

Abstract
Although B cells are traditionally known for their role in propagating proinflammatory immune responses, their immunosuppressive effects have only recently begun to be appreciated. How these regulatory B cells (Bregs) suppress the immune response remains to be worked out in detail. In this article, we show that Bregs can induce the formation of conventional FoxP3+ regulatory T cells (Tregs), as well as a more recently described CD49b+CD223+ regulatory T-cell subset, known as type 1 regulatory T cells (Tr1s). When Bregs are transferred into mice with experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, they home to the spleen and mesenteric lymph nodes, leading to an expansion of Tregs and Tr1 in vivo. Tregs and Tr1s are also found in greater proportions in the CNS of mice with EAE treated with Bregs and are correlated with the remission of symptoms. The discovery that Bregs induce the formation of regulatory T-cell subsets in vivo may herald their use as immunosuppressive agents in adoptive cellular therapies for autoimmune pathologies.
SIGNIFICANCE STATEMENT Although B cells are traditionally known for their role in propagating proinflammatory immune responses, their immunosuppressive effects have only recently begun to be appreciated. How regulatory B cells (Bregs) suppress the immune response remains to be fully understood. In this article, we show that Bregs can induce the formation of conventional regulatory T cells (Tregs) as well as type 1 regulatory T cells (Tr1s). When Bregs are transferred into mice with experimental autoimmune encephalomyelitis (EAE), they home to secondary lymphoid organs, leading to an expansion of Tregs and Tr1s in vivo. Tregs and Tr1s are also found in greater proportions in the CNS of mice with EAE treated with Bregs and are correlated with the remission of symptoms.
Introduction
Regulatory B cells (Bregs) have been ascribed important anti-inflammatory functions in a multitude of autoimmune conditions in both humans and mice, including colitis, arthritis, and neuroinflammatory disorders (Mauri et al., 2003; Evans et al., 2007; Blair et al., 2010; Ray et al., 2011; Yang et al., 2012). Bregs are functionally defined by their ability to inhibit overt inflammatory responses via the production of immunosuppressive cytokines, predominately interleukin (IL)-10, transforming growth factor-β (TGF-β), and, more recently, IL-35 (Tian et al., 2001; Fillatreau et al., 2002; Shen et al., 2014; Rosser and Mauri, 2015). A functional definition remains the current standard of classifying Bregs due to a lack of consensus regarding subset-defining cell surface marker expression or a lineage-specific transcription factor (Rosser and Mauri, 2015; Tedder, 2015). The role that IL-10-secreting Bregs play in disease has been most well documented in experimental autoimmune encephalomyelitis (EAE), a murine model of the human demyelinating disease, multiple sclerosis (MS; Ray et al., 2014). Various groups, including our own, have shown that the adoptive transfer of Bregs into mice with EAE can lead to durable remission of symptoms (Matsushita et al., 2008; Rafei et al., 2009; Ray and Basu, 2014). However, the basic mechanisms that underscore how Bregs may induce EAE remission in vivo remain unknown. The fact that IL-10 and MHC-II have been reported to be critical for Breg-mediated immunosuppression in EAE have steered the investigation toward interactions between Bregs and CD4+ T cells, which initiate and drive EAE through cognate antigen recognition by MHC-II and the pathologic secretion of IL-17 in a particular subset of these cells [T helper type 17 (Th17) cells; Rafei et al., 2009; Yoshizaki et al., 2012].
While B cells have traditionally been thought of as augmenting proinflammatory responses of CD4+ T cells, Bregs have been reported to suppress interferon (IFN)-γ-secreting Th1 effector functions in favor of Th2-like responses (Lund and Randall, 2010). Bregs have also been reported to dampen overt inflammation by inducing FoxP3+ CD4+ regulatory T cells (Tregs) in transplant models of islet allografts and collagen-induced arthritis (Carter et al., 2011; Lee et al., 2014). Further, Bregs have been shown to induce the formation of IL-10-secreting, FoxP3− regulatory CD4+ T cells, known as type 1 regulatory T cells (Tr1s), in mouse models of lupus and collagen-induced arthritis (Gray et al., 2007; Blair et al., 2010). However, Breg induction of regulatory T-cell function in EAE has not been demonstrated.
In an effort to elucidate the effect of Bregs on the induction of regulatory T-cell responses in vivo, we sought to determine how Breg treatment of mice with myelin oligodendrocyte glycoprotein (MOG)35–55-induced EAE alters the CD4+ T-cell compartment in a green fluorescent protein (GFP)/IL-10 reporter mouse model. Using a recombinant, bacterially derived fusion protein consisting of granulocyte-macrophage colony-stimulating factor (GMCSF) and IL-15 (GIFT15; Rafei et al., 2009; Pennati et al., 2014), we show that GIFT15 can convert splenic B cells into IL-10-secreting Bregs (GIFT15 Bregs). In addition to IL-10 secretion, we report for the first time that GIFT15 Bregs also secrete IL-27, a cytokine that is critical for the induction of Tr1s (Awasthi et al., 2007; Murugaiyan et al., 2009; Gagliani et al., 2013). In accordance with this observation, GIFT15 Bregs are capable not only of inducing IL-10-producing CD25+FoxP3+ Tregs, but also CD49b+CD223+ Tr1s, in an antigen-specific manner. GIFT15 Bregs home to the spleen and mesenteric lymph nodes (MLNs) when adoptively transferred into mice and induce the remission of disease in mice with EAE. Treatment of these mice with GIFT15 Bregs correlated with increased Treg and Tr1 subsets in the spleen, MLN, and CNS. This is the first report to demonstrate that IL-10-secreting Bregs are capable of inducing regulatory CD4+ T-cell responses in vivo in an autoimmune, neuroinflammatory disorder.
Materials and Methods
Ethics statement
All animal experiments were approved by the Institutional Animal Care and Use Committee at Emory University and were performed using accepted veterinary standards. C57BL/6, B6(Cg)-Il10tm1.1Karp/J (Vert-X) mice were purchased from The Jackson Laboratory. C57BL/6-Tg(Act-EGFP)C14-Y01-FM131 Osb mice were a gift from Dr. Masaru Okabe (Osaka University, Osaka, Japan) and are maintained by Dr. David Archer at Emory University. The luciferase-expressing (luc+) transgenic C57BL/6 mice (B6-L2G85) were a gift from Dr. Edmund K. Waller, Emory University, Atlanta, GA, USA. Female mice between 6 and 20 weeks of age were used, and were killed using CO2.
Generation of GIFT15 Bregs and phenotyping
B cells were isolated from the spleens of wild-type C57BL/6 mice by negative selection (Stem Cell Technologies) and stimulated with 10 μg/ml recombinant bacterial-derived GIFT15 for 72–96 h. GIFT15 Bregs were assayed by flow cytometry of surface marker expression (CD1d, CD5, CD19, CD21, CD24, CD38, CD40, CD86, IgM, H-2kb, I-Ab, PD-L1, PD-1, and their respective isotype controls, BD Biosciences; and CD80, IgD, and PD-L2, eBioscience). Briefly, B cells or GIFT15 Bregs were harvested from culture, incubated with CD16/32 (Fc Block) antibodies for 30 min, followed by cell surface marker staining with fluorochrome-conjugated antibodies for an additional 30 min at 4°C before flow cytometry analysis. Supernatant from untreated, and GMCSF-, IL-15-, and GIFT15-treated B cells were analyzed for cytokine secretion (IL-10, IL-27, and TGF-β, eBioscience; IL-35, BioLegend; and TNFSF18/glucocorticoid-induced tumor necrosis factor receptor (TNFR)-related protein (GITR) ligand, RayBiotech).
