBS) and fixed in 2% paraformaldehyde overnight at 4°C. Muscles were then separated into small bundles and blocked (5% horse serum, 5% BSA, 2% Triton X‐100 in PBS) overnight at 4°C before being incubated in mouse monoclonal anti‐synaptophysin (ab8049, dilution 1:25; Abcam, Cambridge, MA, USA) antibody overnight at 4°C. Muscle bundles were washed and incubated with AF594‐conjugated goat anti‐mouse secondary antibody together with Alexa488‐conjugated alpha‐bungarotoxin (B‐13422, dilution 1:500; Life Technologies) overnight at 4°C, and then mounted with prolong gold medium.
Images were captured using a LSM710 confocal microscope (Zeiss) with a 63× objective, with quantification of NMJ fragmentation performed using Image J. NMJs were classified into three categories, according to the number of postsynaptic fragments observed (Fig. 1 B and C); 1 = normal NMJ without fragmentation; 2–4 fragments = light fragmentation with pretzel‐like postsynapse; >5 fragments = intermediate to severe fragmentation.
Figure 1. Neurotrypsin overexpression and associated changes in Sarco mice.
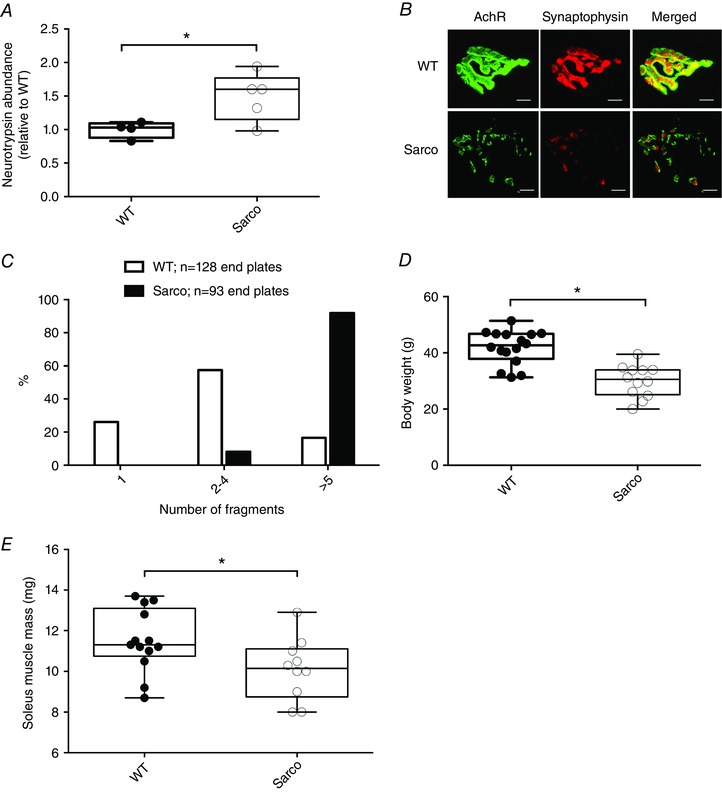
A, quantification of neurotrypsin (relative to WT) in Sarco mice and WT mice (unpaired t test, P = 0.04). B, representative images of neuromuscular junctions from WT and Sarco mice and (C) quantification of NMJ fragmentation in Sarco mice and WT mice. Changes in (D) body mass (exact Wilcoxon rank sum, P < 0.0001; WT, n = 16, and Sarco, n = 12) and (E) soleus muscle mass in Sarco mice (n = 13) compared to WT mice (n = 10) (exact Wilcoxon rank sum, P = 0.03). Scale bars = 10 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
Mitochondrial protein content
Mitochondrial content was assessed by western blotting for voltage‐dependent anion channel (VDAC) and representative oxidative phosphorylation (OXPHOS) subunits in both mice and humans, whereas neurotrypsin content was quantified in mouse spinal cords. Ten to 55 mg of muscle/spinal cord tissue was homogenized in a MM400 robot homogenizer (Retsch) with 10× w/v of extraction buffer (50 mm Tris base, 150 mm NaCl, 1% Triton X‐100, 0.5% sodium deoxycolate, 0.1% sodium dodecyl sulphate and 10 μl ml–1 of Protease Inhibitor Cocktail; Sigma). After 2 h of gentle agitation at 4°C, samples were centrifuged at 12,000 g for 20 min at 4°C. The supernatant was removed and protein concentration assessed by the Bradford assay. Protein samples were then diluted in 4 × Laemli buffer to give a final protein concentration of 2 μg μl–1 before boiling at 95°C for 5 min. Immunoblotting was performed with 15 μg of tissue protein (20 μg of protein for mouse tissue), which was loaded onto a 12% acrylamide gel, electrophoresed by SDS‐PAGE and then transferred to polyvinylidene fluoride membranes (Life Sciences), blocked 5% (w/v) semi‐skimmed milk for 1 h at room temperature and probed overnight at 4°C with the following primary antibodies (diluted in 5% BSA); mouse monoclonal anti‐VDAC (ab14734, dilution 1:1000; Abcam), mouse monoclonal Total OXPHOS Cocktail (ab110413, dilution 1:2000; Abcam) and rabbit polyclonal to anti‐neurotrypsin antibody‐catalytic domain (ab59454, dilution 1:1000; Abcam). Because the OXPHOS cocktail was less sensitive towards CIV after boiling in human samples, this complex was quantified using mouse monoclonal CIV (A21348, dilution 1:1000; Life Technologies). Ponceau staining was performed to normalize protein loading. After washing, membranes were subsequently incubated with HRP‐conjugated secondary antibodies (Abcam; diluted in 5% milk) for 1 h at room temperature. Protein bands were detected using SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA) and imaged with a G‐Box chem imaging system. Identification and quantification of protein bands was performed using GeneTools software (Syngenem, Cambridge, UK).
Mitochondrial function investigations
Manual isolation of muscle fibres was performed as described previously (Picard et al. 2010). Briefly, small bundles (3–6 mg) of myofibres were separated using needles and then chemically permeabilized for 30 min in buffer A with 50 μg ml–1 of saponin. Unless otherwise stated, all permeabilization and wash steps were performed at 4°C with gentle agitation.
Mitochondrial respiration
Myofibres for the assessment of mitochondrial respiration were washed 3 × 10 min in buffer B (2.77 mm CaK2EGTA, 7.23 mm K2EGTA, 1.38 mm MgCl2, 3 mm K2HPO4, 0.5 mm DTT, 20 mm imidazole, 100 mm KMES, 20 mm taurine and 2 mg ml–1 BSA, pH 7.3). Next, 3–6 mg of wet weight fibres were incubated at 37°C with continuous stirring in 2 ml of buffer B in an Oxygraph‐2 K (Oroboros, Innsbruck, Austria). Respiration was recorded after addition of the substrates: glutamate (G) (10 mm) + malate (m) (5 mm), ADP (2 mm), succinate (succ) (10 mm), antimycin A (AA) (10 μm), ascorbate (10 mm) + N,N,N‧,N'‐tetramethyl‐p‐phenylenediamine dihydrochloride (TMPD) (1 mm). Cytochrome c (5 mm) was added after succ to confirm mitochondrial outer membrane integrity. As reported previously (Picard et al. 2010), TMPD‐stimulated respiration values were corrected for auto‐oxidation by TMPD. From these measurements the acceptor control ratio (ACR) was calculated by taking the quotient of respiration with G+M and ADP, to provide an indication of the degree of mitochondrial coupling. Because mitochondrial cristae density can be independently regulated relative to mitochondrial volume (Suarez et al. 1991), the results were expressed relative to the content of a subunit of CIII (a cristae bound protein) to provide an indication of the intrinsic capacity of the respiratory chain, and relative to VDAC (an outer membrane protein) to represent the respiratory capacity per mitochondrion as a whole. Note that the remarkable heterogeneity in half‐life of individual mitochondrial proteins (Karunadharma et al. 2015) highlights the potential for significant variation in stoichiometry of individual mitochondrial proteins within a given mitochondrion and this could have an impact on the viability of our normalizations.
