

Abstract
Hydrocephalus is a brain disorder derived from CSF accumulation due to defects in CSF clearance. Although dysfunctional apical cilia in the ependymal cell layer are causal to the onset of hydrocephalus, mechanisms underlying proper ependymal cell differentiation are largely unclear. SNX27 is a trafficking component required for normal brain function and was shown previously to suppress γ-secretase-dependent amyloid precursor protein and Notch cleavage. However, it was unclear how SNX27-dependent γ-secretase inhibition could contribute to brain development and pathophysiology. Here, we describe and characterize an Snx27-deleted mouse model for the ependymal layer defects of deciliation and hydrocephalus. SNX27 deficiency results in reductions in ependymal cells and cilia density, as well as severe postnatal hydrocephalus. Inhibition of Notch intracellular domain signaling with γ-secretase inhibitors reversed ependymal cells/cilia loss and dilation of lateral ventricles in Snx27-deficient mice, giving strong indication that Snx27 deletion triggers defects in ependymal layer formation and ciliogenesis through Notch hyperactivation. Together, these results suggest that SNX27 is essential for ependymal cell differentiation and ciliogenesis, and its deletion can promote hydrocephalus pathogenesis.
SIGNIFICANCE STATEMENT Down's syndrome (DS) in humans and mouse models has been shown previously to confer a high risk for the development of pathological hydrocephalus. Because we have previously described SNX27 as a component that is consistently downregulated in DS, we present here a robust Snx27-deleted mouse model that produces hydrocephalus and associated ciliary defects with complete penetrance. In addition, we find that γ-secretase/Notch modulation may be a candidate drug target in SNX27-associated hydrocephalus such as that observed in DS. Based on these findings, we anticipate that future study will determine whether modulation of a SNX27/Notch/γ-secretase pathway can also be of therapeutic interest to congenital hydrocephalus.
Keywords: cilia, ependymal cell, γ-secretase, hydrocephalus, SNX27, trafficking
Introduction
As a prevalent birth defect, hydrocephalus affects approximately one in every 500 newborns. It is primarily triggered by the obstruction of CSF flow by obstruction of CSF channels or by excessive CSF secretion or defects in CSF absorption. Cumulative evidence suggests that genetic factors play an important role in the development of hydrocephalus, but the underlying genetic components and their associated molecular mechanisms remain largely unknown (Zhang et al., 2006). Moreover, hydrocephalus-associated mutations that may further define mechanisms underlying its pathophysiological mechanisms have yet to be identified in either humans or model systems.
Cilia are organellar protrusions on the apical surface of eukaryotic cells and their dysfunction is associated with a variety of human diseases such as kidney cysts, hydrocephalus, and cystic fibrosis (Oishi et al., 2006; Fliegauf et al., 2007; Valente et al., 2014). Multiciliated ependymal cells line cerebral ventricles, airways, and reproductive tracts, comprising a cilial layer that beats in a synchronized fashion to drive fluid flow across ventricles. During early stages of development in the mammalian embryonic brain, radial glial cells differentiate into ependymal cells to form an ependymal monolayer lining the ventricular lumen (Spassky et al., 2005). Upon maturation of the ependymal layer, cilia beat in a coordinated manner at the apical ependymal cell surface to facilitate CSF circulation (Del Bigio, 2010). Although cilia and ependymal cell function have been characterized previously, the molecular mechanisms underlying cilium growth and ependymal cell differentiation and maturation are still not clear.
As the sole SNX family member comprising a PDZ domain, SNX27 is an endosomal sorting factor highly expressed in brain. SNX27 has been reported to participate in the trafficking of multiple transmembrane receptors. Our results showed that cognitive impairment in Snx27-deficient mice resemble similar cognitive deficits found in Down's syndrome (DS) (Wang et al., 2013); which occurs at least in part through SNX27 trafficking interactions with ionotropic glutamate receptors to maintain proper synaptic function. In addition to its role in regulating protein trafficking, we also found that SNX27 deletion enhances PS1/γ-secretase complex formation and γ-secretase activity to elevate β-amyloid and Notch intracellular domain (NICD) generation (Wang et al., 2014b).
Interestingly, an essential role for Notch signaling has been established in the differentiation of various multiciliated cell types, including ependymal cells, in brain ventricles (Liu et al., 2007; Carlén et al., 2009; Zhao et al., 2009; Marcet et al., 2011; Lin et al., 2013; Brooks and Wallingford, 2014). In mammalian cells, γ-secretase-dependent cleavage of Notch receptors generates a transcriptionally active NICD, thereby activating downstream Notch signaling pathways to inhibit the differentiation of multiciliated cells. To this point, upstream components and mechanisms of the Notch pathway that are physiologically involved in modulating multiciliated cell differentiation have been poorly characterized and how these mechanisms might be linked to developmental and pathological abnormalities derived from these integrated mechanisms remain largely unclear.
Here, we describe a pathophysiological role for SNX27 in mediating ciliogenesis in the brain ventricular ependymal layer and demonstrate that SNX27 deletion in mouse models impairs cilium development, leading to defective CSF flow and severe hydrocephalus. Defects in ependymal cell layer formation with SNX27 deletion was found to be caused by severe impairment of neural progenitor cell differentiation to terminal ependymal cell types, leading to pathological ventricle dysfunction and hydrocephalus. Given that abnormal Notch hyperactivation is linked to the suppression of multiciliated cell differentiation, we questioned whether modulation of Notch activation could be integrated into SNX27-dependent ependymal cell defects. Remarkably, we find that γ-secretase inhibitor treatment reverses aberrant dilation of lateral ventricles and partially restores the ciliated ependymal cell layer in Snx27-null mice. Together, these results implicate a pathophysiological role for SNX27-mediated γ-secretase inhibition in facilitating proper ependymal cell differentiation through suppression of the Notch pathway and aberrant Notch hyperactivation through SNX27 deletion leads to improper ependymal cell differentiation and hydrocephalus.
Materials and Methods
Antibodies.