In vitro coculture
B cells and CD4+ T cells were isolated by negative selection (STEMCELL Technologies) per manufacturer protocol. CD4+ T cells were placed into coculture with freshly isolated splenic B cells (derived from C57BL/6 mice by negative selection) or GIFT15 Bregs at a ratio of 2:1. CD4+ T cells were stimulated with anti-CD3/28 DynaBeads (ThermoFisher Scientific) and analyzed by flow cytometry for GFP/IL-10 expression 72 h postculture. In MOG35–55 peptide restimulation cocultures, cells from the spleens or MLNs of Vert-X mice exhibiting symptoms of EAE (clinical score, 1–2) were placed into culture with B cells or GIFT15 Bregs in the presence of MOG35–55 peptide (10 μg/ml; Sigma-Aldrich; see below for MOG35–55-induced EAE) at a ratio of 2:1. CD4+ T cells were analyzed by flow cytometry for GFP/IL-10, CD25, FoxP3, CD49b, and CD223 expression 72 h postculture.
Western blot analysis
Cells were extracted in lysis buffer (Cell Signaling Technology) supplemented with 1 mm phenylmethylsulfonyl fluoride and 1× protease inhibitor (ThermoFisher Scientific). Samples were separated by SDS-PAGE and immunoblotted for phospho-STAT3 (1:1000), phospho-STAT5 (1:1000), phospho-Akt (1:500), phospho-IKB (1:500), phospho-p38 (1:500), phospho-JNK (1:500), phospho-Erk1/2 (1:500), STAT3 (1:2000), STAT5 (1:2000), and Erk1/2 (1:1000). All antibodies were obtained from Cell Signaling Technology.
Adoptive transfer of GIFT15 Bregs or B cells
A donor mouse was killed, and the spleen was removed. Lymphocytes were prepared as a single-cell suspension, and B cells were isolated with a STEMCELL Technologies kit. After culture for 4–5 d in complete R10 media with the addition of recombinant GIFT15 (10 ng/ml), the cells were collected and washed twice in PBS. Each mouse received 2 million cells in 0.2 ml of PBS. Recipient mice were injected intravenously by tail vein injection using a small-gauge (28 ga) needle.
Biodistribution of GIFT15 Bregs
GIFT15 Bregs were generated by coculturing CD19+ B cells purified from B6/L2G85 mice splenocytes in complete R10 media with 10 ng/ml recombinant mouse GIFT15 at a cell density of 0.5 × 106 cells/ml for 4–5 d. A total of 5 × 106 B6-L2G85-GIFT15-Bregs or B6-L2G85-B cells were intravenously injected into syngeneic EAE C57BL/6 mice with a clinical score of at least 1 or 2. The mice were injected subcutaneously with luciferin (150 mg/kg body weight) before imaging on the In Vivo Imaging System (IVIS; Xenogen) in the core facility at the Winship Cancer Institute. Alternatively, GIFT15 Bregs were generated from GFP B cells and infused into EAE mice. After 1 week, the animals were killed, and MLN, spleen, and CNS were harvested for analysis. White cells were isolated and analyzed by flow cytometry for the presence of GFP.
Cells isolation and flow cytometry procedures for MLN, spleen, and CNS leukocytes
Spleens and mesenteric lymph nodes were dissected postmortem and collected in RPMI medium (Lonza). For the preparation of splenocytes and lymph node cells, organs were mashed through a 70 μm cell strainer (BD Biosciences), as previously described (Domingues et al., 2010), and erythrocytes from spleens were lysed using Red Cell Lysis Buffer (Sigma-Aldrich).
At different time points, mice (n = 10) were perfused intracardially with ice-cold Dulbecco's PBS (DPBS) without Ca2+ and Mg2+. Brains and spinal cords were extracted and immediately homogenized with a plunger in RPMI medium. The cell suspension was centrifuged at 300 × g for 5 min at room temperature. The supernatant was aspirated, and cells were gently resuspended in 37% Percoll (GE Healthcare). The cell suspension was underlaid beneath 80% Percoll and centrifuged at 600 × g for 25 min, with slow acceleration and deceleration rates. The cell ring at the interphase was collected and mixed thoroughly with DPBS containing 2% FBS (FACS buffer). Cells were then centrifuged at 300 × g for 5 min and washed twice with FACS buffer.
For the detection of cell surface markers, cells were stained in FACS buffer with the different fluorochrome-labeled monoclonal antibodies. Cells were incubated for 30 min on antibodies at 4°C. Flow cytometry analysis and data acquisition were performed using a FACSCanto II Cell Analyzer (BD Biosciences), and the data analysis was performed with FACSDiva Software (BD Biosciences).
Experimental autoimmune encephalomyelitis induction and analysis
We induced and scored EAE, as previously described (Nicholson et al., 1995; Miller et al., 2010). Briefly, EAE was induced by subcutaneous injection of 50 μg of MOG35-55 (Sigma-Aldrich) emulsified in complete Freund's adjuvant (Difco, BD) containing 5 mg/ml H37Ra Mycobacterium tuberculosis. On day 0 and 2, 100 ng of pertussis toxin (Sigma-Aldrich) was administered by intraperitoneal injection. Three independent experiments were conducted (n = 6). Mice were sex and age matched (6–10 weeks of age), and we scored them blinded for the daily treatment group.
EAE clinical scores were graded as follows: 0, normal; 1, flaccid tail; 2, hindlimb weakness; 3, flaccid tail with paralysis of one front or one hindleg; 4, complete hindlimb paralysis and partial front leg paralysis; 5, tetraplegia, moribund, or death. Mice with a clinical score ≥0.5 were included. Mice that did not show any symptoms (clinical score, 0) on the first day of treatment were excluded. Randomization was performed to ensure the same starting average clinical score for each group.
Histopathology
Spinal cords and brains from mice intracardially perfused with 4% (w/v) paraformaldehyde were dissected and postfixed overnight. Paraffin-embedded sections were stained with hematoxylin and eosin (H&E) or Luxol Fast Blue (LFB), and were examined by light microscopy. Semiquantitative analysis of inflammation and demyelination were performed in a blinded manner.
Statistical analysis
All statistical analyses were performed using Prism 6 Software. Unless otherwise indicated, tests of statistical significance were conducted using a two-tailed Student's t test. Data display normal variance. p Values <0.05 were considered to be statistically significant.
