

Abstract
PPARγ is one of the three isoforms of the Peroxisome Proliferator-Activated Receptors (PPARs). PPARγ is activated by thiazolidinediones such as pioglitazone and is targeted to treat insulin resistance. PPARγ is densely expressed in brain areas involved in regulation of motivational and emotional processes. Here, we investigated the role of PPARγ in the brain and explored its role in anxiety and stress responses in mice. The results show that stimulation of PPARγ by pioglitazone did not affect basal anxiety, but fully prevented the anxiogenic effect of acute stress. Using mice with genetic ablation of neuronal PPARγ (PPARγNestinCre), we demonstrated that a lack of receptors, specifically in neurons, exacerbated basal anxiety and enhanced stress sensitivity. The administration of GW9662, a selective PPARγ antagonist, elicited a marked anxiogenic response in PPARγ wild-type (WT), but not in PPARγNestinCre knock-out (KO) mice. Using c-Fos immunohistochemistry, we observed that acute stress exposure resulted in a different pattern of neuronal activation in the amygdala (AMY) and the hippocampus (HIPP) of PPARγNestinCre KO mice compared with WT mice. No differences were found between WT and KO mice in hypothalamic regions responsible for hormonal response to stress or in blood corticosterone levels. Microinjection of pioglitazone into the AMY, but not into the HIPP, abolished the anxiogenic response elicited by acute stress. Results also showed that, in both regions, PPARγ colocalizes with GABAergic cells. These findings demonstrate that neuronal PPARγ is involved the regulation of the stress response and that the AMY is a key substrate for the anxiolytic effect of PPARγ.
SIGNIFICANCE STATEMENT Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) is a classical target for antidiabetic therapies with thiazolidinedione compounds. PPARγ agonists such as rosiglitazone and pioglitazone are in clinical use for the treatment of insulin resistance. PPARγ has recently attracted attention for its involvement in the regulation of CNS immune response and functions. Here, we demonstrate that neuronal PPARγ activation prevented the negative emotional effects of stress and exerted anxiolytic actions without influencing hypothalamic–pituitary–adrenal axis function. Conversely, pharmacological blockade or genetic deletion of PPARγ enhanced anxiogenic responses and increased vulnerability to stress. These effects appear to be controlled by PPARγ neuronal-mediated mechanisms in the amygdala.
Keywords: amygdala, anxiety, conditional PPARγ knock-out mice, pioglitazone, PPARγ, stress
Introduction
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptor proteins that regulate gene expression as ligand-activated transcription factors (Michalik et al., 2006). Three closely related PPAR isoforms have been identified (alpha, delta, and gamma), which are transcribed from different genes and differ from each other in tissue distribution, ligand specificity, and physiological role (Berger and Moller, 2002; Breidert et al., 2002; Chang et al., 2007; Tontonoz and Spiegelman, 2008). Although all PPARs were initially identified in peripheral tissue, there is now evidence of their abundant distribution in the brain (Schnegg and Robbins, 2011).
Among the three isoforms, PPARγ has the highest expression in the CNS, where it has been identified in neurons, astrocytes, and glial cells (Moreno et al., 2004). Putative natural ligands of PPARγ are polyunsaturated fatty acids and arachidonic acid metabolites such as 15-deoxy-delta12,14-prostaglandin J2 and hydroxyoctaedecadienoic acid (Forman et al., 1995). Potent synthetic agonists of PPARγ are molecules that belong to the class of thiazolidinediones (TDZs) such as pioglitazone and rosiglitazone (Lehmann et al., 1995). TDZs have glucose-sensitizing properties and are used clinically to treat type 2 diabetes and insulin resistance (Kersten et al., 2000).
In the brain, relatively high PPARγ expression levels have been found in areas involved in the regulation of motivation and emotion, including the caudate putamen, nucleus accumbens, septum, ventral tegmental area, and hippocampus (HIPP) (Moreno et al., 2004; Gofflot et al., 2007; de Guglielmo et al., 2015). Moreover, PPARγ appears to be linked to stress modulation because its receptor expression is increased in the rat cortex after stress and its activation attenuates the stress response in rodents (García-Bueno et al., 2005). PPARγ activation also attenuates affective symptoms in depressed and bipolar patients (Kemp et al., 2014; Zeinoddini et al., 2015).
It is known that chronic stress induces inflammatory responses in the CNS (Raison et al., 2006; García-Bueno et al., 2008b) and stimulation of the neuroimmune system may contribute to the exacerbation of depression and anxiety disorders (Koo et al., 2010; Christoffel et al., 2011; Tanaka et al., 2012). Reduction of proinflammatory cytokines and the anti-inflammatory response by microglia inhibition after PPARγ stimulation has been proposed as a mechanism for the antidepressant and neuroprotective effects of TDZs (Carta et al., 2011; Kemp et al., 2014). However, a growing body of evidence indicates an important role of PPARγ in the modulation of neuronal-mediated mechanisms. For example, recent microarray studies have shown that treatment with PPARγ agonists modulates preferentially genes linked to synaptic transmission and neuronal functions in the amygdala (AMY) and HIPP (Searcy et al., 2012; Ferguson et al., 2014), whereas inhibition of alcohol drinking and heroin self-administration appear to be mediated by the ability of PPARγ to inhibit dopaminergic activity through the facilitation of presynaptic inhibitory GABAergic transmission within the ventral tegmental area (Stopponi et al., 2011; de Guglielmo et al., 2015).