Antibodies used for immunohistochemistry were as follows: mouse anti-SNX27 (1:200; Abcam), rabbit anti-S100β (1:100; Abcam), mouse anti-acetylated-α-tubulin (1:1000; Sigma-Aldrich), mouse anti-GFAP (1:50; Cell Signaling Technology), rabbit anti-BLBP (1:200; Abcam), rabbit anti-DCX (1:400; Abcam), mouse anti-PAX6 (1:50; Developmental Studies Hybridoma Bank), rabbit anti-CUX1 (1:100; pAb; Millipore), rat anti-CTIP2 (1:500; Abcam), and mouse anti-NeuN (1:100; Millipore).
The antibodies used for Western blot analysis were as follows: SNX27 polyclonal antibodies and SNX27 monoclonal antibodies (described previously; Balana et al., 2011; Cai et al., 2011), rabbit anti-S100β (1:1000; Abcam), mouse anti-GFAP (1:500; Cell Signaling Technology), mouse anti-PAX6 (1:500; Developmental Studies Hybridoma Bank), rabbit anti-BLBP (1:1000; Abcam), rabbit anti-DCX (1:1000; Abcam), rabbit anti-HES1 (1:500; Abcam), mouse anti-NeuN (1:1000; Millipore), rabbit anti-Vangl1 (1:1000; Abcam), rabbit anti-Vangl2 (1 μg/ml; Millipore), goat anti-Fizzled 3 (1:1000; R&D Systems), goat anti-Celsr2 (1:200; R&D Systems), mouse anti-GFAP (1:1000; Cell Signaling Technology), rabbit anti-N-Cadherin (1:1000; Millipore), mouse anti-β-tubulin (1:5000; Sigma-Aldrich), and mouse anti-β-actin (1:5000; Sigma-Aldrich).
Constructs.
pCI-neo (Promega) and pDMyc-SNX27 used in this study have been described previously (Wang et al., 2013). SNX27 and SNX27ΔPDZ were inserted into the pRK5mGST vector (Huang et al., 2013) to generate GST-tagged constructs.
Mouse strains.
Snx27+/+, Snx27+/−, and Snx27−/− mice (Cai et al., 2011) were generated by crossing heterozygotes on C57BL/6 and 129SV mixed backgrounds to produce F1 hybrid background animals. Both males and females were used for biochemical experiments; only male mice were used for EM and histological experiments.
All procedures involving animals were performed under the guidelines of Xiamen University and the Sanford-Burnham Medical Research Institute Institutional Animal Care and Use Committee.
Immunoprecipitation.
293T cells transfected with different expression constructs were lysed in NP-40 buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 1% NP-40, supplemented with protease inhibitors). Lysates were immunoprecipitated using mouse IgG; antibodies against Snx27, Vangl1, Vangl2, and Celsr2; and Trueblot IP beads (eBioscience), followed by Western blotting.
GST pull-down assay.
GST-SNX27 and SNX27 deletion constructs were transfected into HEK293T cells and lysates were probed with glutathione Sepharose. Precipitates were washed three times in lysis buffer and boiled in SDS-PAGE loading buffer. Eluates and lysate inputs were immunoblotted for either Vangl1 or Vangl2.
Electron microscopy.
Mice were perfused initially with saline, followed by 4% paraformaldehyde and 1.5% glutaraldehyde in 0.1 m Na cacodylate buffer. Cortical tissues were dissected to reveal the desired ventricles and fixed by immersion overnight at 4°C. The tissues were washed in buffer, postfixed in 1% osmium tetroxide, and subsequently dehydrated in a graded ethanol series.
For transmission electron microscopy (TEM) examination, the brain tissues were treated in propylene oxide and embedded in EMbed 812/Araldite resin (Electron Microscopy Sciences). Thick sections (2 μm) were cut, mounted on glass slides, and stained in toluidine blue for general assessment by light microscopy. Subsequently, 70 nm thin transverse sections were prepared, mounted on copper slot grids precoated with Parlodion, and stained with uranyl acetate and lead citrate before examination on a Philips CM100 electron microscope at 80 kV. Images were documented on a Megaview III CCD camera (Olympus).
For SEM examination, dehydrated tissue pieces were treated with hexamethyldisilazane (Electron Microscopy Sciences), desiccated, and mounted onto SEM stubs with carbon tape. Samples were then sputter coated with iridium (EMS model 150T S) at 10 μA to a thickness of 6–7 nm for subsequent examination and documentation using a Hitachi S-4800 SEM operating at 5 kV.
Compound E rescue experiment.
Compound E (Adipogen) was dissolved in 5% DMSO + 95% corn oil. Pregnant Snx27+/− mice at embryonic day 15.5 (E15.5) were intraperitoneally injected with compound E (10 nmol/g/d) until birth and elimination of hydrocephalus was assessed in postnatal day 3 (P3) mice. An equivalent amount of DMSO + corn oil was administered to the control group.
Immunohistochemistry and data analyses.
Snx27−/− or Snx27+/− mice and Snx27+/+ littermate controls were anesthetized and fixed by intracardial perfusion with 4% PFA. Whole brains were excised and postfixed in 4% PFA overnight. Tissue blocks were embedded in paraffin and 5 μm sections were cut and stained with Nissl (cresyl violet). Immunostained sections were scanned on the Aperio ScanScope slide scanner (Leica) and analyzed using ImageScope software.
Primary murine embryonic fibroblast (MEF) cell culture.
Primary MEFs were isolated from E13.5 mouse embryos as described previously (Diril et al., 2012). Briefly, the head and the visceral organs were removed, the embryonic tissue was chopped into fine pieces with a razor blade and trypsinized for 15 min at 37°C, and tissue/cell clumps were dissociated by pipetting. Cells were plated in a 10 cm culture dish and grown in DMEM (Invitrogen) supplemented with 10% FBS (Sigma-Aldrich) and 1% penicillin/streptomycin (Invitrogen). Primary MEFs were cultured in a humidified incubator with 5% CO2 and 3% O2.
Cell surface biotinylation assay.
Surface biotinylation was performed following a previously described protocol (Liu et al., 2009).
Human brain specimens.
Human brain samples used in this project was provided by University of California–San Diego and were analyzed with institutional permission under California and National Institutes of Health guidelines.
TUNEL staining.
An in situ cell death detection kit was purchased from Roche and labeling reactions were performed according to the manufacturer's instructions. TUNEL staining was performed on cryosections.
Statistical analyses.