Mitochondrial ROS production
Myofibres for ROS analysis were washed 3 × 10 min in buffer Z (110 mm KMES, 35 mm KCL, 1 mm EGTA, 3 mm MgCL2, 10 mm K2HPO4 and 0.5 mg per ml BSA, pH 7.3). ROS emission was determined by recording the oxidation of amplex red to the fluorescent compound resorufin, catalysed by H2O2 and fatty acid hydroperoxides released from the mitochondria. The fluorescence signal was measured using an F‐2500 fluorescence spectrophotometer (Hitachi, Tokyo, Japan) at an excitation/emission wavelength of 563/587 nm. Between 3 and 6 mg of fibre bundles were added to 600 μl of solution Z, with 5.5 μm amplex red and 1 U per ml HRP in a magnetically stirred cuvette at 37°C. The substrates added were: G + M (5 mm), succ (5 mm), ADP (0.1 mm), ADP (1 mm) and AA (10 μm). ROS emission was determined using a standard curve constructed on the same day of the experiment using known concentrations of H2O2.
Determining the contribution of denervation to mitochondrial ROS emissions
Amplex Red probe not only detects H2O2, but also fatty acid hydroperoxides, with the latter being a ROS species specific to denervation (Bhattacharya et al. 2009). As such, in both aged humans and neurotrypsin over‐expressing mice, the contribution of denervation in the modulation of mitochondrial function was assessed by blocking the denervation‐induced increase in mitochondrial fatty acid hydroperoxide production using the cytoplasmic phospholipase A2 (cPLA2) inhibitor, arachidonyl trifluoromethyl ketone (AACOCF3), as carried out previously by Bhattacharya et al. (2009). Permeabilized muscle fibres were incubated with AACOCF3 (20 μm) and its vehicle control, ethanol (ETOH), in buffer Z for 3 × 10 min. The amplex red signal was measured as mentioned above in the absence (ETOH vehicle only) or presence of AACOCF3 to estimate the rate of endogenous ROS production in the form of fatty acid hydroperoxides.
Determination of the sensitivity of the mitochondrial permeability transition pore (mPTP) to a calcium challenge
Fibres for the determination of mPTP function were washed 3 × 10 min in solution C (80 mm KMES, 50 mm Hepes, 20 mm taurine, 0.5 mm DTT, 10 mm MgCl2 and 10 mm ATP, pH 7.3 at 4°C) and then had myosin extracted using solution D (800 mm KCL, 50 mm Hepes, 20 mm taurine, 0.5 mm DTT, 10 mm MgCl2 and 10 mm ATP, pH 7.3) at 4°C without agitation. Myofibres were then washed 3 × 10 min in CRC solution (250 mm sucrose, 5 μm EGTA‐Tris Base and 10 mm Tris‐MOPS, pH 7.4). Approximately 4–6 mg of permeabilized fibres were added to 600 μl of CRC solution containing glutamate (5 mm), malate (2.5 mm), phosphate (10 mm), oligomycin (0.5 nm) and Calcium Green™‐5 N, Hexapotassium Salt (0.001 mm) (Life Technologies). The Ca2+ uptake by the mitochondria was measured by monitoring the reduction in free Ca2+ in the solution via the decrease in fluorescence observed when Ca2+ was liberated from calcium‐green. The time point where the Ca2+ in the solution began to increase was taken as the time to pore opening, and the amount of Ca2+ up taken by the mitochondria prior to this was designated as the calcium retention capacity (CRC). Fluorescence was detected using an F‐2500 fluorescence spectrophotometer (Hitachi) at an excitation/emission wavelength of 505/535 nm. The Ca2+ concentration in the solution was determined using a calibration curve of known Ca2+ concentrations performed on the day of the experiment.
Analysis of all mitochondrial function experiments was performed using bespoke software created in‐house using Igor Pro Software (Wavemetrics; https://www.wavemetrics.com).
Transcript analysis
Total RNA was extracted from muscle samples using the RNeasy Lipid Tissue Mini Kit (Qiagen, Valanecia, CA, USA). RNA concentration and purity (A 260/A 280 ratios >1.8) were assessed using a NanoDrop‐2000 spectrophotometer (Thermo Scientific). RNA (1 μg) was reverse transcribed to cDNA using a qScript™ cDNA Synthesis Kit (Quanta Biosciences, Beverly, MA, USA). To quantify the genes for MuSK, HSP27, ACh receptor (AchR) and Nav1.4, primers were designed using Primer3Plus (http://primer3plus.com) (Table 1) with the TATA box binding protein being used as an endogenous control. The cDNA was amplified at 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 55°C for 60 s, using Power SYBR® Green PCR Master Mix (Life Technologies). All real time PCRs were performed on a StepOnePlusTM Real‐Time PCR system (Life Technologies), with samples run in triplicate along with a melt curve analysis to assess primer dimer formation or contamination. The comparative threshold cycle (CT) method was used to calculate fold changes in expression. Relative fold changes in gene expression were presented as 2−ΔΔCT and normalized to control subjects.
Table 1.
Primer sequences used for quantitative PCR experiments on human vastus lateralis
| Gene name | Sequence | NCBI reference number |
|---|---|---|
| AChR ε | F 5′‐ ATACTGAGAACGGCGAGTGG ‐3′ | NM_000080.3 |
| R 5′‐ GATGGAGACCGTGCATTTCT ‐3′ | ||
| MuSK | F 5′‐GCCTTCAGCGGAACTGAG AAA‐3′ | PMID: 17192614 |
| R 5′‐ GGCTGGGGGTAGGATTCCA ‐3′ | ||
| HSP 27 | F 5′‐ GGACGAGCATGGCTACATCT ‐3′R 5′‐ GACTGGGATGGTGATCTCGT ‐3′ | NM_001540.3 |
| Nav1.4 | F 5′‐ CTTCATTGGCGTCATCATTG ‐3′ | NM_000334.4 |
| R 5′‐ CACGAGGTCATACACCATGC ‐3′ | ||
| TBP | F 5′‐ TATAATCCCAAGCGGTTTGC ‐3′ | NM_001172085.1 |
| R 5′‐ GCTGGAAAACCCAACTTCTG ‐3′ |
Statistical analysis
Descriptive statistics summarize all study variables of interest. Graphs (boxplots) show individual data points along with medians and the interquartile range and data in the text are reported as the mean ± SD (Barde & Barde, 2012). When we compared two groups with no ‘conditions’ (e.g. human CRC or WT vs. sarco mice): differences between age groups, categorized as young (YA) vs. old, old active (OA) vs. old inactive (OI) or old inactive (OI) vs. very old inactive (VOI), were investigated using the exact version of the Wilcoxon rank sum test for independent samples. When we compared the same group under two different conditions (e.g. Sarco mice ROS production with ethanol vs. AACOCF3), the exact version of Wilcoxon signed rank test for paired observations was used.
When we compared groups and the ‘conditions’ (e.g. mitochondrial respiration with substrate additions): differences between age and activity groups, categorized as young vs. old, old active vs. old inactive or old inactive vs. very old inactive, were investigated using ANOVA. The mixed model was used to account for the fact that all subjects were exposed to each of the different conditions: GM+ADP, succ and ascorbate + TMPD. Thus, we needed to account for the correlation between observations within subjects. The correlation was modelled assuming an unstructured correlation (Littell et al. 2006). An interaction between age and activity groups and conditions (substrates and inhibitors) was also investigated. The mean respiration rate was compared between groups separately by condition. Post hoc multiple comparisons were performed by calculating all‐pair of means contrasts and adjusting P values for multiplicity of testing. We used the SAS macro %SimTests with the unconstrained step‐down method, which is a closed testing procedure to account for logical dependencies between the hypotheses (Westfall et al. 1999). Degrees of freedom were adjusted using the Kenward–Roger correction (Kenward & Roger, 1997) because of imbalances between the groups with respect to the number of subjects.
Assumptions of the ANOVA model (randomness of errors, homogeneity of variance, normality, presence of outliers) were investigated with graphical analysis of residuals. When residuals did not show evidence of following a Normal distribution and/or homogeneity of variance, response values were log‐transformed (natural logarithm) to ensure the data followed an approximately Normal distribution and/or to stabilize the variance.
All analyses were performed using SAS, version 9.3 (SAS Institute Inc., Cary, NC, USA). P < 0.05 (two‐sided) was considered statistically significant.