Results
Recombinant GIFT15-induced regulatory B-cell phenotype
We have previously reported that a fusion protein consisting of GMCSF and IL-15 possesses the ability to convert splenic B cells into IL-10-secreting Bregs (GIFT15 Bregs; Rafei et al., 2009). Beyond IL-10 secretion, we sought to determine whether published cell surface markers, CD1d and CD5, were expressed in GIFT15 Bregs. To do so, we used the GFP/IL-10 (Vert-X) reporter mouse model (Madan et al., 2009) to specifically identify IL-10-secreting GIFT15 Bregs. In contrast to wild-type C57BL/6-derived B cells, a subset of splenic B cells derived from Vert-X mice became GFP/IL-10+ after 72 h of GIFT15 treatment in vitro (Fig. 1A). Consistent with previous reports, this subset of GFP/IL-10+ GIFT15 Bregs expressed higher surface levels of CD1d and CD5 compared with GFP/IL-10− GIFT15 Bregs (p = 0.0004 and p = 0.0006, respectively; Fig. 1A). Compared with GMCSF and IL-15-treated B cells, GIFT15 Bregs also had differential expression of coinhibitory molecules and their ligands, such as PD-1 (CD279), PD-L1 (CD273), and PD-L2 (CD274). We found that, compared with control cytokine treatment, GIFT15 Bregs uniformly upregulated PD-1 (p < 0.0001) expression and expressed higher levels of PD-L1 (p = 0.03), but not of PD-L2 (Fig. 1B). GIFT15 Bregs secrete high levels of IL-10, which we verified by ELISA (Fig. 1C). We also detected the secretion of IL-27 by GIFT15 Bregs, but did not detect any IL-35, TGF-β, or TNFSF18, a soluble ligand for GITR (Fig. 1C). In an effort to find a Breg subset-defining surface marker signature, we compared GFP/IL-10+ GIFT15 Bregs with their GFP/IL-10− counterparts based on their expression of markers related to maturation (IgM, IgD, and CD38), antigen presentation (MHC-I, H-2kb; MHC-2, I-Ab), costimulation (CD24, CD80, CD86, and CD40), and components of the B-cell coreceptor complex (CD19 and CD21), as well as CD23, a negative regulator of B-cell receptor signaling. GFP/IL-10+ GIFT15 Bregs expressed the more costimulatory CD80 and CD21, but less surface IgD compared with their GFP/IL-10− counterparts (Fig. 2A). However, in comparison, splenic B cells treated with GMCSF and IL-15, GIFT15 Bregs, expressed higher levels of antigen presentation (H-2kb and I-Ab) and costimulatory markers (CD24, CD40, CD80, and CD86; Fig. 2B). GIFT15 Bregs also expressed less surface IgM and B-cell receptor inhibitory CD23 compared with control cytokine-treated splenic B cells (Fig. 2B). To determine how GIFT15 may induce conversion of splenic B cells into GIFT15 Bregs, GIFT15 signaling was investigated. The activation status of signal transduction and activator of transcription (STAT) proteins, which are the canonical molecules by which GMCSF and IL-15 transduce their signal, was assayed. Treatment of mouse splenocytes with GIFT15 showed that, distinct from GMCSF and IL-15 treatment, GIFT15 induces STAT3 phosphorylation and activation in the absence of STAT5 activation with delayed kinetics (Fig. 3A). Biochemically, GIFT15 drives STAT3 phosphorylation later than GMCSF and IL-15 treatment, and STAT3 remains phosphorylated for at least 48 h post-treatment in the absence of STAT5 phosphorylation (Fig. 3B). In addition to this unopposed STAT3 activation event, we interrogated other noncanonical signaling pathways that have been associated with GMCSF and γ-chain signaling (Fig. 3C). Fifteen minutes post-treatment, both GMCSF and IL-15, and GIFT15 treatment induced phosphorylation of Erk1/2 (p44/42 MAPK), but this effect was transient. Both GMCSF and IL-15, and GIFT15 also induced sustained Akt phosphorylation.
Figure 1.
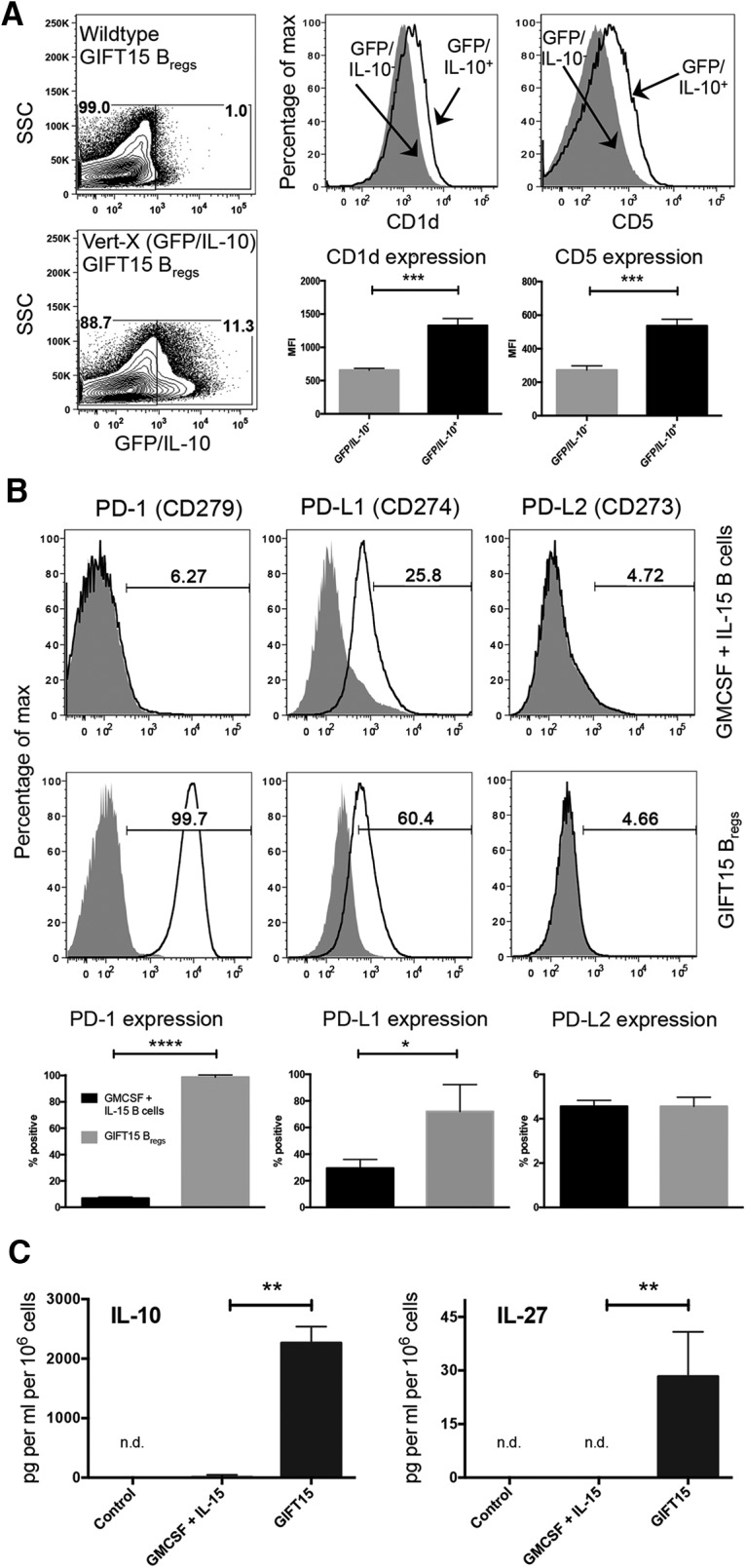
Phenotype of GIFT15 Bregs and cell-signaling profile. A, GFP/IL-10 expression in GIFT15-treated C57BL/6 and Vert-X B cells after 3 d. Comparison of CD1d and CD5 expression on GFP/IL-10− and GFP/IL-10+ Bregs by mean fluorescence intensity. B, Surface expression of PD-1, PD-L1, and PD-L2 on GMCSF- and IL-15-treated B cells compared with GIFT15 Bregs. Shaded histograms represent isotype staining, and solid black lines represent the indicated surface marker staining. C, IL-10 (n = 2) and IL-27 (n = 4) levels in GIFT15 Breg cultured media compared with controls. Flow cytometry plots are representative of a biological replicate from two to four independent experiments. Bar graphs in A–C display mean ± SD values. p Values were calculated using a two-tailed Student's t test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. n.d., Not determined; NS, no stimulation.
Figure 2.

GIFT15 Bregs surface marker expression. A, GFP gating was determined by generating GIFT15 Bregs from wild-type C57BL/6 mice as a control for background fluorescence. IgD, IgM, H-2Kb, I-Ab, CD80, CD86, CD40, PD-L1, PD-L2, PD-1, CD21, CD23, CD24, and CD38 expression levels were compared between GFP/IL-10− and GFP/IL-10+ populations of GIFT15 Bregs. B, Expression of the Figure 2A markers was also assessed between GMCSF- and IL-15-treated splenic B cells compared with GIFT15 Bregs generated from wild-type C57BL/6 mice.
Figure 3.