Statistical analyses were determined using GraphPad Prism. Data distribution was assessed by a Kolmogorov–Smirnoff nonparametric test of equality. Differences between two means were assessed by paired or unpaired t test. Differences among multiple means were assessed, as indicated, by one-way ANOVA, followed by Tukey's post hoc test. Error bars represent SEM. Null hypotheses were rejected at the 0.05 level.
Results
Snx27−/− mice develop progressive hydrocephalus
We have observed previously that Snx27−/− mice demonstrate a slower growth rate at which most pups are viable ∼2–3 weeks after birth (Cai et al., 2011). Snx27−/− mouse brains are notably smaller compared with brains from Snx27+/+ pups at weaning. To characterize the temporal aspects of developmental dysfunction with Snx27 deletion, we compared developing brain mass between P0 and P14 in wild-type and Snx27 knock-out murine pups (Fig. 1A). Snx27−/− mouse brain was comparatively reduced in mass compared with Snx27+/+ at P3, P7, and P14, with marginal differences at P0. This indicates that SNX27-dependent developmental dysfunction occurs primarily during postnatal development. At P18, all Snx27−/− mice develop a dome-shaped skull typically observed in hydrocephalus (Fig. 1B). Histological examination of Snx27−/− mice at varying postnatal stages display enlarged lateral ventricles and abnormal brain morphology (Fig. 1C). Interestingly, Snx27−/− mouse brains are slightly smaller than those of Snx27+/+ at birth (P0) with no noticeable morphological abnormalities (data not shown). However, progressive ventricular dilation in Snx27−/− brain became apparent at P3 and increased in width with progressive severity during postnatal development (Fig. 1C). Together, these results suggest that Snx27 deletion results in developmental brain dysfunction and distinct hydrocephalus characteristics during postnatal development.
Figure 1.

Severe postnatal hydrocephalus in Snx27−/− mice. A, Brain weight of Snx27+/+ and Snx27−/− mice at multiple postnatal time points. Data represent mean ± SEM. n = 3. p-values were calculated using a nonparametric t test, *p < 0.05. B, Comparison of Snx27+/+ and Snx27−/− mice at P18. Note that Snx27−/− animals display a dome-shaped skull typically observed in progressive hydrocephalus. Scale bar, 2 cm. C, Coronal sections of P3 (top) and P14 (bottom) mouse brains were stained with Nissl. Quantitative analysis of P3 (top) and P14 (bottom) lateral ventricles (LV) and brain sections from Snx27+/+ and Snx27−/− mice. Data represent mean ± SEM. n = 3. p-values were calculated using two-tailed Student's t test, *p < 0.05. Scale bar, 1 mm.
Hydrocephalus in Snx27−/− brains is accompanied by loss of ependymal cells and impaired ciliogenesis
Because the obstruction of CSF aqueduct channels could be a causative factor in SNX27-dependent hydrocephalus, we examined a series of sections of Snx27−/− mouse brains and failed to observe stenosis or constriction within aqueduct channels (Fig. 2). The obstruction of CSF channels is not involved in Snx27 null-induced hydrocephalus, so we examined other physiological irregularities that could contribute to the hydrocephalus phenotype observed. Because denudation or ependymal cell loss can accompany hydrocephalus of various etiologies (Sarnat, 1995), we next investigated whether SNX27 expression coincides with the ependymal cell layer. Using a SNX27 monoclonal antibody, we observed that SNX27 is enriched in S100β-expressing cells lining lateral ventricles in human brain tissue (Fig. 3A), indicating that SNX27 is highly expressed in ependymal cells.
Figure 2.
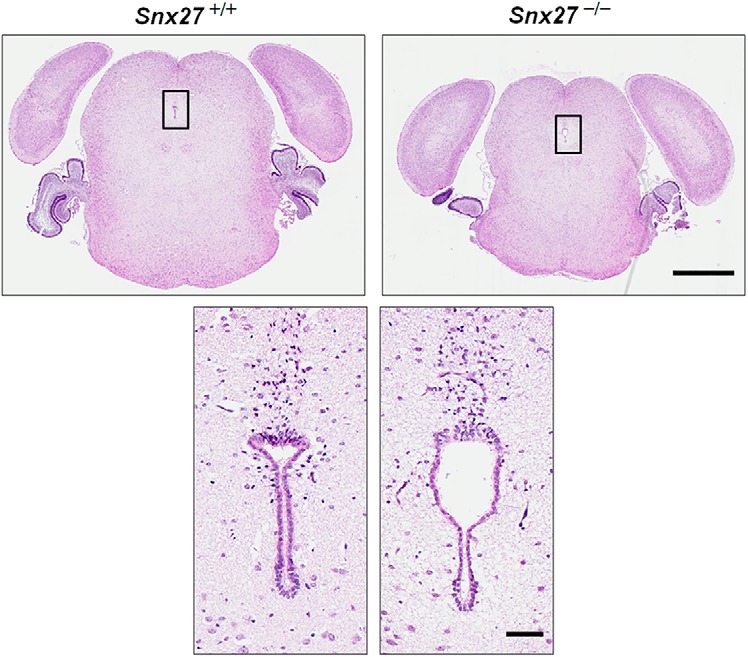
Hydrocephalus is not caused by aqueductal stenosis in Snx27−/− mice. H&E staining of Snx27+/+ and Snx27−/− mouse brains at P3 showing that no aqueductal stenosis was found. Scale bars: top, 1 mm; bottom, 100 μm.
Figure 3.