Results
Mouse model data
Sarco mouse as a model of sporadic denervation
To assist us in understanding the impact of sporadic denervation, independent of ageing, on mitochondrial function in skeletal muscle, we used a genetic model of unstable neuromuscular junctions: the neurotrypsin over‐expressing mouse. Previous studies have shown that transgenic over‐expression of the endogenous protease for neural agrin, neurotrypsin, leads to dispersion of the postsynaptic AChRs and sporadic denervation, similar to the changes seen with normal ageing (Bolliger et al. 2010; Butikofer et al. 2011; Vrijbloed et al. 2011). Consistent with prior studies, we found 49% higher levels of neurotrypsin in spinal cords of Sarco mice (Fig. 1 A). Similarly, this elevation of neurotrypsin resulted in markedly disrupted endplate architecture in soleus muscle, based upon a reduced superimposition of pre‐ and postsynaptic structures (Fig. 1 B) and increased fragmentation of postsynaptic AChR clusters (Fig. 1 C). Furthermore, we also observed that Sarco mice had lower body mass (–27.7%) and lower soleus muscle mass (–12.1%) vs. WT (Fig. 1 D and E, respectively). Collectively, these results confirm previous findings and support the utility of the Sarco mouse as a model of sporadic denervation.
Impact of sporadic denervation on myofibre size and size distribution
Mean myofibre size in soleus muscle was not different between WT (mean ± SD: 1507 ± 206 μm2) and Sarco mice (1789 ± 416 μm2) but, similar to a previous report on this model (Butikofer et al. 2011), there was a small shift in fibre size distribution showing both more small and more larger fibres in Sarco mice compared to WT (data not shown). Taken together with the lower muscle mass noted above, our results are consistent with a reduced fibre number in Sarco mice, as reported previously in neurotrypsin over‐expressing mice (Butikofer et al. 2011).
Impact of sporadic denervation in mice on mitochondrial content and function
The content of a marker of mitochondrial outer membrane proteins VDAC was increased in the soleus muscle of Sarco mice (Fig. 2 A). By contrast, although there was no change in some respiratory chain proteins (complex I subunit NDUFB8, core II subunit of complex III, complex IV subunit I or complex V alpha subunit) (Fig. 2 B, D, E and F), one protein subunit decreased (complex II 30 kDa protein) in the soleus muscle of Sarco mice (Fig. 2 C), suggesting that sporadic denervation causes a selective accumulation of mitochondrial outer membrane protein and a reduction in some electron transport chain proteins (e.g. CII 30 kDa subunit content). In saponin‐permeabilized myofibre bundles in Sarco vs. WT mice, there was no difference in mitochondrial respiration under any of the conditions studied when expressed per myofibre bundle wet weight or respiratory chain core II subunit of complex III (Fig. 3 A and B). These results suggest that the intrinsic capacity of the respiratory chain is maintained with sporadic denervation. On the other hand, after normalizing respiration to a marker of outer mitochondrial membrane protein (VDAC; to provide a more general indictor of intrinsic organelle respiratory function), the complex I+II (G + M, + ADP, +succ) and complex IV (ascorbate + TMPD) respiration was significantly reduced in Sarco mice (mixed ANOVA: group effect, P = 0.01 after omission of one statistical outlier), suggesting an impaired intrinsic mitochondrial respiratory capacity with sporadic denervation. In the presence of the one statistical outlier, the group effect for this comparison was not significant (P = 0.06), although post hoc tests showed complex I+II and complex IV respiration remained significantly lower in Sarco vs. WT mice (Fig. 3 C). There was no change in a marker of mitochondrial coupling, the ACR in Sarco mice (Fig. 3 D). Collectively, our analysis shows that sporadic denervation has a modest depressive effect on mitochondrial respiratory function.
Figure 2. Mitochondrial content changes in mouse soleus muscle.
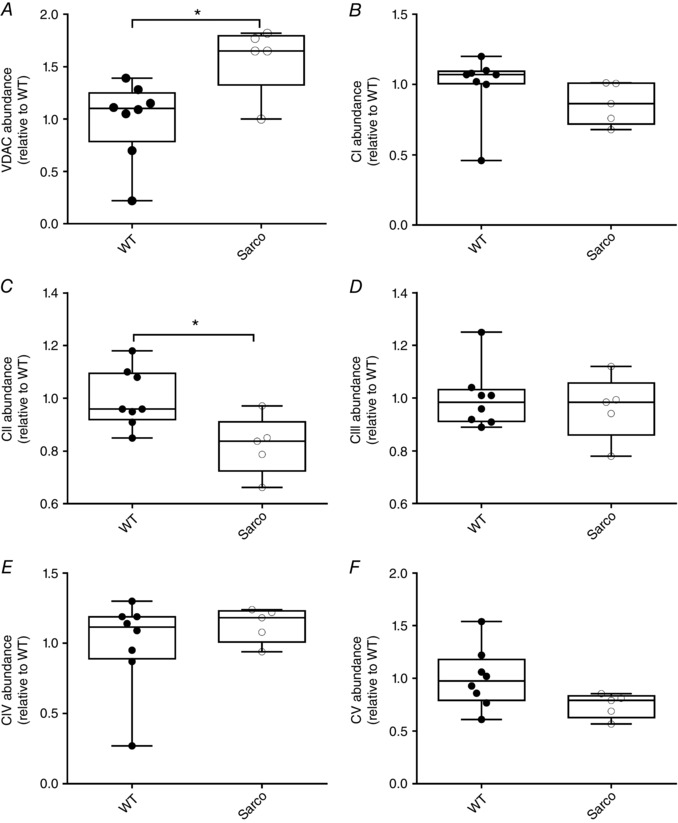
A, VDAC content was increased in Sarco mice (exact Wilcoxon rank sum, P = 0.04). B, conversely, complex I content was not altered in in Saco mice compared to WT mice. C, although there was an increase in complex II content (exact Wilcoxon rank sum, P = 0.04), with none of the other subunits showing changes (D), (E) and (F) (in all experiments, WT, n = 8, Sarco mice, n = 5, unless stated otherwise).
Figure 3. Mitochondrial function changes in mouse soleus muscle.
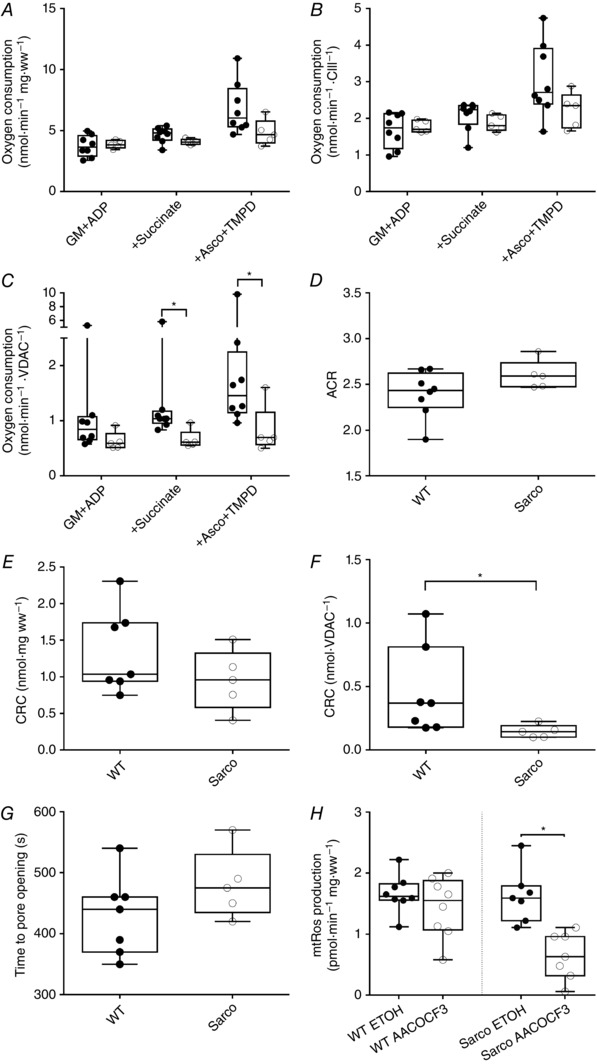
Mitochondrial oxygen consumption in Sarco mice was not altered before (A) or after (B) normalization to CIII (ANOVA mixed model with adjusted post hoc comparisons). C, after normalization with VDAC, mitochondrial respiration was significantly decreased after the addition of succ and ascorbate + TMPD in Sarco mice (ANOVA mixed model with adjusted post hoc comparisons; following log transformation without significant outlier: P = 0.001 for succ and P = 0.02 for ascorbate + TMPD; with outlier: P = 0.045 for succ and P = 0.04 for ascorbate + TMPD). D, ACR was determined by taking the quotient of respiration with G+M and ADP. There was no change in the ACR between WT and Sarco mice (exact Wilcoxon rank sum). E, mitochondrial CRC was measured in response to a calcium challenge in permeabilized myofibres. Although there was no difference in CRC before normalization, the CRC (F) was decreased (exact Wilcoxon rank sum, P = 0.01) without altering the time taken to mPTP opening (G) in Sarco mice (WT, n = 7; Sarco, n = 5). H, application of the cPLA2 inhibitor, AACOCF3, reduced the ROS signal from Sarco mice, suggesting that sporadic denervation contributes to this signal, (WT, n = 8; Sarco, n = 7; P = 0.02; Wilcoxon signed rank) (in all experiments, WT, n = 8, Sarco mice, n = 5 unless stated otherwise).