Cell-signaling profile. A, STAT3 and STAT5 phosphorylation following treatment with GIFT15 or GMCSF and IL-15 at 15 min and 5 h. B, STAT3 and STAT5 phosphorylation time course. C, GIFT15 activation of major MAPK (p38/Erk/Jnk) and PI3K/Akt signaling pathways.
GIFT15 Bregs induce IL-10 expression in activated CD4+ T cells in vitro
To determine the immunoregulatory effects of GIFT15 Bregs, we used an in vitro coculture system. CD4+ T cells isolated from naive Vert-X mice were placed into coculture with splenic B cells or GIFT15 Bregs to determine whether GIFT15 Bregs could induce IL-10 expression by CD4+ T cells. In steady-state conditions, without cognate antigen or polyclonal activation, neither B cells nor GIFT15 Bregs induced IL-10 expression in CD4+ T cells. CD4+ T cells gained the ability to produce IL-10 when stimulated with anti-CD3/28 beads in the presence of GIFT15 Bregs, but not in the presence of splenic B cells (p = 0.02; Fig. 4A). We next sought to determine whether more physiological stimulation of CD4+ T cells by cognate antigen could recapitulate this effect. GIFT15 Bregs or splenic B cells were cocultured with CD4+ T cells derived from the spleens or MLNs of Vert-X mice with EAE and restimulated with MOG35–55 peptide (Fig. 4B). Compared with anti-CD3/28 bead stimulation, we were unable to detect significantly greater levels of IL-10 expression in MOG35–55 restimulated CD4+ T cells in the presence of GIFT15 Bregs compared with splenic B cells. However, we detected significantly greater proportions of CD4+ T cells that were conventional CD25+FoxP3+ Tregs from both the spleens and MLNs of these mice when cocultured with GIFT15 Bregs (p = 0.001 and p = 0.01, respectively; Fig. 4B,C). Further, a significantly higher proportion of CD4+ T cells from the MLNs of Vert-X mice with EAE were of the CD49b+CD223+ Tr1 phenotype when cocultured with GIFT15 Bregs compared with splenic B cells (p = 0.04; Fig. 3C).
Figure 4.
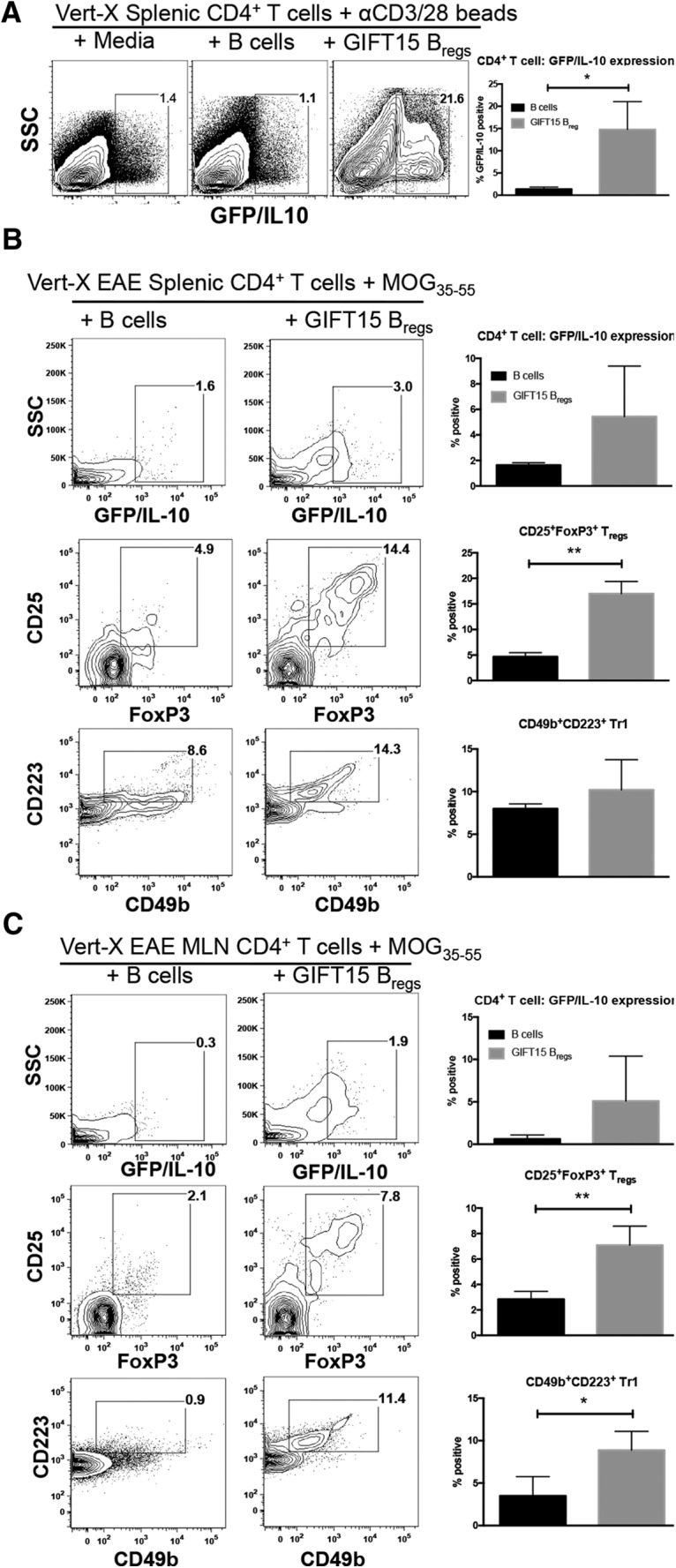
GIFT15 Bregs induce IL-10 expression in activated CD4+ T cells and enhance ex vivo formation of CD25+FoxP3+ Tregs from the spleen and CD49b+CD223+ Tr1s from the MLNs of mice with EAE. A, αCD3/28 Dynabead-stimulated Vert-X CD4+ T cells cultured with splenic B cells or GIFT15 Bregs. B, CD4+ T-cell analysis from the spleens of Vert-X mice with EAE, with a clinical score of 1 or 2, cultured with splenic B cells or GIFT15 Bregs following MOG35–55 peptide stimulation. C, CD4+ T-cell analysis from the MLNs of Vert-X mice with EAE, with a clinical score of 1 or 2, cultured with splenic B cells or GIFT15 Breg following MOG35–55 peptide stimulation. Flow cytometry plots are representative of a biological replicate from three independent experiments. Bar graphs in A–C display mean ± SD values across triplicate samples. p Values were calculated using the two-tailed Student's t test. *p < 0.05; **p < 0.01.
Dynamic distribution of GIFT15 Bregs
Homing of GIFT15 Bregs was first studied after a single intravenous infusion in EAE mice followed by in vivo imaging with the use of luciferase-expressing transgenic B cells. Purified B cells from B6-L2G85 mouse splenocytes were stimulated with recombinant mouse GIFT15 to generate B6-L2G85-GIFT15 Bregs, which were tail vein injected into EAE C57BL/6 mice (5 × 106 cells per mouse). Alternatively, nonstimulated B cells (5 × 106) were injected as a control. Mice were then imaged using a Xenogen IVIS bioluminescent imager for 3 min at small binning. Pseudocolored scale shows by day 2 varying light emittance throughout the gut (Fig. 5A). By day 5, the highest light intensity of the B6-L2G85-GIFT15 Bregs was localized in the spleen. No signal was detectable after 10 d. Minor diffuse light was transiently noticeable in the gut of mice treated with B6-L2G85 B cells during the first 7 d (Fig. 5A). The disease score did not affect the distribution of B6-L2G85 B cells or B6-L2G85-GIFT15 Bregs into EAE C57BL/6 mice.
Figure 5.