Here, we explored the role of PPARγ in the regulation of stress responses and anxiety at the neurocircuitry and mechanistic levels. Using outbred mice, we studied the effect of PPARγ activation on anxiety. To confirm that the effect of pioglitazone was mediated by PPARγ, in a subsequent experiment, we coadministered the selective receptor antagonist GW-9662 (Leesnitzer et al., 2002) with pioglitazone. We then tested basal and stress-induced anxiogenic response in conditional knock-out (KO) mice carrying a genetic ablation of PPARγ in neurons but sparing the receptor in glial cells. We then mapped neuronal activation after a mild stress exposure in KO and wild-type (WT) mice. We identified the AMY as a key area for PPARγ modulation of anxiety and acute stress responses. To confirm this hypothesis, we activated PPARγ selectively in the AMY and HIPP of stressed and nonstressed WT mice and then tested for anxiety. Combining immunohistochemistry and in situ hybridization through RNAscope multiplex fluorescent technology, we found that PPARγ receptors in these regions are heavily expressed in glutamic acid decarboxylase 2 (GAD2)-positive cells, suggesting their ability to regulate GABAergic transmission.
Materials and Methods
Subjects
Experiments were performed using male CD1 (Charles River Laboratories) and mice with neuron-specific PPARγ deletion (PPARγNestinCre KO) kindly provided by Dr K. Niswender (Vanderbilt University, Nashville, TN). To obtain the conditional inactivation of PPARγ in neuronal cells, transgenic mice expressing the Cre recombinase under the control of the rat Nestin (Nes) promoter were bred to homozygous PPARγloxP/loxP mice. The resulting heterozygous F1 offspring (PPARγ+/loxP) were either positive or negative for Nes–Cre. From matings of PPARγ+/loxP with PPARγ+/loxP Nes–Cre mice, F2 mice of the desired genotypes (PPARγloxP/loxP Nes–Cre (PPARγNestinCre) and PPARγloxP/loxP) were obtained, which were then intercrossed to obtain F3 animals. PPARγloxP/loxP mice were used as control littermates for conditional PPARγ mice. The mice used for this study were on a C57BL/6J background (Jones et al., 2002).
Mice were ∼8 weeks of age at the beginning of the experiments and were housed in groups of 5 and kept in a normal light/dark (LD) cycle (08:00/20:00) at constant temperature (20–22°C) and humidity (45–55°), with food and water provided ad libitum.
Each experiment was conducted with independent groups of mice during the dark phase of the cycle. Animals were treated in accordance with the guidelines of the European Community Council Directive for Care and Use of Laboratory Animals.
Drugs
Pioglitazone was prepared from Actos (30 mg) and suspended in distilled water for oral administration via intragastric gavage. The drug (30 mg/kg concentration) was administered orally at a volume of 10 ml/kg body weight. For intra-AMY and intra-HIPPocampal microinjections, pioglitazone was purchased from Molcan and dissolved in 15% DMSO and 10% Cremophor. The final volume was adjusted adding physiological saline solution. Pioglitazone (5 μg/0.6 μl) was administered in a volume of 0.3 μl per each site with a stainless steel injector protruding beyond the cannula tip 1.00 mm and 0.5 mm for the AMY and the HIPP, respectively (de Guglielmo et al., 2015). GW9662 was from Tocris Bioscience and was dissolved in 5% DMSO, 5% Tween 20, and 90% distilled water. The compound was administered at a dose of 5 mg/kg and injected at the volume of 10 ml/kg intraperitoneally. Pioglitazone at doses <20 mg/kg given peripherally was not effective in the mouse forced swimming test (Sadaghiani et al., 2011), whereas, it showed efficacy in various behavioral models at doses as high as 30 mg/kg (Stopponi et al., 2011; de Guglielmo et al., 2014; de Guglielmo et al., 2015). Similarly, previous studies indicated that 5 mg/kg GW9662 is sufficient to reverse the effect of 30 mg/kg pioglitazone selectively (Stopponi et al., 2011; de Guglielmo et al., 2014; de Guglielmo et al., 2015). Drugs were freshly prepared before administration.
Elevated plus maze (EPM) test
Basal anxiety behavior of PPARγNestinCre KO and WT mice was tested in the EPM, which was elevated 50 cm from the floor in the center of a darkened room with three light bulbs placed in each corner of the room. Each open arm of the EPM measured 27 × 7 cm and the closed arms measured 27 × 7 × 13.5 cm. At the beginning of each trial, mice were placed in the center of the EPM facing one of the closed arms and left undisturbed in the room for the remaining time of the trial. Each trial lasted 5 min. Mice were recorded using an Ethovision videotracking system (Noldus Information Technology). The percentage of time spent in open arms [%OAT = 100 × time in open arm/(time in “open arm” + time in “closed” arm)] was considered an index of anxiety behavior and the total number of entries was considered a measure of locomotion. After each trial, the maze was cleaned with water and dried using paper towels.
Open-field test
Mice were tested in an open field (Med Associates) placed in the center of a darkened room to assess locomotor activity and anxiety behavior. At the beginning of each trial, mice were placed in the center of the field and left undisturbed in the room for the remaining period of the trial. The trial lasted 5 min and was recorded using the Ethovision videotracking system. Parameters were set as follows: the open field was divided into a rim zone constituting the outer 6 cm of the field and a center zone including the rest of the field. The parameters measured were total distance traveled, time spent in the central zone, and number of rearings. After each trial, the field was cleaned with water and dried using paper towels.
LD exploration test
The LD exploration test for anxiety-related behavior consisted of an open-topped rectangular Plexiglas box (45 cm × 30 cm × 30 cm) that was partitioned into a small (18 cm × 30 cm) area and a large (27 cm × 30 cm) area with a door (7.5 cm × 7.5 cm) in the center of the partition at floor level. The small compartment was painted black and kept dark, whereas the large compartment was painted white and was brightly illuminated with a 60 W (400 lux) light source. Each mouse was placed into the dark compartment facing away from the aperture and allowed to explore the apparatus freely for 5 min. The number of light compartment entries (defined as all 4 paws out of the shelter) and time spent inside the light compartment over a 5 min session were recorded with the Ethovision videotracking system. The test lasted 5 min because this is the time at which behavior is most strongly influenced by novelty (Mozhui et al., 2010).