Exposure of saponin‐permeabilized myofibres to a calcium (Ca2+) challenge showed no impairment when expressed per myofibre bundle wet weight in Sarco mice (Fig. 3 E). By contrast, normalizing to mitochondrial content revealed a significantly reduced CRC in Sarco mice (Fig. 3 F), despite a maintained time to mitochondrial permeability transition pore (mPTP) opening (Fig. 3 G), suggesting that sporadic denervation sensitizes mitochondria to permeability transition through intrinsic changes in the mPTP. These results are consistent with the sensitization to mPTP opening seen with complete denervation following sciatic nerve transection (Csukly et al. 2006).
To further reveal the contribution of sporadic denervation to mitochondrial function changes in Sarco mice, we employed a pharmacological approach to inhibit the denervation‐induced ROS signal, cPLA2 inhibition using AACOCF3, as previously performed by Bhattacharya et al. (2009). Although we did not see any difference in the baseline ROS emission (as measured under ETOH vehicle condition) in Sarco vs. WT mice, treatment with the cPLA2 inhibitor, AACOCF3, yielded a marked reduction in ROS emission relative to ETOH condition only in Sarco mice (Fig. 3 H). This latter observation shows that sporadic denervation plays a significant role in driving mitochondrial ROS emission in Sarco mice even though baseline ROS emission was not different between WT and Sarco mice, providing further proof of concept that this pharmacological assay can be used to detect the influence of sporadic denervation on mitochondrial function.
Human data
We have previously reported on the muscle fibre size, indices of mitochondrial content by western blotting and mitochondrial function of the YA (mean ± SD age: 23.7 ± 2.7 years) and OA (71.2 ± 4.9 years) groups (Gouspillou et al. 2014). The results of the present study represent the addition of two more groups to address the role of a sedentary lifestyle (OI; 64.8 ± 3.1 years) and very advanced age (VOI; 82.5 ± 4.8 years) on these measures. All western blotting for indices of mitochondrial content was performed de novo for the current experiments for all four human subject groups to facilitate comparisons of protein content across all groups. Consistent with the differences in age and self‐reported physical activity levels of these subjects, whole body , an indicator of overall aerobic fitness, was highest in YA, followed by OA, OI and VOI (Table 2).
Table 2.
Characteristics of human participants
| YA (n = 11) | OA (n = 10) | OI (n = 8) | VOI (n = 8) | |
|---|---|---|---|---|
| Age (years) | 23.7 ± 2.7 | 71.2 ± 4.9 | 64.8 ± 3.1 | 82.5 ± 4.8 |
| Height (cm) | 178.1 ± 6.2 | 170.7 ± 3.6 | 172.9 ± 7.3 | 171.6 ± 8.2 |
| Weight (kg) | 79.5 ± 8.3 | 74.7 ± 7.5 | 75.3 ± 13.3 | 77.1 ± 9.3 |
| BMI | 25.1 ± 2.5 | 25.6 ± 2.4 | 25.2 ± 4.1 | 26.2 ± 3.2 |
| (ml kg min)−1 | 42.0 ± 4.1 | 29.4 ± 6.1 | 22.6 ± 3.0 | 20.8 ± 3.1 |
Age groups were very similar in height, weight and body mass index (BMI). In accordance with our classification of the groups according to self‐reported activity levels, was greater in the YA group compared to all of the old (OA, OI, VOI) groups, and the OA group had a greater than the OI group. Data are presented as the mean ± SD.
Impact of ageing on myofibre size and size distribution in humans
Figure 4 A shows representative images of the human vastus lateralis muscle cross‐sections labelled using primary antibodies against MHC isoforms to identify fibre type and laminin to demarcate fibre borders. Muscle fibre size exhibited a decline in the older age groups (Fig. 4 B). Furthermore, there was a dramatic increase in the heterogeneity of fibre size in the oldest group (VOI), characterized by a marked increase in abundance of very small muscle fibres (<2000 μm2) (Fig. 4 C). This is consistent with the marked accumulation of small angular fibres, more than 90% of which are positive for a marker of denervation, in very old rat muscle (Rowan et al. 2011; Rowan et al. 2012), suggesting a significant accumulation of persistently denervated muscle fibres in very old human muscle (VOI; > 75 years).
Figure 4. Myofibre morphology in human muscle.
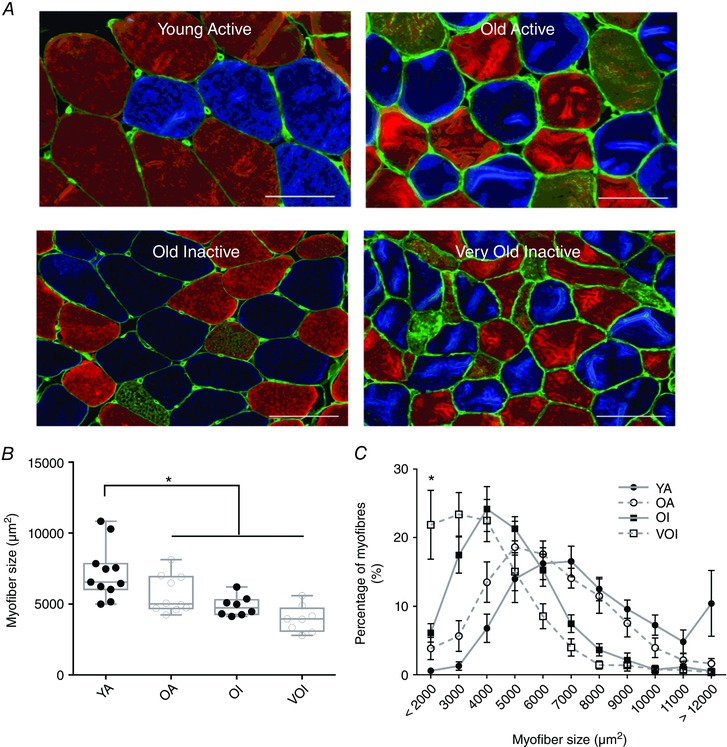
A, transverse muscle sections from YA, OA, OI and VOI men were labelled to visualize the basal lamina and fibre type. Systematic sampling was used to trace a minimum of 200 fibres to give indications of size. B, ANOVA with adjusted post hoc comparisons revealed a significant effect of group status on fibre size, with the YA group having a significantly larger fibre size than the older participants (P < 0.0001). C, fibre size distribution shows a greater accumulation of really small fibres in the VOI group (YA, n = 11; OA, n = 10; OI, n = 8; VOI, n = 8). Scale bars = 100 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
Impact of ageing on mitochondrial content and function in humans
There was no change in a marker of mitochondrial outer membrane proteins with increasing age (VDAC) (Fig. 5 A) or complexes I, II, IV and V (Fig. 5 B, C, E and F). On the other hand, there was a significant reduction in core II subunit of complex III in VOI vs. OI (Fig. 5 D), which is similar to that seen with sporadic denervation in Sarco mice where core II subunit of complex III was unchanged despite an increase in VDAC (i.e. the stoichiometry of outer membrane protein relative to inner membrane protein is altered with very advanced age and sporadic denervation such that there is less inner membrane protein relative to outer membrane protein). Interestingly, although respiratory capacity did not differ between groups when normalized to fibre wet weight (Fig. 6 A), state 3 respiratory capacity with complex I substrates only (GM+ADP) or complex I and II substrates together (+succ) was lower in OI than OA (P < 0.05 when normalized to core II of complex III; P = 0.05 when normalized to VDAC) but not further eroded in VOI (Fig. 6 B and C), suggesting a mild impairment in the intrinsic respiratory capacity of mitochondria with ageing is associated with low physical activity. Unlike that seen in Sarco mice, there was a reduction in a marker of mitochondrial coupling (i.e. the ACR) with age in human muscle and this was independent of physical activity status (Fig. 6 D). Thus, this change in mitochondrial function appears unrelated to sporadic denervation.