Dynamics of GIFT15 Breg biodistribution. GIFT15 Bregs ameliorate EAE, and induce in vivo formation of CD25+FoxP3+ Tregs and CD49b+CD223+ Tr1s in mice with EAE. A, Real-time IVIS imaging with GIFT15-luciferase-Bregs or luciferase-B cells. MOG-immunized C57BL/6 mice were injected with 5 × 106 B6-L2G85-GIFT15 Bregs or B6-L2G85 B cells, and they were imaged every day for 2 weeks. B, Flow cytometry plot of GFP-GIFT15 Bregs or GFP-B cells after adoptive transfer in EAE C57BL/6 mice. Immunized mice were injected with 5 × 106 cells. After 7 d, the animals were killed (n = 5), and spleens, MLNs, and CNSs were collected. Leukocytes were isolated and analyzed by flow cytometry. Data are representative of two independent experiments with n = 5 mice in each group.
To have a better understanding of the migratory patterns of GIFT15 Bregs, transgenic animals expressing enhanced GFP under the chicken β-actin promoter (Okabe et al., 1997) were used as B-cell donors. GIFT15 Bregs generated from these mice (GFP-GIFT15 Bregs) were then adoptively transferred into a wild-type recipient with EAE. After 5 d, the recipient C57BL/6 mice were killed, and the cell contents within the spleen and MLN were analyzed by flow cytometry. GFP-labeled cells were observed in MLNs and spleens of EAE mice that received GFP-GIFT15 Bregs. No signal was detectable in the CNS of any animal in either spleen or MLN collected from the animal treated with GFP-B cells (Fig. 5B).
T-cell dynamics in MLN and spleen after adoptive transfer of GIFT15 Bregs
We have shown that GIFT15 Bregs generated with the mammalian-derived GIFT15 were able to reverse EAE in mice (Rafei et al., 2009). To verify whether this therapeutic effect was reproducible by GIFT15 Bregs generated with bacterial-derived GIFT15 (Pennati et al., 2014), we injected EAE C57BL/6 mice with 2 × 106 GIFT15 Bregs every 10 d and followed the disease score over time. Clinical attenuation of the disease score was observed in mice treated with GIFT15 Bregs (Fig. 6A). Complete and stable remission was achieved 1 month after adoptive transfer. In mice treated with B cells (control group), there was no suppression or clinical amelioration of EAE disease (Fig. 6A).
Figure 6.

GIFT15 Bregs ameliorate EAE and induce in vivo formation of CD25+FoxP3+ Tregs and CD49b+CD223+ Tr1s in mice with EAE. A, The evolution of clinical score in MOG35–55-immunized Vert-X mice treated with GIFT15 Bregs or B cells (2 × 106 cells/mouse, n = 10 per group). Data are reported as the mean ± SEM. The data are representative of two different experiments. B, Two-parameter contour plots show CD3 vs CD4, Treg (CD25 vs FoxP3), and Tr1 (CD49b vs CD223) expression in spleen and MLN after 1 week of GIFT15 Breg or B-cell adoptive transfer in EAE mice. The gating strategy was as follows: the lymphocytes were gated according to their side-scatter and forward-scatter properties, and CD4+ cells were gated from lymphocytes. CD4+ cells were stained for Treg or Tr1 phenotype. GFP/IL-10 vs SSC is the frequency of either Tregs or Tr1s, respectively. C, Bar graphs are the mean ± SD values of two independent experiments (biological replicates) with n = 6 per group. p Values were calculated using the two-tailed Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001.
To better understand the effect of GIFT15 Bregs on CD4+ T cells in MLN and spleen, EAE C57BL/6 mice were intravenously infused with GIFT15 Bregs or B cells; organs were collected at 1, 2, and 4 weeks after adoptive transfer; and lymphoid cells were isolated and analyzed by flow cytometry (Fig. 6B).
Within the first week, a higher frequency of CD4+ T cells was observed in the spleens of the animals that received GIFT15 Bregs relative to those of the control animals. The two most well known types of regulatory CD4+ T cells are CD25+ and FoxP3+ (Tregs); and CD49b+, CD223+, and IL-10+ (Tr1s; Pellerin et al., 2014). To determine whether GIFT15 Bregs treatment altered the regulation of CD4+ T-cell content, and if so, whether it was due to differences in Tregs or Tr1 populations, we performed further subset analyses. An increase of Tregs was observed in the GIFT15 Bregs-treated group as well as of Tr1s (p = 0.006, p = 0.001 respectively; Fig. 6C). A moderate increase in CD4+ T cells was also observed in the MLN with an increased frequency of Tr1s (p = 0.03) and Tregs (p = 0.002; Fig. 6C).
After 2 weeks, no major differences were noticeable in the spleen between GIFT15 Breg-treated mice and controls, whereas in the MLN of GIFT15 Breg-treated mice a modest surge of CD4+ T cells was still present. Further analysis showed expansion of both Tregs and Tr1s in the MLNs of mice in the GIFT15 Breg-treated group compared with controls (p = 0.03 and p = 0.05 respectively; Fig. 6C).
At 1 month after adoptive transfer, no major differences were noticeable in the MLNs, whereas in the spleens of the mice that received GIFT15 Bregs, there was a sustained increase in Tr1s (p = 0.02; Fig. 6C).
Leukocyte dynamics in the CNS of EAE mice after adoptive transfer of GIFT15 Bregs
To determine both the extent and the composition of CNS-infiltrating cells, and the subsequent changes in clinically improved animals, spinal cords and brains were examined for leukocyte subset content at 2, 4, and 12 weeks after GIFT15 Bregs adoptive transfer.
Relative CD45.2 surface expression levels can be used to distinguish between microglia (CD45.2dimCD11b+) and infiltrating macrophages (CD45.2highCD11b+). We evaluated the relative dynamics of the CNS microglial and macrophage responses following adoptive transfer of GIFT15 Bregs by assessing the percentage of CD11b+ cells that expressed low or high levels of CD45.2. We did not find major differences in the CNS leukocyte profile in GIFT15 Breg-treated mice relative to controls during the first month (data not shown). In contrast, at 3 months we observed that blood-derived infiltrate macrophages (CD45high) were increased in the CNS of mice treated with B cells (i.e., mice with ongoing EAE), whereas resident microglia (CD45low) content was similar to that observed in naive mice (Fig. 7A). Mice in the GIFT15 Breg-treated group showed a comparable percentage of infiltrated macrophages to that observed in normal mice.
Figure 7.

Profile of infiltrating leukocytes in the CNSs of mice with EAE 3 months after adoptive transfer of GIFT15 Bregs. CNS monuclear cells from naive C57BL/6, MOG35–55-immunized mice with EAE treated with B cells or GIFT15 Bregs were analyzed 3 months after immunization. A, The gated populations represent microglial cells (CD45.2int CD11b+), macrophages (CD45.2hi CD11b+), and leukocytes (CD45.2hi CD11b+). Infiltrating lymphocytes were stained for the presence of CD4+, CD8+, CD25+, FoxP3+, CD49b+, and CD223+. The data are representative of two independent experiments with n = 6 per group. B, The bar graph is the mean ± SD of two independent experiments (biological replicates) with n = 6 per group. C, Frequency of Th1 and Th17 in spleens, MLNs, and CNSs of EAE C57BL/6J mice 1 and 2 weeks after adoptive transfer analyzed by flow cytometry (n = 4). D, H&E stain showed the infiltration of inflammatory cells in white matter. Arrows indicated infiltration of inflammatory cells. Luxol Fast Blue stain showed areas of intact myelin (blue) and demyelination (light blue-pink). Statistical analysis of infiltrating lymphocytes and demyelination score (n = 4). p Values were calculated using the two-tailed Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001.