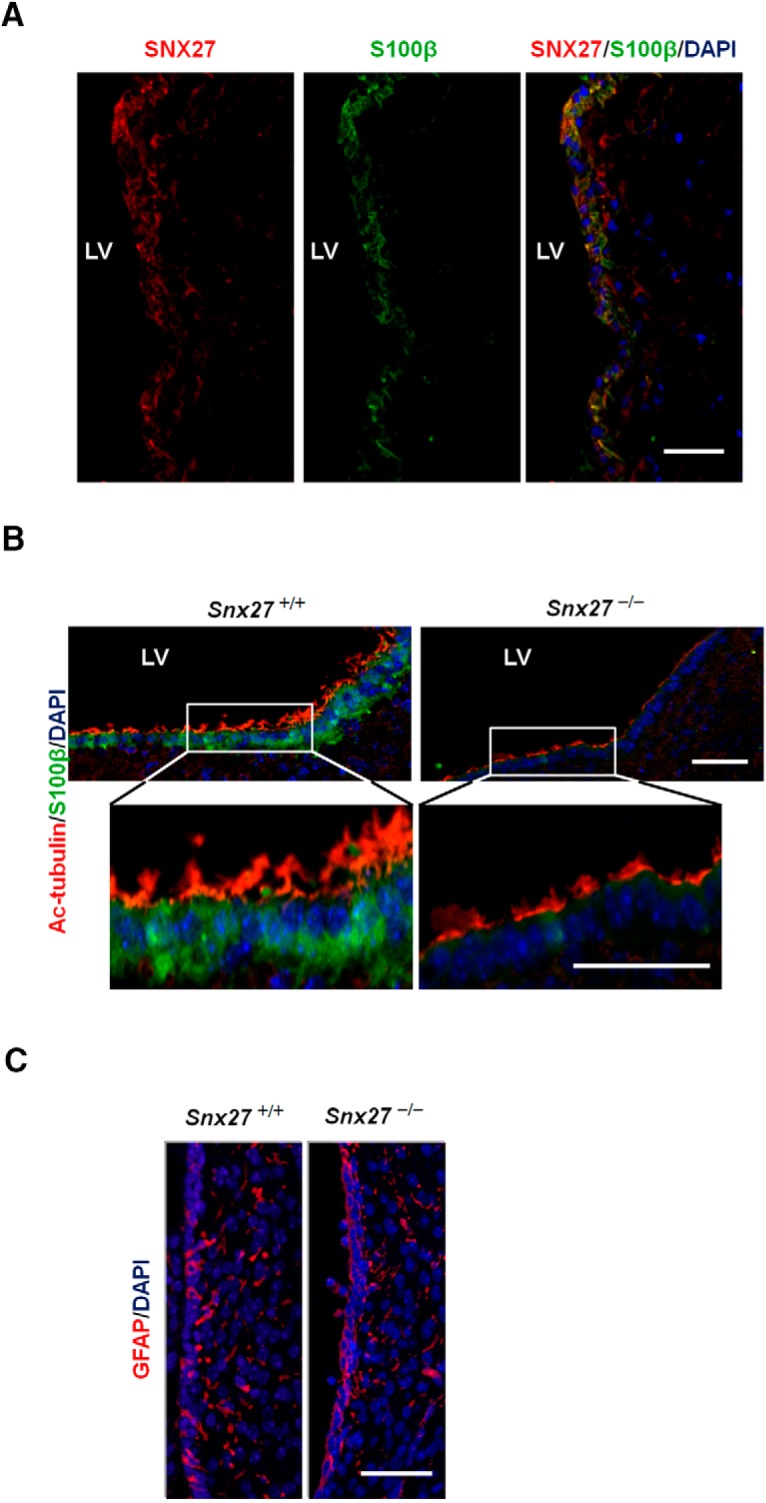
Loss of ependymal cells and cilia in Snx27−/− mice. A, SNX27 expression in the ependymal cell layer. Lateral ventricular surface from a frozen human brain section stained with anti-SNX27 (red), anti-S100β (green, ependymal cell marker), and DAPI (blue, nuclear marker). Scale bar, 50 μm. B, P3 mouse brain sections were stained with anti-acetylated-tubulin (red, cilium marker), anti-S100β (green, ependymal cell marker), and DAPI (blue, nuclear marker). Scale bar, 50 μm. C, P3 mouse brain sections were stained with anti-GFAP (red, astrocyte marker) and DAPI (blue, nuclear marker). Scale bar, 50 μm.
Given that SNX27-enrichment in ependymal cells may implicate its importance in the ependyma, we examined the ventricular surface of P3 Snx27+/+ and Snx27−/− mice by histological analysis and compared differences in distribution of ependymal cells (S100β) and cilia (acetylated α-tubulin). Interestingly, both ependymal cell and cilia density are markedly reduced in ventricular regions in Snx27−/− brains compared with Snx27+/+ brain ventricles (Fig. 3B). However, the density of GFAP-positive cells remained unchanged in Snx27−/− mouse brain, suggesting that astrocytogenesis was unaffected by Snx27 deletion (Fig. 3C). Western blot analysis consistently showed reductions in S100β expression with Snx27 deletion, with little or no change in GFAP levels (see Fig. 8B). This indicates that SNX27 may be important in establishing and/or maintaining the ependyma and its associated ciliary elements.
Figure 8.

Disruption of adherens junctions in the Snx27−/−SVZ. A, TEM of P7 ventricular wall lateral junctions between ependymal cells. Note the shorter length of adherens junction in Snx27−/− mouse brain tissue. Scale bar, 500 nm. B, Western blot analysis of Vangl2, Celsr2, S100β, GFAP, and SNX27 in the cortex of P1 and P18 Snx27+/+ and Snx27−/− mice. Signal intensities derived from immunoblotting were determined and normalized to β-actin. n = 3. p-values were calculated using a nonparametric t test, *p < 0.05. C, Decreased cell surface expression of Vangl2 and Frizzled-3 in Snx27−/− MEF cells. Data represent mean ± SEM. n = 3. p-values were calculated using two-tailed Student's t test, *p < 0.05. D, Coimmunoprecipitation showing that myc-SNX27 interacts with Vangl2, but not Vangl1 and Celsr2. Cell lysates were immunoprecipitated with mouse IgG (mIgG) or Myc antibody and Western blotted with the indicated antibodies. E, The PDZ domain is required for interaction between SNX27 and Vangl2. GST-SNX27, GST-SNX27-PDZ, or GST-SNX27 lacking the PDZ domain (SNX27-ΔPDZ) expressed in HEK293T cells were precipitated with glutathione Sepharose beads and endogenous Vangl2 was detected by immunoblot as indicated.
The ependymal apical layer comprises a motile surface mobilized by a confluent ciliary bed that promotes the flow of CSF through brain ventricles. Because Snx27 deletion affects ependymal and ciliary structure, we analyzed the ultrastructure of the lateral ventricle wall in wild-type and Snx27−/− mouse brain using SEM. At P7, the lateral ventricle wall in Snx27+/+ mice comprised a confluent ependymal cilia layer, whereas cilium coverage in lateral ventricles derived from Snx27−/− mouse brain was comparatively sparse (Fig. 4A). In addition, cilia derived from Snx27−/− brain ventricles were found to be comparatively shorter than those observed in Snx27+/+ mice (Figs. 3B, 4A).