Figure 5. Mitochondrial content changes in human vastus lateralis muscle.
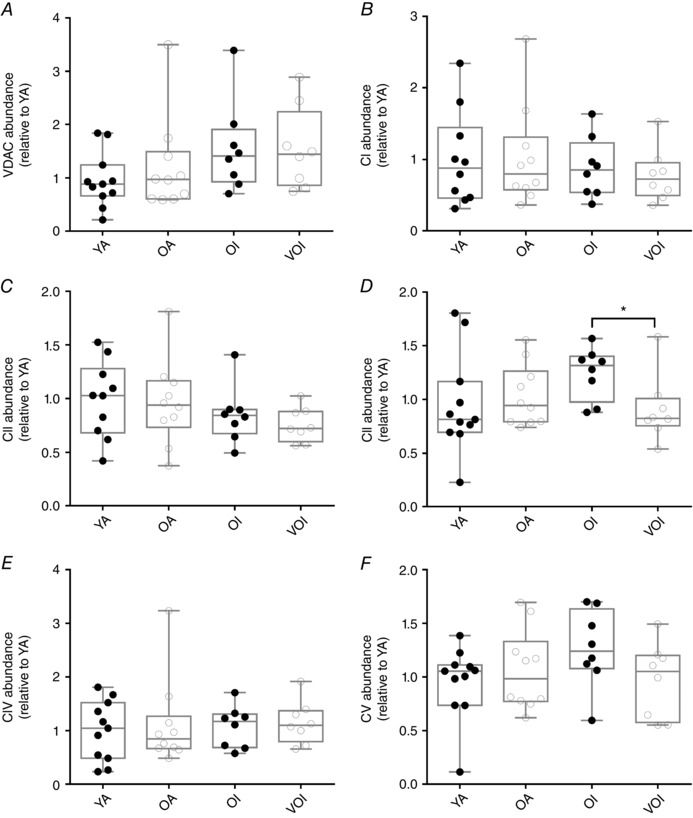
A, VDAC content was similar between groups (YA, n = 11; OA, n = 10; OI, n = 8; VOI, n = 8; exact Wilcoxon rank sum). Although there were no differences in complexes I (B) and II (C), complex III Core II subunit content was lower in VOI compared to OI participants (D). Complexes IV (E) and V (F) also showed no changes (YA, n = 11; OA, n = 10; OI, n = 8; VOI, n = 8; exact Wilcoxon rank sum, P = 0.04).
Figure 6. Mitochondrial function in human vastus lateralis muscle.
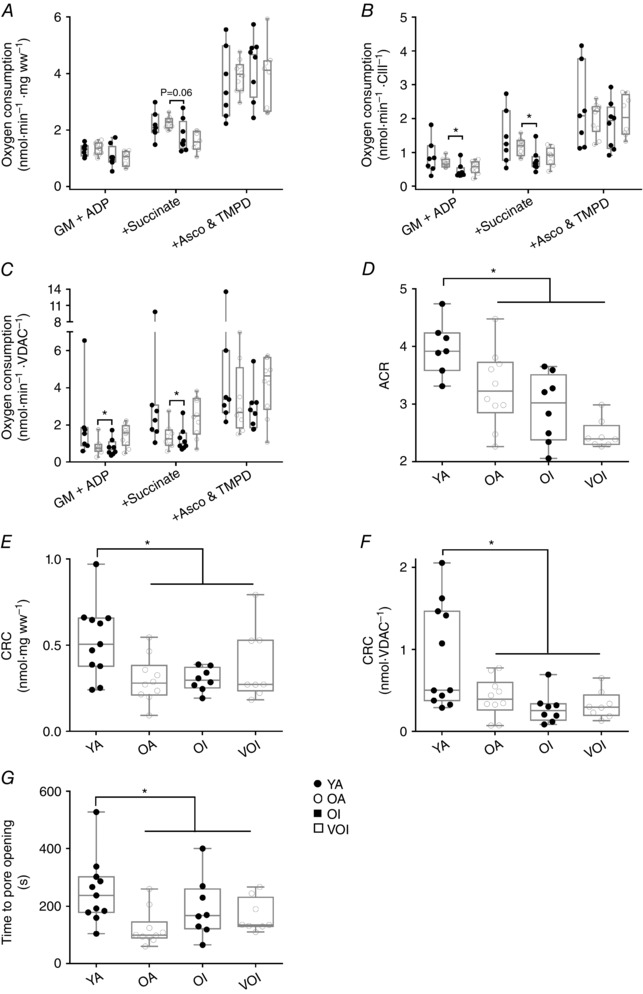
A, oxygen consumption was slightly lower in OI when compared to OA, although this was not significant. B, following normalization to CIII, there was no reduction in oxygen consumption between young and old participants (YA, n = 7; OA, n = 10; OI, n = 8; VOI, n = 8, ANOVA mixed model with adjusted post hoc comparisons following log transformation), although there was a significant reduction between OA and OI participants after the addition of both G+M and succ (P = 0.0074 for both additions). C, following normalization with VDAC, there was still a moderate reduction in respiration in OI participants (ANOVA mixed model with adjusted post hoc comparisons; following log transformation, P = 0.05). D, ACR was determined by taking the quotient of respiration with G+M and ADP. There was a significant decrease in the ACR between young and old groups (YA, n = 7; OA, n = 10; OI, n = 8; VOI, n = 8; exact Wilcoxon rank sum, P = 0.0003). E, in response to a calcium challenge, the calcium retention capacity was significantly lower in old compared to young subjects. F, this reduction persisted following normalization for mitochondrial content. In addition, the time to mPTP opening (G) was significantly lower in older subjects (YA, n = 11; OA, n = 10; OI, n = 8; VOI, n = 8; exact Wilcoxon rank sum, P = 0.004 and P = 0.01 respectively).
Exposure of saponin‐permeabilized myofibres to a Ca2+ challenge revealed a significantly reduced CRC and time to mPTP opening in aged subjects, with this not being impacted by physical activity status (OA = OI), nor was there further erosion in this function at very advanced age (OI = VOI) (Fig. 6 E, F and G). These results suggest that, although increased sensitivity of mitochondria to permeability transition is probably intrinsic to ageing, it does not falter progressively with increasing age once it becomes impaired.
To evaluate the contribution of sporadic denervation to mitochondrial function changes in ageing human muscle, we inhibited cPLA2 with AACOCF3 in saponin‐permeabilized myofibres. Because there was insufficient tissue to conduct this assay on all groups, the results of the present study are based on nine subjects <75 years of age (three from OA and six from OI) and seven subjects >75 years of age (all from VOI), with such an approach permitting us to compare the ages where morphological evidence of sporadic denervation is also contrasting (small fibres were principally a feature of the VOI group who were all >75 years) (see Fig. 4 C). Similar to the impact of sporadic denervation in Sarco mice, we observed that AACOCF3 induced a marked reduction in mitochondrial ROS in subjects >75 years of age but had no effect in subjects <75 years of age (Fig. 7 A), revealing the significant modulation of mitochondrial function by sporadic denervation in very old human muscle. Consistent with the contrasting effect of AACOCF3 in subjects <75 years vs. those >75 years, and the morphological evidence of an accumulation of denervated myofibres in subjects >75 years of age (VOI group; Fig. 4 C), we also found that transcriptional markers of denervation were differentially expressed in subjects >75 years vs. <75 years of age (Fig. 7 B). Collectively, therefore, our results show that sporadic denervation plays an important modulating role in mitochondrial function changes in very old humans (>75 years).
Figure 7. Evidence for denervation and its modulation of mitochondrial function in very old human subjects.