There was no obvious correlation between disease severity and the proportion of GR-1/Ly-6G+, neutrophils (always <1%), in all groups of mice. Lymphocytes were increased in the CNS of B cell-treated mice that showed signs of disease (CD45.2+CD11b− or CD45.2+GR1−).
A detailed analysis of the lymphocyte compartment revealed increases of both CD3+ T cells (fourfold increase) and CD19+ B cells (fivefold increase) in EAE mice. In mice treated with GIFT15 Bregs, the percentages of B and T cells were similar to those in normal mice.
Accumulation of Tregs was detectable in both EAE groups (i.e., mice treated with B cells or GIFT15 Bregs) when compared with normal mice. However, only in the group treated with GIFT15 Bregs was a significant increase in Tr1 frequency observed compared with non-EAE mice (p = 0.006; Fig. 7B).
IFN-γ-producing Th1 cells and IL-17-producing Th17 cells have been shown to drive EAE. To determine whether GIFT15 Bregs have any effect on Th1 and Th17 cells, EAE mice were intravenously infused with GIFT15 Bregs or B cells. One and 2 weeks after adoptive transfer, mice were killed, and their spleens, MLNs, and CNSs were collected and analyzed by flow cytometry. During the first week, a lower frequency of Th1 was observed in the spleens and MLNs of the GIFT15 Breg-treated group compared with the B cell-treated group. Interestingly, the major difference in the CNS of the animal treated with GIFT15 Bregs was a lower frequency of Th17 cells over the 2 week investigation (Fig. 7C).
Similarly, we tested the spleens and MLNs of EAE C57BL/6 mice for the presence of proinflammatory GMCSF-producing B cells, as was recently reported in the blood of MS patients (Li et al., 2015b). We did not observe any significant difference between GIFT15 Breg-treated or B cell-treated mice over 2 weeks after adoptive transfer (data not shown).
Since the most significant features of EAE are inflammation and nerve fiber demyelination, we investigated the effect of GIFT15 Breg adoptive transfer on both of these parameters. Histological analysis of the brain of EAE mice reveled that GIFT15 Breg-treated mice had fewer infiltrating inflammatory cells than B cell-treated EAE mice (Fig. 7D; p = 0.02). No major differences were noticed in the spinal cords of the Breg-treated mice compared with the B cell-treated mice (data not shown).
Discussion
Bregs are a functional subpopulation of B cells that exert their immune-suppressive function via the production of regulatory cytokines, such as IL-10 (Fillatreau et al., 2002), TGF-β (Lee et al., 2011), IL-27 (Vasconcellos et al., 2011), and IL-35 (Wang et al., 2014), and the expression of inhibitory surface molecules that suppress pathogenic T cells and auto-reactive B cells in a cell-to-cell contact-dependent manner (Klinker and Lundy, 2012). Akin to our previously published report (Pennati et al., 2014), we show that recombinant, bacterial-derived GIFT15 is able to convert a subset of splenic B cells into IL-10-secreting Bregs. Consistent with other reports (Matsushita and Tedder, 2011; Li et al., 2015a), we show with the GFP/IL-10 (Vert-X) reporter mouse model that the B cells capable of secreting IL-10 are enriched for the expression of CD1d and CD5. Interestingly, GIFT15 Bregs express high levels of PD-1 and PD-L1, but not PD-L2. Moreover, we demonstrate that, in addition to IL-10 secretion, GIFT15 Bregs also secrete IL-27, but not TGF-β or IL-35. Mechanistically, GIFT15 leads to asymmetrical signaling through the IL-15 receptor complex, which manifests as STAT3 hyperphosphorylation in the absence of STAT5 signaling for up to 48 h after stimulation. In an effort to identify other potential signal transduction pathways activated by GIFT15 stimulation, we interrogated noncanonical pathways of GMCSF and IL-15 signaling. We found that both GIFT15 and control cytokine treatment results in transient activation of Erk and sustained Akt activation.
While IL-10 expression is considered essential to the immunosuppressive properties of GIFT15 Bregs, our result suggests that IL-27 may play a role as well. IL-27 is a member of the IL-6/IL-12 cytokine family and has been shown to have anti-inflammatory properties. In vitro, IL-27 elicited the differentiation of Tr1-like cells (Wang et al., 2011), which express IL-10 and have been more recently described to coexpress CD49b and lymphocyte activation gene 3 (CD22; also known as Lag-3), a negative regulator of T-cell function (Gagliani et al., 2013). The secretion of IL-27 by GIFT15 Bregs may promote the differentiation of Tr1s in vitro and in vivo. Contrary to a recent report describing the role of Breg-derived IL-35, we did not detect this cytokine in media cultured by GIFT15 Bregs. It is possible that the mechanism by which GIFT15 promotes differentiation of Bregs is distinct from the one dependent on IL-35 (Wang et al., 2014). Independent of IL-10, B-cell production of TNFSF18, a soluble ligand for GITR, was reported to be required for the induction of protective Tregs in mice with EAE (Ray et al., 2012). However, we were also unable to detect this molecule in media cultured by GIFT15 Bregs.
Using Vert-X reporter mice, we show that GIFT15 Bregs were able to induce IL-10 expression in activated CD4+ T cells. CD4+ T cells isolated from the spleen of Vert-X mice and cultured with GIFT15 Bregs were not induced to secrete IL-10. However, Vert-X CD4+ T cells stimulated with anti-CD3/28 beads and cocultured with GIFT15 Bregs displayed a dramatic increase in the proportion of cells secreting IL-10. More physiologically, we wished to determine whether CD4+ T cells could respond in a fashion similar to that of their cognate antigen. To test this, CD4+ T cells were isolated from the spleens and MLNs of MOG35–55-immunized Vert-X mice, and stimulated ex vivo with MOG35–55 peptide in the presence of GIFT15 Bregs or splenic B cells. While we did not detect significantly increased IL-10 expression in MOG35–55 peptide-restimulated CD4+ T cells cocultured with GIFT15 Bregs, we did observe that CD4+ T cells from both spleens and MLNs had increased proportions of CD25+FoxP3+ Tregs. Further, in the presence of GIFT15 Bregs we observed significantly greater CD4+ T cells that were CD49b+CD223+ Tr1s compared with CD4+ T cells cocultured with splenic B cells. While there have been reports of Breg induction of conventional Tregs, we believe this is the first report of in vitro induction of Tr1s by Bregs (Chai et al., 2008; Schreiber et al., 2010).
In vivo, adoptively transferred GIFT15 Bregs home to MLN and spleen, leading to an increased frequency of endogenous FoxP3+ and Tr1 CD4+ T cells, which may play a direct role in attenuating EAE. One week after the adoptive transfer of GIFT15 Bregs, we observed an intense increase in the proportion of both Tregs and Tr1s in the spleen. To a lesser extent, but in a more sustained fashion, we observed an expansion of Tregs and Tr1s in MLN. In the CNS, an increase of Tregs and Tr1s was evident only 1 month after adoptive transfer (Fig. 8). While this is the first demonstration of the GIFT15 Breg-dependent increase of Tregs in mice with EAE, the role that Tregs play in the autoimmune pathogenic scenario of EAE is well established (Buc, 2013). Less well characterized is the role of Tr1s in EAE. Several reports indicate that Tr1s are protective through their provision of IL-10 and inhibition of Th17 differentiation in an IL-27-dependent manner (Diveu et al., 2009). Interestingly, in humans, it has been reported that MS patients exhibit impaired Tr1 differentiation and IL-10 secretion by Tr1s (Astier and Hafler, 2007; Martinez-Forero et al., 2008). We hypothesize that adoptive transfer of GIFT15 Bregs induces an expansion or differentiation of regulatory T cells, both Tregs and Tr1s, with IL-10-secreting capabilities, which ultimately relocate to areas of active inflammation within the CNS of EAE mice.
Figure 8.