Restraint stress procedure
Restraint stress was applied by placing the animal in a conic 50 ml plastic tube. The tube diameter was adjusted with plaster tape that was also used to close the posterior opening to avoid free movements to the mice. There was a 0.5–1.0 cm hole at the far end of the plastic tube to allow normal breathing. After 1 h of restraint stress, mice were tested immediately for anxiety-related behavior. For behavioral studies, mice received pioglitazone or the vehicle 12 h and 10 min before they were immobilized in tube for the 1 h restraint. Anxiety behavior was assessed immediately after stress.
Intracranial surgery and histological analysis
For intracranial surgery, mice were anesthetized by intramuscular injection of 100–150 μl of a solution containing tiletamine chlorohydrate (58.17 mg/10 ml) and zolazepam chlorohydrate (57.5 mg/10 ml) and placed into a stereotaxic frame. The skull was exposed and stainless steel guide cannulae (diameter, 0.35 mm; length, 7 mm) were bilaterally implanted to reach the AMY or the HIPP using the following coordinates: (1) AMY: 1.4 mm caudal from the bregma, ±3.0 mm mediolateral and −3.9 mm ventral from the dura; (2) dorsal HIPP, 1.7 posterior to bregma, ± 1.5 mm mediolateral and 1.3 mm ventral to the skull surface (Paxinos and Franklin, 2003). The guide cannulae were fixed to the skull with dental cement and two anchoring screws. Behavioral tests were initiated after full recovery (5–6 d after surgery).
For the intracranial injections, pioglitazone was administered using a Hamilton microsyringe in a volume of 0.3 μl/site by mean of a stainless steel injector 0.5 or 1.0 mm longer than the guide cannula for HIPP and AMY, respectively, so that its tip protruded into the area. After the experiments, to verify the cannula placement, mice were lightly anesthetized with isoflurane and 0.3 μl/site malachite green solution was injected into the area. After the mice were killed, the ink diffusion into the region was evaluated histologically.
The injection sites (see Fig. 6C,F) were confirmed for both AMY and HIPP by comparison with plates taken from a mouse brain atlas (Paxinos and Franklin, 2003). Histological analysis confirmed bilateral injections into the AMY in 41 animals and HIPP in 44 animals. These mice were used for the analysis of the effects of intra-AMYr and hippocampal pioglitazone injections on the anxiety behavior. Mice with incorrectly placed cannulas were excluded from analysis.
Figure 6.

A, Restraint stress significantly decreased the time spent in the light side of the apparatus, whereas intra-AMY administration of pioglitazone reversed the anxiogenic-like effect of stress. B, No differences were detected in the number of entries into the light side of the arena. C, Schematic representation of intra-AMY sites of injection assessed by histological analysis. D, Restraint stress significantly decreased the time spent in the light side of the apparatus, whereas intra-HIPPocampal administration of pioglitazone did not reverse the anxiogenic-like effect of stress. E, No differences were detected in the number of entries into the light side of the arena. F, Schematic representation of intra-HIPP sites of injection assessed by histological analysis at the completion of the experiment. Data are expressed as mean ± SEM (n = 13–15 per group of intra-AMY injections; n = 11 per group of intra-HIPP injections). Difference between restraint stressed mice (S) and nonstressed (NS) controls: *p < 0.05, **p < 0.01. Difference between vehicle and pioglitazone in stressed mice: #p < 0.05.
In situ hybridization
Radioactive labeling of riboprobes and the in situ hybridization procedure has been described previously (Hansson et al., 2006; Bernardi et al., 2014; Sommer et al., 2014).
Riboprobe synthesis
Mouse-specific riboprobes were generated from mouse cDNA templates (PPARγ, position 138 bp and 341 bp on mouse cDNA, gene reference sequence in PubMed database; BC_021798, http://www.ncbi.nlm.nih.gov/Entrez). A total of 200 ng of DNA (PCR template) was used for the synthesis of RNA probes (antisense and sense). The DNA was incubated with transcription buffer (Ambion) in the presence of 12.5 nmol ATP, CTP, and GTP; 50 pmol UTP; 125 pmol [α-35S]UTP (1250 Ci/mmol; PerkinElmer); and 1 unit of RNase inhibitor and 1 unit of RNA polymerase (Roche Molecular Biochemicals) at 37°C for 90 min. This step was followed by digestion of the DNA with RNase-free DNase at 37°C for 20 min. The transcripts were purified using spin columns (illustraTM MicrospinTM S-200 HR Columns; GE Healthcare).