Figure 4.

Ciliogenesis is altered in Snx27-deficient mice. A, SEM micrographs of ependymal cilia lining the surface of lateral ventricles in P7 mouse brains. Scale bars: top: 5 μm; bottom: 2 μm. B, TEM analysis of tangential sections of ependymal cilia from P7 Snx27+/+ and Snx27−/− mouse brain. Scale bar, 100 nm.
We also performed TEM analysis to determine potential substructural differences in cilia derived from wild-type and Snx27−/− ependyma. The ultrastructure of residual Snx27−/− ependymal cilia retained a normal 9 + 2 structure of motile cilia, suggesting that Snx27 deletion has no direct influence on inner cilia structure (Fig. 4B). Rather, given the marked impairment of ciliogenesis in Snx27−/− brain, Snx27 may be required for proper ependymal cell layer formation and the lack of ependymal cilia with Snx27 deletion may perturb normal CSF flow, leading to hydrocephalus.
SNX27 is required for ependymal cell differentiation from radial glia
Reduction of the ependymal cell layer in Snx27−/− ventricles could be caused either by decreased ependymal cell generation or increased cell death. To determine whether Snx27 deficiency can trigger apoptosis in the ependymal layer, we performed costaining with TUNEL and S100β and observed comparable levels of apoptotic cells in Snx27−/− and Snx27+/+ ependyma (Fig. 5), indicating that loss of ventricular ependymal cells in Snx27−/− mouse brain is not caused by cell death. We then investigated whether progenitor and differentiated cell populations differed in ventricular regions with Snx27 deletion. Interestingly, we observed depletion of neural progenitor cells in Snx27−/− mouse cortex at E15.5 compared with Snx27+/+ mice. The density of BLBP-expressing radial glial cells was markedly reduced in Snx27−/− cortex (Fig. 6A,B). Western blot analysis consistently revealed reduced expression of PAX6 and BLBP in E15.5 Snx27−/− cortex (Fig. 6C). These results demonstrate a role for SNX27 in regulating cell fate in the developing brain.
Figure 5.

Apoptosis likely does not contribute to the loss of ventricular ependymal cells in Snx27 −/− mouse brain. A, No apoptotic ependymal cells were detected in P3 Snx27−/− mouse brain using TUNEL staining. Scale bar, 50 μm. B, H&E staining showing loss of ependymal cell layer in P10 Snx27−/− mouse brain. Scale bar, 200 μm.
Figure 6.

Disrupted cell fate in the Snx27−/− SVZ. A, At E15.5, the cortex of Snx27−/− mouse embryos contain markedly fewer BLBP-positive cells (radial glial marker, red color) compared with Snx27+/+ mice. Nuclei are visualized by DAPI staining (blue). Scale bar, 200 μm. B, Magnified view of BLBP-positive cells in the cortex of E15.5 Snx27+/+ and Snx27−/− mice. Note that BLBP-positive cells are primarily localized within the ependymal cell layer and are largely absent in Snx27−/− mouse embryonic cortex. Scale bar, 50 μm. C, Western blot analysis of PAX6, BLBP, NeuN, HES1, and SNX27 in the cortex of E15.5 Snx27+/+ and Snx27−/− mice. Signal intensities from the immunoblots indicated were determined and normalized to β-tubulin. n = 3. p-values were calculated using a nonparametric t test, *p < 0.05. D, Lateral ventricular wall whole-mount IHC staining in P7 Snx27+/+ and Snx27−/− mice. The whole mounts were stained with anti-BLBP (red, radial glial marker) and S100β (green, ependymal cell marker). Scale bar, 20 μm.
Immunohistochemical (IHC) staining of ventricular wall whole mounts from P7 mice showed that cells derived from the ventricular zone (VZ) in Snx27−/− brain remained BLBP-positive, whereas those from Snx27+/+ mice comprised mostly S100β-positive ependymal cells. This suggests that, in Snx27−/− mice, radial glial cells for the most part fail to mature and properly differentiate into ependymal cells (Fig. 6D). Interestingly, IHC staining showed abnormal cortical development in Snx27−/− mouse brains (Fig. 7A–D) because the thickness of the Snx27−/− mouse cortex at P3 was markedly reduced. However, the number of mature neurons was not significantly reduced by Snx27 deletion (Fig. 7B). Together, these results implicate SNX27 in early cell fate determination to establish the ventricular ependyma during postnatal brain development.
Figure 7.

Snx27 deficiency disrupts cortical development. A, Nissl staining revealing the reduction in the thickness of the cortex of P3 Snx27−/− mouse cortex. Scale bar, 100 μm. B, NeuN staining revealing the reduction in the thickness of the cortex of P3 Snx27−/− mouse cortex. Scale bar, 100 μm. C, CUX1 staining revealing the reduction in the thickness of P3 Snx27−/− mouse cortical layers II–IV. Scale bar, 100 μm. D, CTIP2 staining revealing the reduction in the thickness of P3 Snx27−/− mouse cortical layers V–VI. Scale bar, 100 μm.
Snx27 deletion leads to the disorganization of ependymal adherens junctions
The brain ventricle epithelium is stratified in a polar fashion and laterally sealed via adherens junctions, where cilia are distributed on the apical surface (Chenn et al., 1998). Because we have established that Snx27 deletion results in impaired ciliogenesis and malformation of the ependymal layer, we wished to further characterize the structural and molecular nature of ependymal misorganization in Snx27−/− brain to induce hydrocephalus. To this end, we searched for abnormalities within the ultrastructure of Snx27−/− P7 ependyma at by TEM analysis. We observed that the lining of ependymal cells in Snx27+/+ mice was highly polarized with characteristic adherens junctions. Although adherens junctions were present in Snx27−/− mice, the distance spanning these junctions appeared to be reduced with Snx27 deletion (Fig. 8A).