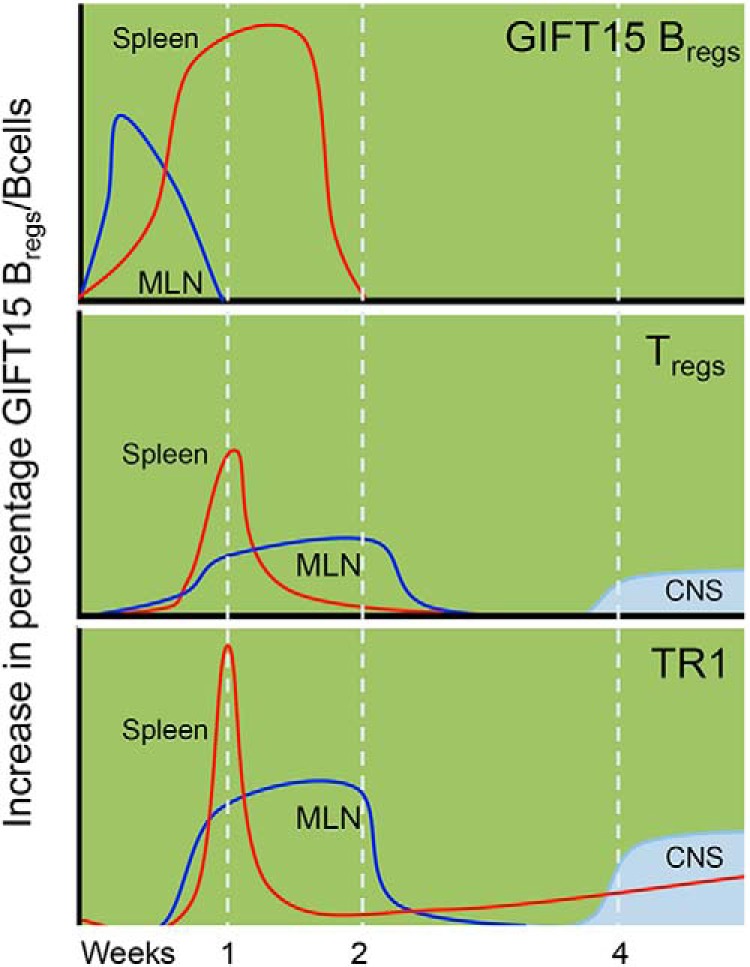
Kinetics of GIFT15 Breg vs B-cell treatment in mice with EAE. Kinetics of occurrence of GIFT15 Bregs, Tregs, and Tr1s over time in spleens, MLNs, and CNSs in MOG35–55-immunized mice adoptively transferred with GIFT15 Bregs.
Mice treated with GIFT15 Bregs had fewer CNS-infiltrating macrophages compared with B cell-treated mice with EAE, with levels similar to that observed in normal, non-EAE C57BL/6 mice. In the CNS of mice with EAE, microglia/macrophage activation has been shown to lead to the secretion of proinflammatory cytokines and antigen presentation. We show here that GIFT15 Breg-treated mice with EAE have fewer CNS-infiltrating macrophages compared with EAE mice treated with B cells, a finding that correlates with their disease remission. Additionally, mice with EAE showed a higher recruitment of lymphocytes in the CNS, both T cells and B cells, a sign of compromised blood–brain barrier function. In contrast, mice treated with GIFT15 Bregs had levels of lymphocyte infiltration that were similar to those of normal, non-EAE mice. Further analysis of CD4+ T cells revealed a moderate increase of Tregs compared with normal mice. An increase in Tregs population in mice with EAE has been previously observed (Koutrolos et al., 2014). Indeed, during EAE, Tregs enter the CNS, where they may locally regulate pathogenic inflammation (Stephens et al., 2005), a phenomenon augmented by the adoptive transfer of GIFT15 Bregs. Consistent with our results, a persistent decrease in autoreactive Th17 in the CNS of the GIFT15 Bregs-treated mice was observed compared with the group adoptively transferred with B cells. Similarly, H&E and LFB staining of brain sections reveal the absence of pathologic autoimmunity in the mice that received GIFT15 Bregs.
We have previously shown that adoptive transfer of syngeneic, but not allogeneic, GIFT15 Bregs can induce EAE remission in an MHC-II- and IL-10-dependent manner (Rafei et al., 2009). These aggregate findings suggest that GIFT15 Bregs likely interact with CD4+ T cells in vivo as part of their physiology. Here, we have shown that GIFT15 Bregs do not directly migrate to an inflamed EAE CNS, but rather home to MLN and spleen, and dissipate within 2 weeks. We show that GIFT15 Bregs home to the MLN and spleen, where they may directly interact with CD4+ T cells in a manner that leads to an augmentation of endogenous IL-10+ Tregs and Tr1s, which latterly home to and accumulate in inflamed CNS, altering the lymphomyeloid brain compartment to a pattern seen in noninflamed normal brain (Fig. 8). These observations support the notion that pharmacological augmentation of autologous B cells to a GIFT15 Breg functionality may allow for adoptive cell therapy of multiple sclerosis (Ray et al., 2014). We and others (Habib et al., 2015) have shown that the endogenous content of circulating Bregs in human subjects with and without MS is surprisingly low and that <1% of blood B cells fulfill a Breg definition.
Conclusion
In our EAE murine model, we administered the equivalent of three doses of 100 million cells/kg intravenously. Extrapolating to human translation, any attempt to collect and enrich a sufficient number of endogenous Bregs from blood would be logistically unfeasible. Therefore, the demonstrated property of GIFT15 to convert ex vivo resting blood B cells to Bregs would foreshadow the possibility of exploiting an autologous augmented B-cell therapy for MS. This strategy differs from the clinical use of alternate autologous suppressor cell types for autoimmune disorders, such as Tregs (Clinical trial reg. no. NCT02428309, clinicaltrials.gov), as we show that Bregs appear to launch a pan-IL-10+ CD4 T-cell response in vivo that is durable and CNS tropic, far outlasting the Bregs initiators. Last, though GIFT15 Bregs are pharmacologically activated, we speculate that their functionality likely reflects that of endogenous Bregs and may provide the insight that Breg biology may play a key role in the physiopathology of maladapted immune responses seen in EAE/MS and possibly other autoimmune disorders.
Footnotes
This work was supported by National Institutes of Health Grant R01-AI-093881. National Multiple Sclerosis Society Grant FG1963A1/1 was awarded to J.L.B.
The authors declare no competing financial interests.