Tissue preparation and brain sections
Fresh brains were collected from PPARγ KO and WT mice in RNase-free conditions and were immediately frozen in dry ice and stored at −80°C until the brains were cryosectioned at 10 μm thickness. Mouse brain sections were brought to room temperature (RT) for fixation in 4% paraformaldehyde in PBS, pH 7.0, for 15 min, followed by washing in PBS, pH 7.4, for 10 min and rinsing twice in sterilized water (5 min). Deproteination of the tissue was accomplished by incubation in 0.1 m triethanolamine, pH 8.0, for 10 min. After 2 further washing steps in PBS, pH 7.4, for 5 min, the tissue was acetylated in 0.1 m triethanolamine, pH 8.0, with 0.25% acetic anhydride for 20 min and then washed again twice in PBS, pH 7.4, for 5 min. The sections were then dehydrated in graded ethanol and air dried. Prehybridization in prehybridization buffer (50% deionized formamide, 50 mm Tris-HCl, pH 7.6, 25 mm EDTA, pH 8.0, 20 mm NaCl, 0.25 mg/ml yeast tRNA, 2.5 × Denhardt's solution; Invitrogen) was performed in humidified chambers at 37°C for 2–4 h. Afterward, the prehybridization solution was drained off the slides and the sections were hybridized with 100 μl of hybridization buffer (50% deionized formamide, 20 mm Tris-HCl, pH 7.6, 10× Denhardt's solution, 5 mg/ml yeast tRNA, 1 mg/ml polyadenylic acid, 10 mm EDTA, pH 8.0, 150 mm DTT, 330 mm NaCl, and 10% dextran sulfate) containing 1 × 106 cpm of either the labeled antisense RNA or sense RNA. Siliconized coverslips were used to cover the sections for the incubation at 55°C overnight in a humidified chamber. After removal of the coverslips by washing with 1× standard saline citrate (SSC, 42°C, 40 min), the slides were washed again twice with 1× SSC and then in 0.5× SSC/50% formamide for 1 h at 42°C, followed by 2 washing steps in 1× SSC for 30 min at 42°C. The sections were then treated with 1 μg/ml RNase in RNase buffer (0.5 m NaCl, 10 mm Tris, pH 8.0, 1 mm EDTA, pH 7.5) for 1 h at 37°C, washed twice in 1× SSC for 30 min at 55°C, and briefly washed in 1×SSC at RT. Finally, the sections were dehydrated in graded ethanol and air-dried. BAS-SR 2025 imaging plates were exposed to the dry slides for 7 d and then scanned with a PhosphorImager (Fuji Typhoon FLA 700). MCID Image Analysis Software (Imaging Research) was used for densitometric analysis: bregma 0.14 mm [lateral septum and bed nucleus of the stria terminalis (BNST) and bregma −1.22 mm (sensory cortex (S1), habenula, laterodorsal thalamic nucleus, central AMY (CeA), and basolateral AMY (BLA) according to the mouse brain atlas; Paxinos and Franklin, 2003]. The mean density values were measured as minimal detectable change (MDC) units per square millimeter and were converted into nano-Curies per gram using [14C] microscales (GE Healthcare Life Sciences).
Histochemistry
Tissue preparation.
Mice were deeply anesthetized by CO2 inhalation and transcardially perfused with 4% paraformaldehyde in PBS. The brains were postfixed for 1 h and cryoprotected in 30% sucrose solution in PBS at 4°C until sectioning (30 μm thickness). All reactions were performed on free-floating sections for c-Fos immunostaining. Sections were rinsed in PBS, treated for 30 min in 1% H2O2 in PBS to reduce background, and incubated in blocking solution [3% normal sheep serum (NSS) in PBS] for 2 h. The sections were then placed in a solution (3% NSS in PBS) containing an antibody directed against c-Fos (1:10.000; Ab-5; Calbiochem) overnight at RT. After several washings in PBS, the sections were incubated in blocking solution (3% NSS in PBS) containing biotinylated goat antirabbit antibody (1:500; Vector Laboratories) for 90 min, followed by a 1 h incubation in an avidin-biotin peroxidase complex solution (Vector Laboratories). Immunoreactivity was visualized as a black reaction product after 5 min in a 0.04% 3,3′ diaminobenzidinetetrahydrochloride solution containing 0.01% H2O2 (Sigma-Aldrich) and 0.1% nickel ammonium sulfate. The sections were mounted on Superfrost1 slides (Fisher Scientific), air dried, dehydrated by serial alcohol rinsing, cleared in xylene, and coverslipped.
RNAscope multiplex fluorescent.
In situ hybridization was performed using the RNAscope Multiplex Fluorescent kit (Advanced Cell Diagnostics) according to the manufacturer's instructions and as described previously (Li et al., 2015).
Briefly, fresh frozen tissues were sectioned, mounted on slides, and fixed in 10% formalin (Thermo Fisher Scientific) for 20 min at 4°C. After 3 washes in 1× PBS, sections were dehydrated in 50%, 70%, and 100% ethanol and then treated with protease solution (pretreatment 4) for 20 min at room temperature. After pretreatment 4, target probes for PPARγ (GenBank accession number: NM_ 001127330.2) and GAD 65 (GAD2) (NM_008078.2) were applied on the slides and incubated at 40°C for 2 h in the HybEZ oven. Next, slides were incubated with preamplifier and amplifier probes (AMP1 at 40°C for 30 min, AMP2 at 40°C for 15 min, and AMP3 at 40°C for 30 min). The slides were then incubated with fluorescently labeled probes by selecting a specific combination of colors: green (Alexa Fluor 448) for PPARγ and red (Atto 550) for GAD2. Finally, brain sections were incubated for 20 s with DAPI (Thermo Fisher Scientific). Fluorescent images of the HIPP and AMY were captured using an LSM700 Zeiss upright confocal microscope.
Plasma corticosterone levels
Blood samples were taken from 1 h restrained and nonrestrained PPARγ KO and WT mice. Plasma was obtained by centrifuging the samples at 1000 × g for 15 min. All plasma samples were stored at −20°C before assaying with a commercially available radioimmunoassay with 126I-labeled rat/mice corticosterone (MP Biomedicals). A gamma counter was used to measure radioactivity of the samples. The values obtained are expressed in nanograms per milliliter.
Statistical analysis
Behavioral analysis and corticosterone levels were analyzed by two-way ANOVA or, when necessary, by Student's t tests. Newman–Keuls post hoc analysis was performed when necessary. For quantification of c-Fos, the number of positively stained nuclei per region of interest was determined bilaterally in two sections for each mouse by using the cell counter analysis macro of ImageJ. All cell counts were performed in regions of interest covering identical areas in square micrometers. For immunohistochemistry and gene expression, the regions of interest were defined by anatomical landmarks as described in the atlas (Paxinos and Franklin, 2003). Because the data had homogenous variances within, but not between, regions, statistical analysis was performed by two-way ANOVA and t test analysis within each brain region for c-Fos and in situ hybridization, respectively. Statistical analysis of the raw data was performed in Statistica 7 (StatSoft).
Results
Pioglitazone reduces acute stress-induced anxiety without affecting basal anxiety levels
The anxiolytic effect of pioglitazone was assessed under basal conditions or after 1 h of restraint stress in the LD test. Fifty CD1 mice were divided into 4 groups (n = 12–13/group) and were treated with pioglitazone (30 mg/kg) or vehicle given twice at 12 h and 10 min before restraint stress (70 min before the LD test).