Several studies have shown that several planar cell polarity (PCP) signaling components such as Vangl2, Frizzled-3, and Celsr1/2/3 are essential for the maintenance of adherens junctions and ciliogenesis (Wang et al., 2013; Boutin et al., 2014). We investigated whether the abundance of these components was perturbed by Snx27 deletion and found that protein expression of Vangl2 was markedly downregulated in both P1 and P18 Snx27−/− cortex (Fig. 8B). Moreover, cell surface biotinylation assays showed reduced cell surface levels of Vangl2 and Frizzled-3 in Snx27−/− MEF cells compared with those in Snx27+/+ MEF cells (Fig. 8C). Interestingly, we also observed a reduction in total Vangl2 in Snx27−/− MEF cells (Fig. 8C).
To determine whether SNX27 regulates Vangl2 cell surface delivery through physical interactions, we transfected myc-tagged SNX27 into HEK293T cells and assayed for interactions between myc-SNX27 and Vangl1/2 by coimmunoprecipitation. We detected an interaction between SNX27 and Vangl2, but not Vangl1 (Fig. 8D). Interestingly, both Vangl2 and Vangl1 comprise canonical E-T-S-V class I PDZ-interacting tail motifs, suggesting that other factors may contribute to binding specificity with SNX27. To confirm whether SNX27 binds to Vangl2 through PDZ-dependent interactions, we performed in vitro binding assays using GST-SNX27, GST-SNX27-PDZ, and GST-SNX27-ΔPDZ immobilized on glutathione Sepharose and found that both GST-SNX27-PDZ domain and full-length GST-SNX27 coprecipitated with Vangl2, whereas no interaction was observed between GST-SNX27-ΔPDZ and Vangl2. These results suggest that SNX27 regulates the cell surface transport of Vangl2 through a PDZ-dependent interaction between SNX27 and Vangl2. Together, these results indicate that Snx27 deletion can reduce adherens tight junctions and PCP components required for ciliogenesis and tight junction formation. Because SNX27 has been well characterized as a transport component, we also show that SNX27 can facilitate cell surface distribution of PCP components such as Vangl2.
Inhibition of Notch signaling blocks Snx27 deletion-associated ependymal cell loss and partially rescues hydrocephalus in Snx27−/− mice
It has been reported that overexpression of the NICD leads to a reduced number of ciliated cells and, conversely, that repression of Notch signaling results in an increased number of precursor cells adopting a multiciliated cell fate (Deblandre et al., 1999; Tsao et al., 2009; Marcet et al., 2011). Although our previous findings indicate that SNX27 deficiency upregulates γ-secretase cleavage of Notch and generation of NICD (Wang et al., 2014b), it was unclear how SNX27-mediated γ-secretase suppression could influence pathophysiological brain development. Because we observed increased expression of the Notch signaling target Hes1 in Snx27−/− brain by immunoblot analysis (Fig. 6C), it appears likely that the absence of SNX27 could aberrantly elevate γ-secretase activity to hyperactivate Notch signaling pathways. We therefore determined whether inhibiting excessive Notch signaling could rescue aberrations in ciliogenesis, ependymal cell developmental dysfunction, and hydrocephalus observed in Snx27−/− mice. Because ependymal cells are established primarily between E14 and E16, we delivered a γ-secretase inhibitor compound E into pregnant Snx27-null mice at E14.5 through intraperitoneal injection daily until birth. We found that γ-secretase inhibitor injection partially reversed the dilation of lateral ventricles (Fig. 9A) and restored the loss of ependymal cells and cilia (Fig. 9B) in Snx27-null mice. Therefore, inhibition of Notch signaling by compound E-mediated γ-secretase inhibition ameliorates the development of hydrocephalus in mice caused by Snx27 deficiency.
Figure 9.

Inhibition of Notch signaling blocks the development of hydrocephalus in Snx27−/− mice. A, Enlarged lateral ventricles caused by Snx27 deficiency were partially rescued by treatment with compound E. Data represent mean ± SEM. n = 3. p-values were calculated using a nonparametric t test, *p < 0.01, **p < 0.001, ***p < 0.0001. Scale bar, 500 μm. B, Snx27 deficiency induced loss of lateral ventricular (LV) ependymal cells and cilia was reversed by compound E treatment. Scale bar, 50 μm.
Discussion
In this study, we observed that Snx27−/− mice develop progressive hydrocephalus during early postnatal development. Although we excluded the possibility of aqueduct stenosis, which is one possible cause for hydrocephalus (Fig. 2), we present evidence that malformation of the ependymal cilium layer with Snx27 deletion is likely causal to the physiological aberrations in the hydrocephalus pathology observed. Together, these results present a model in which SNX27 normally maintains proper differentiation of neural progenitors into ependymal cells through suppression of γ-secretase/Notch and loss of Snx27 results in the aberrant accumulation of premature neurons, impaired ciliogenesis, and hydrocephalus (Fig. 10).
Figure 10.

A working model of Snx27 deficiency and the onset of hydrocephalus. In Snx27−/− mouse brains, radial glial cells cannot differentiate into ependymal cells (S100β+) and the density of ependymal cilia is markedly decreased, resulting in hydrocephalus.
Several cilium-related genes have been implicated in hydrocephalus, suggesting that loss of cilia contributes to the development of hydrocephalus. Genetic deficiency of genes such as Mdnah5 (Ibanez-Tallon et al., 2004), Tg737 (Banizs et al., 2005), Hy3 (Davy and Robinson, 2003), Celsr2/3 (Tissir et al., 2010), Foxj1 (Jacquet et al., 2009), Six3 (Lavado and Oliver, 2011), c-Myb (Malaterre et al., 2008), and Snx27 results in impaired ciliogenesis and hydrocephalus. Interestingly, we observed that Snx27 deletion results in deficiencies in proper ependymal cell differentiation to ependymal cell types with a compensatory increase in differentiation to nonciliated cells (Páez et al., 2007).