References
- Astier AL, Hafler DA. Abnormal Tr1 differentiation in multiple sclerosis. J Neuroimmunol. 2007;191:70–80. doi: 10.1016/j.jneuroim.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32:129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Buc M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediators Inflamm. 2013;2013:963748. doi: 10.1155/2013/963748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, Ehrenstein MR, Flavell RA, Mauri C. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol. 2011;186:5569–5579. doi: 10.4049/jimmunol.1100284. [DOI] [PubMed] [Google Scholar]
- Chai JG, Coe D, Chen D, Simpson E, Dyson J, Scott D. In vitro expansion improves in vivo regulation by CD4+CD25+ regulatory T cells. J Immunol. 2008;180:858–869. doi: 10.4049/jimmunol.180.2.858. [DOI] [PubMed] [Google Scholar]
- Diveu C, McGeachy MJ, Boniface K, Stumhofer JS, Sathe M, Joyce-Shaikh B, Chen Y, Tato CM, McClanahan TK, de Waal Malefyt R, Hunter CA, Cua DJ, Kastelein RA. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. 2009;182:5748–5756. doi: 10.4049/jimmunol.0801162. [DOI] [PubMed] [Google Scholar]
- Domingues HS, Mues M, Lassmann H, Wekerle H, Krishnamoorthy G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS One. 2010;5:e15531. doi: 10.1371/journal.pone.0015531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, Mauri C. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. 2007;178:7868–7878. doi: 10.4049/jimmunol.178.12.7868. [DOI] [PubMed] [Google Scholar]
- Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, Guo B, Herbert DR, Bulfone A, Trentini F, Di Serio C, Bacchetta R, Andreani M, Brockmann L, Gregori S, Flavell RA, Roncarolo MG. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. 2013;19:739–746. doi: 10.1038/nm.3179. [DOI] [PubMed] [Google Scholar]
- Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci U S A. 2007;104:14080–14085. doi: 10.1073/pnas.0700326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib J, Deng J, Lava N, Tyor W, Galipeau J. Blood B cell and regulatory subset content in multiple sclerosis patients. J Mult Scler (Foster City) 2015;2:139. doi: 10.4172/2376-0389.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinker MW, Lundy SK. Multiple mechanisms of immune suppression by B lymphocytes. Mol Med. 2012;18:123–137. doi: 10.2119/molmed.2011.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutrolos M, Berer K, Kawakami N, Wekerle H, Krishnamoorthy G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol Commun. 2014;2:163. doi: 10.1186/s40478-014-0163-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Noh J, Noh G, Choi WS, Cho S, Lee SS. Allergen-specific transforming growth factor-beta-producing CD19+CD5+ regulatory B-cell (Br3) responses in human late eczematous allergic reactions to cow's milk. J Interferon Cytokine Res. 2011;31:441–449. doi: 10.1089/jir.2010.0020. [DOI] [PubMed] [Google Scholar]
- Lee KM, Stott RT, Zhao G, SooHoo J, Xiong W, Lian MM, Fitzgerald L, Shi S, Akrawi E, Lei J, Deng S, Yeh H, Markmann JF, Kim JI. TGF-beta-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol. 2014;44:1728–1736. doi: 10.1002/eji.201344062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Shen C, Liu Y, Li Y, Sun L, Jiao L, Jiao W, Xiao J, Shen C, Qi H, Xu F, Ma L. Impaired function of CD5+CD19+CD1dhi B10 cells on IgE secretion in an atopic dermatitis-like mouse model. PLoS One. 2015a;10:e0132173. doi: 10.1371/journal.pone.0132173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, Moore CS, Michel L, Althekair F, Rajasekharan S, Gommerman JL, Prat A, Fillatreau S, Bar-Or A. Proinflammatory GM-CSF-producing B cells in multiplesclerosis and B cell depletion therapy. Sci Transl Med. 2015b;7:310ra166. doi: 10.1126/scitranslmed.aab4176. [DOI] [PubMed] [Google Scholar]
- Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol. 2010;10:236–247. doi: 10.1038/nri2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, Yogev N, Gu Y, Khodoun M, Hildeman D, Boespflug N, Fogolin MB, Gröbe L, Greweling M, Finkelman FD, Cardin R, Mohrs M, Müller W, Waisman A, Roers A, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. 2009;183:2312–2320. doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Forero I, Garcia-Munoz R, Martinez-Pasamar S, Inoges S, Lopez-Diaz de Cerio A, Palacios R, Sepulcre J, Moreno B, Gonzalez Z, Fernandez-Diez B, Melero I, Bendandi M, Villoslada P. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur J Immunol. 2008;38:576–586. doi: 10.1002/eji.200737271. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Tedder TF. Identifying regulatory B cells (B10 cells) that produce IL-10 in mice. Methods Mol Biol. 2011;677:99–111. doi: 10.1007/978-1-60761-869-0_7. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003;197:489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SD, Karpus WJ, Davidson TS. Experimental autoimmune encephalomyelitis in the mouse. Curr Protoc Immunol Chapter. 2010;15 doi: 10.1002/0471142735.im1501s88. Unit 15.1. [DOI] [PubMed] [Google Scholar]
- Murugaiyan G, Mittal A, Lopez-Diego R, Maier LM, Anderson DE, Weiner HL. IL-27 is a key regulator of IL-10 and IL-17 production by human CD4+ T cells. J Immunol. 2009;183:2435–2443. doi: 10.4049/jimmunol.0900568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/S0014-5793(97)00313-X. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Jenks JA, Bégin P, Bacchetta R, Nadeau KC. Regulatory T cells and their roles in immune dysregulation and allergy. Immunol Res. 2014;58:358–368. doi: 10.1007/s12026-014-8512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennati A, Deng J, Galipeau J. Maltose-binding protein fusion allows for high level bacterial expression and purification of bioactive mammalian cytokine derivatives. PLoS One. 2014;9:e106724. doi: 10.1371/journal.pone.0106724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafei M, Hsieh J, Zehntner S, Li M, Forner K, Birman E, Boivin MN, Young YK, Perreault C, Galipeau J. A granulocyte-macrophage colony-stimulating factor and interleukin-15 fusokine induces a regulatory B cell population with immune suppressive properties. Nat Med. 2009;15:1038–1045. doi: 10.1038/nm.2003. [DOI] [PubMed] [Google Scholar]
- Ray A, Basu S. Regulatory B cells in experimental autoimmune encephalomyelitis (EAE) Methods Mol Biol. 2014;1190:243–255. doi: 10.1007/978-1-4939-1161-5_17. [DOI] [PubMed] [Google Scholar]
- Ray A, Mann MK, Basu S, Dittel BN. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neuroimmunol. 2011;230:1–9. doi: 10.1016/j.jneuroim.2010.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188:3188–3198. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607–612. doi: 10.1016/j.immuni.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Schreiber TH, Wolf D, Tsai MS, Chirinos J, Deyev VV, Gonzalez L, Malek TR, Levy RB, Podack ER. Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J Clin Invest. 2010;120:3629–3640. doi: 10.1172/JCI42933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Roch T, Lampropoulou V, O'Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, Li R, Jouneau L, Boudinot P, Wilantri S, Sakwa I, Miyazaki Y, Leech MD, McPherson RC, Wirtz S, Neurath M, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507:366–370. doi: 10.1038/nature12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LA, Gray D, Anderton SM. CD4+CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor crossreactivity. Proc Natl Acad Sci U S A. 2005;102:17418–17423. doi: 10.1073/pnas.0507454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedder TF. B10 cells: a functionally defined regulatory B cell subset. J Immunol. 2015;194:1395–1401. doi: 10.4049/jimmunol.1401329. [DOI] [PubMed] [Google Scholar]
- Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–1089. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- Vasconcellos R, Carter NA, Rosser EC, Mauri C. IL-12p35 subunit contributes to autoimmunity by limiting IL-27-driven regulatory responses. J Immunol. 2011;187:3402–3412. doi: 10.4049/jimmunol.1100224. [DOI] [PubMed] [Google Scholar]
- Wang H, Meng R, Li Z, Yang B, Liu Y, Huang F, Zhang J, Chen H, Wu C. IL-27 induces the differentiation of Tr1-like cells from human naive CD4+ T cells via the phosphorylation of STAT1 and STAT3. Immunol Lett. 2011;136:21–28. doi: 10.1016/j.imlet.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, Wingfield PT, Kim SH, Egwuagu CE. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20:633–641. doi: 10.1038/nm.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Deng J, Liu Y, Ko KH, Wang X, Jiao Z, Wang S, Hua Z, Sun L, Srivastava G, Lau CS, Cao X, Lu L. IL-10-producing regulatory B10 cells ameliorate collagen-induced arthritis via suppressing Th17 cell generation. Am J Pathol. 2012;180:2375–2385. doi: 10.1016/j.ajpath.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, Spolski R, Poe JC, Leonard WJ, Tedder TF. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature. 2012;491:264–268. doi: 10.1038/nature11501. [DOI] [PMC free article] [PubMed] [Google Scholar]