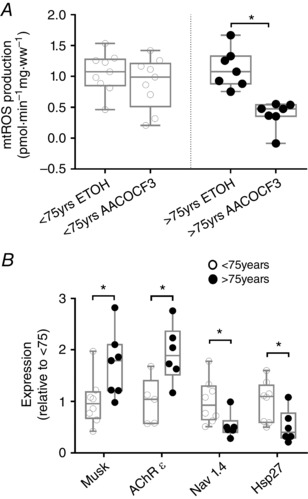
A, application of the cPLA2 inhibitor, AACOCF3, did not reduce the ROS signal from subjects younger than 75 years, suggesting that denervation does not contribute to this ROS signal. However, in subjects older than 75 years, AACOCF3 significantly reduced the ROS signal [<75 years, n = 9 (two assays unsuccessful), >75 years, n = 7 (one assay unsuccessful), P = 0.02; Wilcoxon signed rank]. B, analysis of key transcripts that are altered in response to denervation (exact Wilcoxon rank sum; Musk <75 years, n = 8 (one outlier removed and two samples with insufficient material), >75 years, n = 7 (one sample with insufficient material), P = 0.03; Hsp27 <75 years, n = 8 (three samples with insufficient material), >75 years, n = 6 (two samples with insufficient material), P = 0.02; AChR ε <75 years, n = 5 (one outlier removed, five samples insufficient material), >75 years, n = 6 (two samples with insufficient material), P = 0.02; Nav 1.4 <75 years, n = 8 (three samples with insufficient material), >75 years, n = 6 (two samples with insufficient material)], P = 0.01.
Discussion
The objective of the present study was to evaluate the hypothesis that some alterations in mitochondrial function with ageing are secondary to the sporadic denervation that normally occurs in ageing muscle, particularly in advanced age when muscle atrophy has the greatest probability of impairing mobility and independence (Cruz‐Jentoft et al. 2010). Accordingly, we used a genetic model of sporadic denervation, first, to demonstrate the impact that sporadic denervation can have on mitochondrial function independent of ageing per se and, second, to confirm that the impact of sporadic denervation on ROS emission can be detected pharmacologically by inhibiting cPLA2 activity. Subsequently, we evaluated mitochondrial function in human skeletal muscle across a range of ages and physical activity levels aiming to understand, first, the evolution of mitochondrial function with ageing and the impact of physical activity and, second, the contribution of sporadic denervation to mitochondrial dysfunction in ageing muscle.
Our results show that sporadic denervation in mouse muscle reduces mitochondrial respiratory capacity (based upon a reduced respiration per VDAC) and increases sensitivity to mPTP opening, comprising two changes in mitochondrial function that are well‐known in ageing muscle (Chabi et al. 2008; Marzetti et al. 2008; Picard et al. 2011). Although we did not observe an increase in ROS emission under basal conditions in mice with sporadic denervation, inhibition of cPLA2 activity reduced ROS emission by 60%, supporting the utility of this assay in detecting the influence of sporadic denervation on mitochondrial function (Bhattacharya et al. 2009). By contrast to that observed in sporadically denervated mouse muscle, some changes in mitochondrial function in ageing human muscle occurred prior to the detectable influence of denervation. Specifically, we observed a decline in maximal ADP‐stimulated respiration with both complex I (glutamate, malate) and complex II (succ) substrates in inactive but not active old subjects, as well as a sensitization to mPTP opening in both active and inactive old subjects. This suggests that impaired respiratory capacity with ageing is largely a function of physical inactivity, whereas the sensitization to mPTP opening represents a primary organelle defect worthy of therapeutic targeting. Interestingly, although neither respiration, nor sensitivity to mPTP opening were further eroded in advanced age, the VOI subjects (>75 years of age) were the only group who demonstrated a sensitivity to cPLA2 inhibition and this coincided with an up‐regulation of denervation‐responsive transcripts. As such, our results support our hypothesis that, at very advanced age, some changes in mitochondrial function are secondary to sporadic denervation rather than primary organelle defects.
Pharmacological detection of denervation impact on mitochondrial function
A key measurement in our studies was the use of cPLA2 inhibition to pharmacologically uncloak the influence of sporadic denervation on mitochondrial function in ageing muscle. This is based upon previous work examining the mechanisms of denervation‐induced mitochondrial ROS potentiation. Such work showed that cPLA2 inhibition not only normalizes the ROS signal of mitochondria isolated from surgically denervated muscle, but also reduces the ROS signal of mitochondria isolated from the superoxide dismutase 1 knockout mouse (Bhattacharya et al. 2009), a model that exhibits many features of sporadic denervation (Jang et al. 2010; Kostrominova, 2010; Shi et al. 2014). Notably, the ROS sensitivity of mitochondria from denervated muscle was only seen with the cPLA2 inhibitor, AACOCF3, and not with an inhibitor of the calcium independent isoform of PLA2, bromoenol lactone (Bhattacharya et al. 2009). Thus, although the calcium independent isoform of PLA2 has been implicated in production of muscle ROS (Gong et al. 2006), in the present study, we are using an inhibitor shown to be specific to the cPLA2 isoform responsible for the denervation‐induced increase in mitochondrial ROS.
An important issue that we cannot resolve is why basal ROS was not elevated in either sporadically denervated mouse or VOI human muscle, given that denervation is known to increase mitochondrial ROS (Muller et al. 2007). One factor probably relates to the sporadic nature of the denervation in both situations, meaning that the ROS signal measured in our muscle bundles will be a composite of ROS release from both innervated and denervated muscle fibres. Because the abundance of the denervated fibres is probably several‐fold less than that of the innervated fibres (based upon the fibre size distributions we observed), their signal would be obscured in the measurement. In addition, it is possible that there may be a compensatory reduction in non‐denervation related mitochondrial ROS with prolonged denervation. Although the best we can do is speculate on this issue, it is noteworthy that, in experimentally denervated rat muscle (Dow et al. 2005), it takes ∼2 weeks for muscle fibres to atrophy to the same relative size (1 percentile of the healthy young adult distribution) shown to accumulate in VOI subjects, meaning that the sporadically denervated muscle fibres accumulating in this group have probably been denervated for at least 2 weeks and possibly longer. This issue will require further study.
Role of physical inactivity in skeletal muscle mitochondrial function
Mitochondrial dysfunction is proposed to play a key role in the reduction of muscle aerobic capacity and muscle mass with advancing age (Hepple, 2014). Despite the evidence for a progressive reduction in contraction‐induced muscle aerobic capacity with increasing age (Hepple et al. 2003; Hagen et al. 2004), whether this is secondary to reduced mitochondrial content (Chabi et al. 2008) and/or impaired organelle respiratory capacity (Picard et al. 2011), or even other influences (Siegel et al. 2013), is controversial. Physical activity also plays an important role in that physically active subjects largely preserve their muscle respiratory capacity (Lanza et al. 2008; Safdar et al. 2010; Gouspillou et al. 2014). Consistent with a centrally important influence of physical activity in mitochondrial respiratory changes with ageing, we observed an exacerbation of the age‐related decline of intrinsic mitochondrial respiratory capacity in physically inactive older subjects. By contrast, physical inactivity had no impact on the previously reported sensitization of mitochondria to permeability transition in aged human muscle (Gouspillou et al. 2014), consistent with this being an intrinsic process to ageing. Because a sensitized mPTP is a signal for mitophagy (Twig et al. 2008), the accumulation of mitochondria with a sensitized mPTP in ageing muscle suggests impaired mitophagy. Therefore, based on recent studies showing a reduced Parkin expression in ageing human muscle (Drummond et al. 2014; Gouspillou et al. 2014), as well as evidence of impaired mitochondrial quality control in ageing mouse muscle (Joseph et al. 2013), we expect that strategies aimed at restoring mitochondrial quality control would prove beneficial in correcting a sensitized mPTP in ageing muscle.
Progression of mitochondrial dysfunction in advanced age
Although many studies have examined alterations in mitochondrial function in ageing muscle, there is very little evidence concerning how this progresses with increasing age, particularly in humans at ages when the clinical impact of muscle ageing is most likely to become functionally relevant (e.g. in terms of mobility impairment, falls and physical frailty). For this reason, we included a group of very old physically inactive subjects (>75 years of age) in the current investigation. Interestingly, our results show that, although the expression of some mitochondrial electron transport chain proteins decline at this more advanced age, neither respiratory capacity, nor sensitization to permeability transition were further impaired at very advanced age relative to the younger of the older aged groups (OA and OI). Thus, our results suggest that mitochondrial dysfunction occurs at ages prior to the most significant complications of ageing muscle but does not progress further at very advanced age. How this progression (or lack thereof) relates to the probable efficacy of therapeutic strategies targeting the mitochondrion is key to advancing the current state of knowledge, as is discussed below.