The Notch receptor is a single-pass transmembrane protein involved in multiple important biological processes and is especially critical for embryonic development. More specifically, Notch signaling controls the differentiation of neural progenitor cells to terminal ependymal cells in the VZ during embryonic and postnatal development. Activation of Notch signaling requires cleavage of the Notch receptor by γ-secretase and release of an active NICD, which acts as a transcription factor through translocation to the nucleus. Several studies have demonstrated that functional Notch signaling is crucial for ependymal cell differentiation and ciliogenesis. Genetic deletion of either Lgl1 or Numb, negative Notch regulators, leads to severe hydrocephalus (Klezovitch et al., 2004; Kuo et al., 2006). Although genetic Notch suppressors such as Lgl1 or Numb have been implicated in hydrocephalus, how inhibitors of the γ-secretase complex are integrated in suppressing hydrocephalus has been unclear. Our results described here provide evidence that SNX27 is a suppressor of Notch signaling and that deletion of Snx27 triggers an aberrant hydrocephalus pathophysiology derived at least in part from abnormal Notch hyperactivation (Wang et al., 2014b). This likely occurs through SNX27-dependent suppression of Notch activity through homeostatic inhibition of Notch cleavage and activation by the γ-secretase complex (Wang et al., 2014b). Because we previously described a mechanism for SNX27 disassembly of the γ-secretase complex through interactions with Presenilin 1, it is likely that excessive generation of the transcriptionally active NICD fragment is key in inhibiting ependymal cell differentiation to promote ciliogenesis deficits and hydrocephalus.
The mechanistic link between SNX27 and Notch signaling described here provides a novel targeting pathway for hydrocephalus and other neurological disorders involving SVZ formation and CSF clearance. For example, recent evidence indicates that sleep promotes CSF exchange to facilitate clearance of the proteotoxic Alzheimer's disease-associated Aβ peptide (Xie et al., 2013). This suggests that perturbations in SNX27/Notch signaling may impede clearance of certain toxicity factors cleared by the CSF. The overabundant differentiation of radial glial cells into premature neurons with Snx27 deletion also indicates an imbalance in differentiated cell types that may result from perturbations of SNX27/Notch pathway.
Because specific pharmacological treatments for hydrocephalus are currently scarce, drug-targeting strategies that may restore CSF balance in affected individuals would be extremely beneficial. In severe cases in which hydrocephalus is induced pathologically through defects in ciliogenesis, it may be possible that specific inhibition of Notch signaling or upstream activation of SNX27-mediated γ-secretase inhibitory mechanisms may be viable molecular nodes for future targeting strategies. We previously reported an important role of Snx27 deficiency in the pathogenesis of DS (Wang et al., 2013). Moreover, researchers found that both individuals with DS (Torfs and Christianson, 1998) and a DS mouse model (Yu et al., 2010) had a higher risk of developing hydrocephalus. Therefore, SNX27 deficiency may also contribute to the onset of hydrocephalus in DS.
Footnotes
This work was supported in part by the National Natural Science Foundation of China (Grant 81571176 to X.W. and Grants U1405222 and 91332114 to H.X.), the Thousand Young Talents Program of China, the Fundamental Research Funds for the Chinese Central Universities (Grant 20720150061 to X.W.), the National Institutes of Health (Grants R01 AG038710, R01 AG021173, R01 NS046673, R01 AG030197, and R01 AG044420 to H.X.), and the Alzheimer's Association (H.X), the Global Down Syndrome Foundation (Grant DSADIIP-13-283543, to H.X.), the BrightFocus Foundation (H.X.), and the Cure Alzheimer's Fund (H.X.). We thank B. Ranscht, C. Kintner, D. Hanein, W.C. Mobley, and C. Wu for fruitful discussion; G. Garcia, A. Keeton, and L. Lacarra for technical help; E. Masliah for providing human brain specimens; and M. Wood and T. Fassel for technical help with electron microscopy.
The authors declare no competing financial interests.
References
- Balana B, Maslennikov I, Kwiatkowski W, Stern KM, Bahima L, Choe S, Slesinger PA. Mechanism underlying selective regulation of G protein-gated inwardly rectifying potassium channels by the psychostimulant-sensitive sorting nexin 27. Proc Natl Acad Sci U S A. 2011;108:5831–5836. doi: 10.1073/pnas.1018645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banizs B, Pike MM, Millican CL, Ferguson WB, Komlosi P, Sheetz J, Bell PD, Schwiebert EM, Yoder BK. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development. 2005;132:5329–5339. doi: 10.1242/dev.02153. [DOI] [PubMed] [Google Scholar]
- Boutin C, Labedan P, Dimidschstein J, Richard F, Cremer H, André P, Yang Y, Montcouquiol M, Goffinet AM, Tissir F. A dual role for planar cell polarity genes in ciliated cells. Proc Natl Acad Sci U S A. 2014;111:E3129–E3138. doi: 10.1073/pnas.1404988111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks ER, Wallingford JB. Multiciliated cells. Curr Biol. 2014;24:R973–R982. doi: 10.1016/j.cub.2014.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Loo LS, Atlashkin V, Hanson BJ, Hong W. Deficiency of sorting nexin 27 (SNX27) leads to growth retardation and elevated levels of N-methyl-D-aspartate receptor 2C (NR2C) Mol Cell Biol. 2011;31:1734–1747. doi: 10.1128/MCB.01044-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlén M, Meletis K, Göritz C, Darsalia V, Evergren E, Tanigaki K, Amendola M, Barnabé-Heider F, Yeung MS, Naldini L, Honjo T, Kokaia Z, Shupliakov O, Cassidy RM, Lindvall O, Frisén J. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci. 2009;12:259–267. doi: 10.1038/nn.2268. [DOI] [PubMed] [Google Scholar]
- Chenn A, Zhang YA, Chang BT, McConnell SK. Intrinsic polarity of mammalian neuroepithelial cells. Mol Cell Neurosci. 1998;11:183–193. doi: 10.1006/mcne.1998.0680. [DOI] [PubMed] [Google Scholar]
- Davy BE, Robinson ML. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum Mol Genet. 2003;12:1163–1170. doi: 10.1093/hmg/ddg122. [DOI] [PubMed] [Google Scholar]