Two-way ANOVA revealed a significant overall difference between groups with a main effect of restraint stress (F(1,46) = 17.6; p = 0.0001), accompanied by a main pioglitazone effect (F(1,46) = 6.4; p = 0.014) and a significant stress × treatment interaction (F(1,46) = 5.4; p = 0.024). Newman–Keuls post hoc tests showed an increase in stress-induced anxiety (p = 0.0002) and an effect of pioglitazone in reducing it (p = 0.001; Fig. 1A). No effect of pioglitazone was observed in the group tested under basal conditions (p = 0.884). No significant effect of restraint and treatment on number of entries into the light side was observed (F(1,46) = 3.0, p = 0.089 and F(1,46) = 0.6, p = 0.433; Fig. 1B).
Figure 1.

A, Restraint stress significantly decreased the time spent in the light side of the compartment. Pretreatment with pioglitazone (30 mg/kg) reversed the anxiogenic-like effect of stress. B, No differences were detected in the number of entries into the light side of the arena. The data represent the mean ± SEM values (n = 12–13 per group). Difference between vehicle nonstressed and vehicle stressed: ***p < 0.001. Difference between vehicle stressed and pioglitazone stressed: ## p < 0.01.
To evaluate whether a single pioglitazone administration is sufficient to exert an anxiolytic action, we performed an additional experiment in which we injected the PPARγ agonist only once 10 min before restraint stress (70 min before the LD test).
One-way ANOVA revealed a significant overall difference between groups (F(2,34) = 8.12; p = 0.0013). The Newman–Keuls test revealed that restraint stress significantly reduced the time spent in the light compartment compared with unrestrained control mice (p = 0.0012). Pretreatment with pioglitazone did not significantly reverse the anxiogenic effect of restraint stress (data not shown). No significant effect of stress and treatment on number of entries into the light side was observed (F(2,34) = 3.21; p = 0.053).
GW9662 blocks the anxiolytic effect of pioglitazone on stress-induced anxiety
The effect of pioglitazone, GW9662, and their combination was tested on anxiety induced by restraint stress. CD1 mice were divided into five groups (n = 12–13/group). Animals were subjected to restraint stress for 1 h. Treatments with pioglitazone (0 and 30 mg/kg), GW9665 (0 and 5 mg/kg), or their combination were given twice 12 h and 10 min before restraint stress (70 min before the LD test). GW9662 preceded the administration of pioglitazone. An additional group was not subjected to restraint and served as a control. To control for the effect of stress, we compared restrained and nonrestrained animals using a t test. Results showed a significant difference between groups, with stressed mice spending less time in the light side of LD compared with nonstressed mice (t(25) = 4.72, p = 0.000076). At this point, to evaluate the effect of treatments, we used a two-way factorial ANOVA. Results showed no main effect for pioglitazone (F(1,48) = 3.61; p = 0.063) or GW9662 (F(1,48) = 0.68; p = 0.41), but a significant drug × drug interaction (F(1,48) = 7.2; p = 0.009). Newman–Keuls post hoc tests showed that pioglitazone reversed stress-induced anxiety (p = 0.011) and GW9662 significantly blocked it (p = 0.042; Fig. 2A). No significant effect of restraint (t(25) = 1.90, p = 0.068) or administration of pioglitazone (F(1,48) = 1.38; p = 0.24), GW9662 (F(1,48) = 0.21; p = 0.64), or both (F(1,48) = 2.8; p = 0.10) was observed on the number of entries into the light side (Fig. 2B).
Figure 2.

A, Restraint stress significantly decreased the time spent in the light side of the apparatus. Pretreatment with pioglitazone (PIO, 30 mg/kg) reversed the anxiogenic-like effect of stress and GW9662 (GW) blocked the anxiolytic-like effect of pioglitazone. B, No differences were detected in number of entries into the light side of the arena. The data represent the mean ± SEM values (n = 12–13 per group). ***p < 0.001; *p < 0.05. S, Stress; NS, no stress.
Analysis of PPARγ expression in conditional PPARγNestinCre KO mice
For studying the contribution of neuronal PPARγ in anxiety-related behavior, we used PPARγNestinCre KO mice. We verified the neuronal deletion of PPARγ using in situ hybridization (ISH) and demonstrated a significant effect of genotype on PPARγ expression in all of the following brain areas analyzed in PPARγNestinCre KO and WT animals (n = 7 per group): septum (t = 15.53, df = 12, p = 0.0000); BNST (t = 68.95, df = 12; p = 0.0000); S1 (t = 16.82, df = 12; p = 0.001); BLA (t = 51.05, df = 11; p = 0.0000); CeA (t = 12.99, df = 12; p = 0.003); habenula (t = 35.9, df = 12; p = 0.0000); and laterodorsal thalamic nucleus (t = 33.58, df = 12; p < 0.0000) (Fig. 3A). The distribution pattern of PPARγ mRNA expression of the septum and BNST was analyzed in a brain section taken 0.14 mm anterior to the bregma, whereas the pattern of PPARγ gene expression of BLA, CeA, S1, habenula, and thalamus was analyzed in a brain section taken 1.22 mm from the bregma (Fig. 3B). In summary, consistent with previously published data, residual PPARγ expression was still detectable in KO mice, indicating receptor deletion in neurons, but not in other cell types (i.e., microglia, oligodendrocytes; Sarruf et al., 2009).
Figure 3.