Sporadic denervation as a key modulator of mitochondrial function in ageing muscle
The presence and influence of persistent sporadic denervation of muscle fibres in ageing muscle was first identified morphologically through the presence of small angular fibres at very advanced age in both human (Scelsi et al. 1980; Lexell & Taylor, 1991) and animal models of muscle ageing (Ansved & Larsson, 1990; Rowan et al. 2011). More recently, the importance of denervation in ageing muscle has been reflected in muscle microarray analyses showing alterations in denervation‐responsive transcripts (Ibebunjo et al. 2013) and in situ labelling studies showing marked atrophy and induction of proteolytic pathways in persistently denervated myofibres of ageing muscle (Purves‐Smith et al. 2012; Rowan et al. 2012). Although surgical denervation studies have clearly shown that denervation is an important modulator of mitochondrial function by inducing an increase in ROS emission (Muller et al. 2007) and sensitizing the mitochondria to permeability transition (Csukly et al. 2006), the role of sporadic denervation in modulating mitochondrial function in ageing muscle had not been considered previously and is much more challenging to isolate because muscle samples may contain both denervated and innervated fibres.
When addressing this gap in knowledge, we first used a genetic model of sporadic denervation aiming to understand how sporadic denervation could alter mitochondrial function when measured at the whole tissue level, noting that the saponin‐permeabilized myofibre system employed will harvest a mixture of denervated and innervated muscle fibres when both are present in a muscle. This allowed us to reveal the influence of sporadic denervation without the influence of ageing. The key results from this model are that sporadic denervation can modulate mitochondrial function in the form of reduced mitochondrial respiratory capacity and sensitization to permeability transition, and also that the influence of sporadic denervation can be unmasked by pharmacologically inhibiting cPLA2 activity when monitoring the ROS emission response.
Second, we examined mitochondrial function in ageing human muscle across a range of ages and physical activity levels, and used morphological (the presence of very small muscle fibres), pharmacological (cPLA2 inhibition using AACOCF3) and transcriptional criteria (changes in MuSK, AChR subunit ε, Nav1.4 and Hsp27) to evaluate the contribution of sporadic denervation in these groups. Our results reveal for the first time that sporadic denervation becomes an important modulator of mitochondrial function in humans aged >75 years, as revealed by the marked attenuation of mitochondrial ROS with cPLA2 inhibition, and this is also shown to track the appearance of small fibres typical of denervation (Dow et al. 2005; Purves‐Smith et al. 2012; Rowan et al. 2012) and the transcriptional changes associated with denervation (Yang et al. 1991; Adams et al. 1995; Ma et al. 2011; Ibebunjo et al. 2013). In addition, our results show that the sensitization of mitochondria to permeability transition with ageing reported previously in physically active older subjects (Gouspillou et al. 2014) occurs prior to morphological, pharmacological and transcriptional evidence of denervation. Thus, although the results from the genetic model of sporadic denervation show that this can also occur as a consequence of sporadic denervation, our results in ageing humans show that the sensitization of mitochondria to permeability transition in ageing muscle is probably a primary organelle defect that occurs prior to persistent sporadic denervation.
In conclusion, the results of the present study support the view that therapeutic intervention for ageing muscle atrophy should aim to restore normal sensitivity to mitochondrial permeability transition, provided that this occurs prior to the accumulation of persistently denervated muscle fibres (i.e. <75 years of age). However, at very advanced age (>75 years), disturbances in mitochondrial function that are secondary to sporadic denervation render the mitochondrion a less viable therapeutic target because this could prevent a physiologically beneficial atrophy of sporadically denervated muscle fibres.
Additional information
Competing interests
Sally Spendiff, Madhusudanarao Vuda, Gilles Gouspillou, Sudhakar Aare, Anna Perez, José A. Morais, Robert T. Jagoe, Marie‐Eve Filion, Robin Glicksman, Sophia Kapchinsky, Norah MacMillan, Charlotte H. Pion, Mylène Aubertin‐Leheudre, Stefan Hettwer, José A. Correa, Tanja Taivassalo and Russell T. Hepple declare that they have no conflicts of interest.
Author contributions
SS, MV, GG, SA, AP, MF, RG and NM performed the experiments. JM and RJ performed the muscle biopsies. ML, CP and SK performed the subject recruitment and biometric data collection. SS, GG, SH, TT and RTH were involved in the experimental design. JC performed the statistical analysis of the data. SS, MV and RTH drafted the manuscript. SS, MV, GG, SA, AP, MF, RG, NM, JM, RJ, ML, CP, SK, SH, TT, JC and RTH revised the manuscript. RTH conceived the initial study. All authors have seen and approved of the final manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors gratefully acknowledge the support of the Canadian Institutes of Health Research (MOPs 125986 and 119583 to RTH), the Canada Foundation for Innovation (Leaders Opportunity Fund to RTH) and the Quebec Network for Research on Aging (Pilot Grant to MAL). Dr S. Spendiff was supported by a Postdoctoral Fellowship award from the Research Institute of the McGill University Health Centre.
Translational perspective.
Mitochondria are frequently implicated in the age‐related atrophy of skeletal muscle. However, the degree to which any mitochondrial alterations in ageing muscle represent a primary organelle defect worthy of therapeutic targeting vs. a consequence of other changes occurring with ageing is an important consideration when advancing to effective treatments. For example, sporadic denervation, which results in the accumulation of severely atrophied angular fibres in muscle at very advanced age (>75 years in humans), is a probable contributor to mitochondrial dysfunction. Indeed, because the changes in mitochondrial function with denervation are involved in mediating the atrophy of denervated muscle fibres, attempting to ‘correct’ mitochondrial changes in ageing muscle could exacerbate matters. In the presence of significant myofibre denervation, preventing atrophy of denervated fibres would place a greater burden on the remaining innervated fibres. We tested the hypothesis that, although some alterations of mitochondrial function in aged muscle are attributable to a primary organelle defect, in advanced age, mitochondrial dysfunction would be impacted by denervation. Our results support the view that therapeutic intervention for ageing muscle atrophy should aim to restore mitochondrial quality control as a means of restoring normal sensitivity to permeability transition, provided that this occurs prior to the accumulation of denervated muscle fibres. However, at very advanced age, disturbances in mitochondrial function that are secondary to denervation render the mitochondrion as a less viable therapeutic target because this could prevent a physiologically beneficial atrophy of denervated muscle fibres.
Acknowledgements
The authors wish to thank Dr Harry Rossiter (David Geffen School of Medicine at UCLA) for insightful feedback on this paper.