- Deblandre GA, Wettstein DA, Koyano-Nakagawa N, Kintner C. A two-step mechanism generates the spacing pattern of the ciliated cells in the skin of Xenopus embryos. Development. 1999;126:4715–4728. doi: 10.1242/dev.126.21.4715. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR. Ependymal cells: biology and pathology. Acta Neuropathol. 2010;119:55–73. doi: 10.1007/s00401-009-0624-y. [DOI] [PubMed] [Google Scholar]
- Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, Kaldis P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci U S A. 2012;109:3826–3831. doi: 10.1073/pnas.1115201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Huang TY, Michael S, Xu T, Sarkeshik A, Moresco JJ, Yates JR, 3rd, Masliah E, Bokoch GM, DerMardirossian C. A novel Rac1 GAP splice variant relays poly-Ub accumulation signals to mediate Rac1 inactivation. Mol Biol Cell. 2013;24:194–209. doi: 10.1091/mbc.E12-07-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez-Tallon I, Pagenstecher A, Fliegauf M, Olbrich H, Kispert A, Ketelsen UP, North A, Heintz N, Omran H. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum Mol Genet. 2004;13:2133–2141. doi: 10.1093/hmg/ddh219. [DOI] [PubMed] [Google Scholar]
- Jacquet BV, Salinas-Mondragon R, Liang H, Therit B, Buie JD, Dykstra M, Campbell K, Ostrowski LE, Brody SL, Ghashghaei HT. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development. 2009;136:4021–4031. doi: 10.1242/dev.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klezovitch O, Fernandez TE, Tapscott SJ, Vasioukhin V. Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev. 2004;18:559–571. doi: 10.1101/gad.1178004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CT, Mirzadeh Z, Soriano-Navarro M, Rasin M, Wang D, Shen J, Sestan N, Garcia-Verdugo J, Alvarez-Buylla A, Jan LY, Jan YN. Postnatal deletion of Numb/Numblike reveals repair and remodeling capacity in the subventricular neurogenic niche. Cell. 2006;127:1253–1264. doi: 10.1016/j.cell.2006.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A, Oliver G. Six3 is required for ependymal cell maturation. Development. 2011;138:5291–5300. doi: 10.1242/dev.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Liu B, Yang X, Yue X, Diao L, Wang J, Chang J. Genetic deletion of Rnd3 results in aqueductal stenosis leading to hydrocephalus through up-regulation of Notch signaling. Proc Natl Acad Sci U S A. 2013;110:8236–8241. doi: 10.1073/pnas.1219995110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pathak N, Kramer-Zucker A, Drummond IA. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development. 2007;134:1111–1122. doi: 10.1242/dev.02806. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang YW, Wang X, Zhang H, You X, Liao FF, Xu H. Intracellular trafficking of presenilin 1 is regulated by beta-amyloid precursor protein and phospholipase D1. J Biol Chem. 2009;284:12145–12152. doi: 10.1074/jbc.M808497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaterre J, Mantamadiotis T, Dworkin S, Lightowler S, Yang Q, Ransome MI, Turnley AM, Nichols NR, Emambokus NR, Frampton J, Ramsay RG. c-Myb is required for neural progenitor cell proliferation and maintenance of the neural stem cell niche in adult brain. Stem Cells. 2008;26:173–181. doi: 10.1634/stemcells.2007-0293. [DOI] [PubMed] [Google Scholar]
- Marcet B, Chevalier B, Luxardi G, Coraux C, Zaragosi LE, Cibois M, Robbe-Sermesant K, Jolly T, Cardinaud B, Moreilhon C, Giovannini-Chami L, Nawrocki-Raby B, Birembaut P, Waldmann R, Kodjabachian L, Barbry P. Control of vertebrate multiciliogenesis by miR-449 through direct repression of the Delta/Notch pathway. Nat Cell Biol. 2011;13:693–699. doi: 10.1038/ncb2241. [DOI] [PubMed] [Google Scholar]
- Oishi I, Kawakami Y, Raya A, Callol-Massot C, Izpisúa Belmonte JC. Regulation of primary cilia formation and left-right patterning in zebrafish by a noncanonical Wnt signaling mediator, duboraya. Nat Genet. 2006;38:1316–1322. doi: 10.1038/ng1892. [DOI] [PubMed] [Google Scholar]
- Páez P, Bátiz LF, Roales-Buján R, Rodríguez-Pérez LM, Rodríguez S, Jiménez AJ, Rodríguez EM, Pérez-Fígares JM. Patterned neuropathologic events occurring in hyh congenital hydrocephalic mutant mice. J Neuropathol Exp Neurol. 2007;66:1082–1092. doi: 10.1097/nen.0b013e31815c1952. [DOI] [PubMed] [Google Scholar]
- Sarnat HB. Ependymal reactions to injury: a review. J Neuropathol Exp Neurol. 1995;54:1–15. doi: 10.1097/00005072-199501000-00001. [DOI] [PubMed] [Google Scholar]
- Spassky N, Merkle FT, Flames N, Tramontin AD, García-Verdugo JM, Alvarez-Buylla A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Qu Y, Montcouquiol M, Zhou L, Komatsu K, Shi D, Fujimori T, Labeau J, Tyteca D, Courtoy P, Poumay Y, Uemura T, Goffinet AM. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci. 2010;13:700–707. doi: 10.1038/nn.2555. [DOI] [PubMed] [Google Scholar]
- Torfs CP, Christianson RE. Anomalies in Down syndrome individuals in a large population-based registry. Am J Med Genet. 1998;77:431–438. [PubMed] [Google Scholar]
- Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development. 2009;136:2297–2307. doi: 10.1242/dev.034884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Rosti RO, Gibbs E, Gleeson JG. Primary cilia in neurodevelopmental disorders. Nat Rev Neurol. 2014;10:27–36. doi: 10.1038/nrneurol.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med. 2013;19 doi: 10.1038/nm.3117. 473-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Huang T, Bu G, Xu H. Dysregulation of protein trafficking in neurodegeneration. Mol Neurodegener. 2014a;9:31. doi: 10.1186/1750-1326-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Huang T, Zhao Y, Zheng Q, Thompson RC, Bu G, Zhang YW, Hong W, Xu H. Sorting nexin 27 regulates Abeta production through modulating gamma-secretase activity. Cell Rep. 2014b;9:1023–1033. doi: 10.1016/j.celrep.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010;19:2780–2791. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Williams MA, Rigamonti D. Genetics of human hydrocephalus. J Neurol. 2006;253:1255–1266. doi: 10.1007/s00415-006-0245-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Suh H, Gage FH. Notch keeps ependymal cells in line. Nat Neurosci. 2009;12:243–245. doi: 10.1038/nn0309-243. [DOI] [PubMed] [Google Scholar]