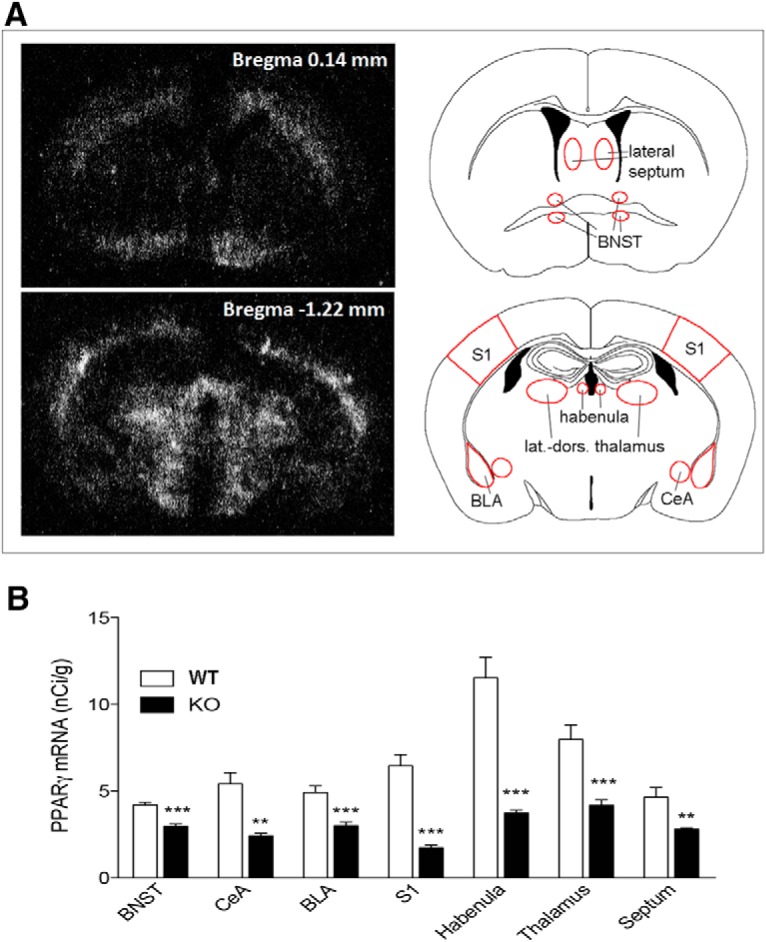
A, Dark-field photomicrographs from autoradiograms depicting the pattern of PPARγ expression of coronal mouse brain section at the bregma level 0.14 mm (top) and 1.22 mm (bottom). B, PPARγ mRNA levels of PPARγNestinCre KO and WT mice in the BNST, CeA, BLA, S1, habenula, thalamus, and septum. The data represent the mean ± SEM (n = 7 per group). **p < 0.01; ***p < 0.001.
PPARγNestinCreKO animals exhibit an anxiogenic-like phenotype and blockade of PPARγ elicits a significant anxiogenic-like response in WT, but not in KO, mice
A group of PPARγ KO (n = 20) and a WT (n = 20) mice were used for this experiment. Each group was subdivided in 2 subgroups (n = 10) and treated with the selective PPARγ antagonist GW9662 (5 mg/kg, i.p.) or vehicle given 12 h and 1 h before the behavioral tests. In the analysis of time spent in the light side of the compartment, overall factorial ANOVA revealed a significant main effect of genotype (F(1,36) = 23.4; p = 0.0000), a significant main effect of treatment (F(1,36) = 5.1; p = 0.020), and a significant genotype × treatment interaction (F(1,36) = 4.7; p = 0.039). Post hoc analysis showed that PPARγ KO mice spent less time in the light side of the LD compartment compared with WT mice (p = 0.0001). GW9662 administration significantly (p = 0.003) reduced the time spent in the light side for WT, but not PPARγ KO, mice (p = 0.75; Fig. 4A). Similarly, for the number of entries into the light side, overall ANOVA revealed a significant effect of genotype (F(1,36) = 21.9; p = 0.0001), treatment (F(1,36) = 6.1; p = 0.019), and a genotype × treatment interaction (F(1,36) = 5.3; p = 0.025). Post hoc analysis showed that there were fewer entries into the light side by PPARγ KO mice than by WT mice (p = 0.0009). GW9662 administration significantly (p = 0.010) reduced the number of entries into the light side of the arena for WT, but not PPARγ KO, mice (p = 0.685; Fig. 4B).
Figure 4.

A, B, PPARγNestinCre KO mice and WT animals treated with GW9662 spent significantly less time (A) and displayed fewer entries into the light side of the apparatus (B) compared with controls in the light dark test. C, PPARγNestinCre KO mice and WT mice treated with GW9662 spent significantly less time in the open arms compared with controls in the EPM. D, No differences among groups were observed in total number of entries in the EPM. E, PPARγNestinCre KO mice and WT mice treated with GW9662 spent significantly less time in the central zone of the open-field arena. F, No differences among groups were observed in the total distance traveled in the open field. The data represent the mean ± SEM (n = 10 per group). Difference between PPARγNestinCre KO and WT mice: ##p < 0.01, ###p < 0.001 and difference between GW9662-treated mice and control: **p < 0.01, ***p < 0.001.
For the EPM test, each group was subdivided in 2 subgroups (n = 9/10) and treated with GW9662 (5 mg/kg, i.p.) or vehicle 12 h and 1 h before EPM test. Analysis of percentage of time spent in the open arms showed a significant main effect of genotype (F(1,33) = 15.98; p = 0.0003), a significant main effect of treatment (F(1,33) = 7.351; p = 0.01), and a significant genotype × treatment interaction (F(1,33) = 15.35; p = 0.0004). Post hoc analysis showed that PPARγ KO mice spent less time in the open arms of the EPM compared with WT mice (p = 0.0002). GW9662 administration significantly (p = 0.0002) reduced the time spent in the open arms for WT, but not PPARγ KO, mice (p = 0.40; Fig. 4C). ANOVA also showed a significant genotype difference in the number of entries into open arms (F(1,33) = 5.872; p = 0.021). There was no significant effect of the treatment (F(1,33) = 0.953; p = 0.34), but a significant interaction genotype × treatment: (F(1,33) = 4.76; p = 0.036) (data not shown). Overall ANOVA did not reveal a significant effect of the genotype (F(1,33) = 0.97; p = 0.329) for either treatment (F(1,33) = 0.97; p = 0.668) or the genotype × treatment interaction (F(1,33) = 0.38; p = 0.668) in the total number of entries (Fig. 4D).