References
- Adams L, Carlson BM, Henderson L & Goldman D (1995). Adaptation of nicotinic acetylcholine receptor, myogenin, and MRF4 gene expression to long‐term muscle denervation. J Cell Biol 131, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansved T & Larsson L (1990). Effects of denervation on enzyme‐histochemical and morphometrical properties of the rat soleus muscle in relation to age. Acta Physiol Scand 139, 297–304. [DOI] [PubMed] [Google Scholar]
- Barde MP & Barde PJ (2012). What to use to express the variability of data: standard deviation or standard error of mean? Perspect Clin Res 3, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom J ( 1975). Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 35, 609–616. [PubMed] [Google Scholar]
- Bhattacharya A, Muller FL, Liu Y, Sabia M, Liang H, Song W, Jang YC, Ran Q & Van Remmen H (2009). Denervation induces cytosolic phospholipase A2‐mediated fatty acid hydroperoxide generation by muscle mitochondria. J Biol Chem 284, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolliger MF, Zurlinden A, Luscher D, Butikofer L, Shakhova O, Francolini M, Kozlov SV, Cinelli P, Stephan A, Kistler AD, Rulicke T, Pelczar P, Ledermann B, Fumagalli G, Gloor SM, Kunz B & Sonderegger P (2010). Specific proteolytic cleavage of agrin regulates maturation of the neuromuscular junction. J Cell Sci 123, 3944–3955. [DOI] [PubMed] [Google Scholar]
- Butikofer L, Zurlinden A, Bolliger MF, Kunz B & Sonderegger P (2011). Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J 25, 4378–4393. [DOI] [PubMed] [Google Scholar]
- Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A & Hood DA (2008). Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 7, 2–12. [DOI] [PubMed] [Google Scholar]
- Cruz‐Jentoft AJ, Landi F, Topinkova E & Michel JP (2010). Understanding sarcopenia as a geriatric syndrome. Curr Opin Clin Nutr Metab Care 13, 1–7. [DOI] [PubMed] [Google Scholar]
- Csukly K, Ascah A, Matas J, Gardiner PF, Fontaine E & Burelle Y (2006). Muscle denervation promotes opening of the permeability transition pore and increases the expression of cyclophilin D. J Physiol 574, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow DE, Dennis RG & Faulkner JA (2005). Electrical stimulation attenuates denervation and age‐related atrophy in extensor digitorum longus muscles of old rats. J Gerontol A Biol Sci Med Sci 60, 416–424. [DOI] [PubMed] [Google Scholar]
- Drummond MJ, Addison O, Brunker L, Hopkins PN, McClain DA, LaStayo PC & Marcus RL (2014). Downregulation of E3 ubiquitin ligases and mitophagy‐related genes in skeletal muscle of physically inactive, frail older women: a cross‐sectional comparison. J Gerontol A Biol Sci Med Sci 69, 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong MC, Arbogast S, Guo Z, Mathenia J, Su W & Reid MB (2006). Calcium‐independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J Appl Physiol (1985) 100, 399–405. [DOI] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, Barbat‐Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin‐Leheudre M & Hepple RT (2014). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 28, 1621–1633. [DOI] [PubMed] [Google Scholar]
- Hagen JL, Krause DJ, Baker DJ, Fu M, Tarnopolsky MA & Hepple RT (2004). Skeletal muscle aging in F344BN F1‐hybrid rats: I. Mitochondrial dysfunction contributes to the age‐associated reduction in VO2max . J Gerontol A Biol Sci Med Sci 59A, 1099–1110. [DOI] [PubMed] [Google Scholar]
- Hepple RT (2014). Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci 6, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepple RT, Hagen JL, Krause DJ & Jackson CC (2003). Aerobic power declines with aging in rat skeletal muscles perfused at matched convective O2 delivery. J Appl Physiol 94, 744–751. [DOI] [PubMed] [Google Scholar]
- Hutter E, Skovbro M, Lener B, Prats C, Rabol R, Dela F & Jansen‐Durr P (2007). Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell 6, 245–256. [DOI] [PubMed] [Google Scholar]
- Ibebunjo C, Chick JM, Kendall T, Eash JK, Li C, Zhang Y, Vickers C, Wu Z, Clarke BA, Shi J, Cruz J, Fournier B, Brachat S, Gutzwiller S, Ma Q, Markovits J, Broome M, Steinkrauss M, Skuba E, Galarneau JR, Gygi SP & Glass DJ (2013). Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol Cell Biol 33, 194–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A & Van Remmen H (2010). Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 24, 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph AM, Adhihetty PJ, Wawrzyniak NR, Wohlgemuth SE, Picca A, Kujoth GC, Prolla TA & Leeuwenburgh C (2013). Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 8, e69327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunadharma PP, Basisty N, Chiao YA, Dai DF, Drake R, Levy N, Koh WJ, Emond MJ, Kruse S, Marcinek D, Maccoss MJ & Rabinovitch PS (2015). Respiratory chain protein turnover rates in mice are highly heterogeneous but strikingly conserved across tissues, ages, and treatments. FASEB J 29, 3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent‐Braun JA & Ng AV (2000). Skeletal muscle oxidative capacity in young and older women and men. J Appl Physiol 89, 1072–1078. [DOI] [PubMed] [Google Scholar]
- Kenward MG & Roger JH (1997). Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 53, 983–997. [PubMed] [Google Scholar]
- Kostrominova TY (2010). Advanced age‐related denervation and fibre‐type grouping in skeletal muscle of SOD1 knockout mice. Free Radic Biol Med 49, 1582–1593. [DOI] [PubMed] [Google Scholar]
- Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP & Nair KS (2008). Endurance exercise as a countermeasure for aging. Diabetes 57, 2933–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexell J & Taylor CC (1991). Variability in muscle fibre areas in whole human quadriceps muscle: effects of increasing age. JAnat 174, 239–249. [PMC free article] [PubMed] [Google Scholar]
- Littell RC, Milliken GA, Stroup WW, Wolfinger RD & Schabenberger O (2006). SAS® for Mixed Models. SAS Institute Inc, Cary, NC. [Google Scholar]
- Ma CH, Omura T, Cobos EJ, Latremoliere A, Ghasemlou N, Brenner GJ, van Veen E, Barrett L, Sawada T, Gao F, Coppola G, Gertler F, Costigan M, Geschwind D & Woolf CJ (2011). Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest 121, 4332–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, Richardson A, Huang TT, Epstein CJ & Van RH (2006). Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind‐limb skeletal muscle during aging. Mech Ageing Dev 127, 298–306. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S & Leeuwenburgh C (2008). Age‐related activation of mitochondrial caspase‐independent apoptotic signalling in rat gastrocnemius muscle. Mech Ageing Dev 129, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A & Van Remmen H (2007). Denervation‐induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 293, R1159–R1168. [DOI] [PubMed] [Google Scholar]
- Picard M, Ritchie D, Thomas MM, Wright KJ & Hepple RT (2011). Alterations in intrinsic mitochondrial function with aging are fibre type‐specific and do not explain differential atrophy between muscles. Aging Cell 10, 1047–1055. [DOI] [PubMed] [Google Scholar]
- Picard M, Ritchie D, Wright KJ, Romestaing C, Thomas MM, Rowan SL, Taivassalo T & Hepple RT (2010). Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibres. Aging Cell 9, 1032–1046. [DOI] [PubMed] [Google Scholar]
- Purves‐Smith FM, Solbak NM, Rowan SL & Hepple RT (2012). Severe atrophy of slow myofibres in aging muscle is concealed by myosin heavy chain co‐expression. Exp Gerontol 47, 913–918. [DOI] [PubMed] [Google Scholar]
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R & Sandri M (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 29, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan SL, Purves‐Smith FM, Solbak NM & Hepple RT (2011). Accumulation of severely atrophic myofibres marks the acceleration of sarcopenia in slow and fast twitch muscles. Exp Gerontol 46, 660–669. [DOI] [PubMed] [Google Scholar]
- Rowan SL, Rygiel K, Purves‐Smith FM, Solbak NM, Turnbull DM & Hepple RT (2012). Denervation causes fibre atrophy and myosin heavy chain co‐expression in senescent skeletal muscle. PLoS ONE 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Hamadeh MJ, Kaczor JJ, Raha S, Debeer J & Tarnopolsky MA (2010). Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE 5, e10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scelsi R, Marchetti C & Poggi P (1980). Histochemical and ultrastructural aspects of M. vastus lateralis in sedentary old people (age 65–89 years). Acta Neuropathol Acta Neuropathologica 51, 99–105. [DOI] [PubMed] [Google Scholar]
- Shi Y, Ivannikov MV, Walsh ME, Liu Y, Zhang Y, Jaramillo CA, Macleod GT & Van Remmen H (2014). The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PLoS ONE 9, e100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS & Marcinek DJ (2013). Mitochondrial‐targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez RK, Lighton JRB, Brown GS & Mathieu‐Costello O (1991). Mitochondrial respiration in hummingbird flight muscle. Proc Natl Acad Sci U S A 88, 4870–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE & Shirihai OS (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrijbloed JW, Hettwer S, Kucsera S & Fariello RG (2011). Animal model for agrin‐dependent sarcopenia: the sarco mouse. Journal of Nutrition, Health & Aging 15, A500. [Google Scholar]
- Westfall PH, Tobias RD, Rom RD, Wolfinger RD & Hochberg Y (1999). Multiple Comparisons and Multiple Tests Using the SAS System. SAS Institute Inc, Cary, NC. [Google Scholar]
- Yang JS, Sladky JT, Kallen RG & Barchi RL (1991). TTX‐sensitive and TTX‐insensitive sodium channel mRNA transcripts are independently regulated in adult skeletal muscle after denervation. Neuron 7, 421–427. [DOI] [PubMed] [Google Scholar]