In the open-field test (n = 9/10 mice), ANOVA showed a significant effect of genotype in the time spent in the center zone (F(1,33) = 10.01; p = 0.0033) and a significant effect of treatment (F(1,33) = 7.9; p = 0.008), but no significant genotype × treatment interaction (F(1,33) = 2.389; p = 0.1316). Post hoc analysis showed that PPARγ KO mice spent less time in the center zone (p = 0.006), whereas GW9662 reduced the time in the center zone in WT only (p = 0.0042; Fig. 4E). ANOVA revealed no significant effect of the genotype (F(1,33) = 0.9044; p = 0.3485), no significant effect of treatment (F(1,33) = 0.0274; p = 0.8694), and no significant genotype × treatment interaction (F(1,33) = 0.4944; p = 0.4868) in total distance traveled (Fig. 4F).
c-Fos analysis reveals a different pattern of stress-induced neuronal activation in PPARγ KO and WT mice
c-Fos immunoreactivity was assessed in PPARγ KO and WT mice (n = 3–4 per group) after exposure to the bright illuminated and novel environment as in the light dark exploration test (Fig. 5). Rodents show an innate aversion for bright illuminated compartments and the contemporary exposure to light and a novel environment acts as a mild stressor (Bourin and Hascoët, 2003). For each brain region, data were collected bilaterally from at least two separate sections. Brains were collected 90 min after the LD test (stressed group). The control group (nonstressed) remained in their home cages. The results for all regions analyzed are reported in Figure 5. In the paraventricular nucleus of the thalamus (PVA), two-way ANOVA did not show a main effect of genotype (F(1,9) = 1.25; p = 0.290), but there was a significant main effect of stress (F(1,9) = 9.3; p = 0.015) without a significant genotype × stress interaction (F(1,9) = 0.2; p = 0.65). Post hoc analysis showed a significant increase in the number of c-Fos-positive cells after stress in both WT (p = 0.033) and KO mice (p = 0.042). In the paraventricular nucleus of the hypothalamus (PVN), ANOVA revealed no main effect of genotype (F(1,9) = 0.03; p = 0.857), a significant effect of stress (F(1,9) = 48.9; p = 0.0000), and a significant genotype × stress interaction (F(1,9) = 9.5; p = 0.013). Post hoc analysis showed a significantly higher number of c-Fos-positive cells after stress in both WT (p = 0.0004) and KO mice (p = 0.022). Moreover, the data revealed that, under basal conditions, KO mice expressed a higher level of c-Fos compared with WT mice (p = 0.045).
Figure 5.

Top, Representative images of c-Fos immunohistochemistry in PPARγNestinCre KO and WT mice under basal conditions or after exposure to a mild stress (n = 3–4 per group). Scale bar, 100 μm. Bottom, c-Fos expression levels in the PVA, PVN, AMY, and HIPP. Data are expressed as mean ± SEM of the number of c-Fos-positive cells: *p < 0.05; **p < 0.01; ***p < 0.001 stress versus no stress; °p < 0.05 KO no stress versus WT no stress; #p < 0.05 KO stress versus WT stress.
In the AMY region, ANOVA showed no effect of genotype (F(1,9) = 0.37; p = 0.558), a significant effect of stress (F(1,9) = 5.7; p = 0.031), and a significant genotype × stress interaction (F(1,9) = 10.6; p = 0.0098). Newman–Keuls post hoc test showed a significantly higher level of c-Fos-positive cells in WT mice after stress (p = 0.015), but not in KO mice (p = 0.50). PPARγ KO stressed mice exhibited a significantly lower expression level of c-Fos compared with WT stressed mice (p = 0.019). In the HIPP, the analysis showed no effect of genotype (F(1,9) = 0.2; p = 0.661), but there was a significant difference between groups in the main effect of stress (F(1,9) = 7.1; p = 0.025) and a significant genotype × stress interaction (F(1,9) = 6.6; p = 0.030). Post hoc analysis showed that c-Fos expression was increased only in WT after stress (p < 0.020). c-Fos did not change in KO stressed mice compared with nonstressed control mice (p = 0.944), but showed a trend close to significance with lower expression in KO mice after stress compared with WT stressed mice (p = 0.063).
PPARγ WT and KO animals show a similar endocrine response to acute stress
Plasma corticosterone levels were detected in PPARγ KO and WT mice under basal conditions and after 1 h of restraint stress. Mice were divided into 4 groups (n = 7–8/group) and corticosterone levels were assessed. WT mice showed a corticosterone level of 65.5 ± 8.0 ng/ml that was increased to 124. 6 ± 8.0 ng/ml after stress. KO basal costicosterone was 72.1 ± 8.7 ng/ml and increased to 151.1 ± 18.5 after stress. Two-way ANOVA did not show a significant overall difference between groups with main effect of genotype (F(1,25) = 1.39; p = 0.248), but revealed a significant main effect of stress (F(1,25) = 26.04; p = 0.0000) without a significant genotype × stress interaction (F(1,25) = 0.5; p = 0.460). The Newman–Keuls test showed an increase of corticosterone levels in stressed mice compared with the baseline in both WT (p = 0.013) and KO (p = 0.001) mice.
Intra-AMY, but not intra-HIPPocampal, infusion of pioglitazone reduces stress-induced increase in anxiety-like behavior
Restraint-stressed WT mice (n = 13 mice per group) were treated with pioglitazone or vehicle twice 12 h and 10 min before stress directly into the AMY (2.5 μg/side/0.3 μl). A third group of mice (n = 15) not subjected to restraint stress but injected into the AMY with vehicle served as a control. Anxiety-like behavior was monitored by the LD test. One-way ANOVA revealed a significant overall difference between groups (F(2,37) = 4.8; p = 0.013). Newman–Keuls test revealed that restraint stress significantly reduced the time spent in the light compartment compared with unrestrained control mice (p = 0.014). Pretreatment with pioglitazone reversed the anxiogenic effect of restraint stress (p = 0.035; Fig. 6B). ANOVA showed no overall effect of the treatment in the number of entries into the light side (F(2,37) = 0.62; p = 0.541; Fig. 6C). When the other two groups of mice (n = 9–10/group) were tested for anxiety-like behavior in LD in response to intra-AMY pioglitazone infusion under basal conditions (nonstressed), no drug effect was detected in the time spent in the light side (t(17) = 0.36, NS) or in the total number of entries in the light side (t(17) = 0.81, NS).
When pioglitazone or its vehicle was infused into the HIPP of stressed mice (n = 11/group), compared with nonstress controls (n = 11), ANOVA revealed a significant overall difference between groups (F(2,30) = 7.9; p = 0.0016). Newman–Keuls test showed that restraint stress significantly reduced the time spent in the light compartment compared with unrestrained control mice (p = 0.0067). Pretreatment with pioglitazone did not modify the effect of stress (p = 0.035; Fig. 6E). ANOVA showed no overall effect of the treatment in the number of entries into the light side (F(2,30) = 1.7; p = 0.20; Fig. 6C).
PPARγ is largely expressed in GAD2 cells
PPARγ expression was analyzed in the AMY (Fig. 7) and HIPP (Fig. 8). The RNAscope multiplex fluorescent technique with simultaneous detection of PPARγ transcript expression and GAD2 and DAPI staining allowed us to analyze the colocalization of PPARγ in GABAergic cells. After counterstaining the sections with DAPI, we found the most PPARγ expression in GAD2 cells in both areas.
Figure 7.

Representative images (40× magnification) of the AMY for: GAD2 (red) + PPARγ (green) (A); PPARγ + DAPI (blue) (B); GAD2 + DAPI (C), and merge of PPARγ + GAD2 + DAPI (D). E, Illustration of the region of the AMY sampled for RNAscope ISH (Paxinos and Franklin, 2003). Arrows indicate representative cells showing PPARγ + GAD2 + DAPI colocalization.
Figure 8.

Representative images (40× magnification) of the HIPP for: GAD2 (red) + PPARγ (green) (A); PPARγ + DAPI (blue) (B); GAD2 + DAPI (C), and merge of PPARγ + GAD2 + DAPI (D). E, Illustration of the region of the HIPP sampled for RNAscope ISH (Paxinos and Franklin, 2003). Arrows indicate representative cells showing PPARγ + GAD2 + DAPI colocalization.
Discussion
In this study, we show that genetic deletion of neuronal PPARγ enhances the emotional response to acute stress and exacerbates anxiety. PPARγ in the AMY appears to play a major role. Initially, we found that systemic PPARγ activation by the selective agonist pioglitazone inhibited the anxiogenic effect of a stressor in outbred mice. To confirm that the effect of pioglitazone was mediated by selective activation of PPARγ, we pretreated the mice with the selective receptor antagonist GW9662 and found that it fully reversed pioglitazone anxiolytic effect. Based on these findings, we hypothesized that neuronal PPARγ is involved in the regulation of anxiety-related behavior associated with stress. To further explore the relationship between PPARγ regulation and anxiety, we investigated stress response in PPARγNestinCre KO mice. The genetic deletion of PPARγ in the brains of KO mice was confirmed by ISH data showing a marked reduction of receptor transcript expression in several regions involved in emotional control, including the septum, BNST, BLA, CeA, habenula, and thalamus. Consistent with previously published data, residual PPARγ expression was still detectable in KO mice, indicating receptor deletion in neurons but not in other cell types (i.e., microglia, oligodendrocytes; Sarruf et al., 2009).
Neuronal PPARγ is involved in anxiety-related behaviors
In a battery of tests to explore anxiety-related behaviors, neuronal PPARγNestinCre KO mice exhibited higher innate anxiety responses compared with WT mice. Notably, in the open-field test, we observed that PPARγ KO mice, despite spending less time in the center or the arena, showed no differences in the total distance traveled. Moreover, the two mouse lines did not differ in the total number of arm entries into the EPM, excluding the possibility that anxiety tests could have been influenced by differences in locomotor behavior. This result is consistent with previous pharmacological data revealing that PPARγ activation by pioglitazone and rosiglitazone did not result in locomotor impairment or reduction in muscle strength in rodents (Morgenweck et al., 2010; Sadaghiani et al., 2011; de Guglielmo et al., 2014). Altogether, these results suggest a specific involvement of PPARγ in the modulation of anxiety and indicate that this effect is mediated by neuronal mechanisms.
Our finding of a neuronal contribution of PPARγ to anxiety-related behavior has important implications because, given evidence that the stress/anxiety response is associated with neuroimmune signaling and increased expression of cytokines (Raison et al., 2006; García-Bueno et al., 2008a; Leonard and Myint, 2009; Hou et al., 2013), one possibility would have been that the PPARγ system modulates anxiety through anti-inflammatory mechanisms (Feinstein, 2003; García-Bueno et al., 2005; Heneka and O'Banion, 2007; Woster and Combs, 2007; Yuan et al., 2015). In contrast to this hypothesis, we found a distinct anxiogenic-like phenotype in neuron-specific PPARγ KO mice, suggesting that this negative affective state is linked to the lack of PPARγ in neuronal cells.
To further validate whether the specific blockade of PPARγ would increase anxiety, we administered the PPARγ antagonist GW9662 to WT and KO mice that were then tested for an anxiety-like response. As expected, blockade of PPARγ exerted a marked anxiogenic-like effect in WT mice, whereas no effects were observed in KO mice.
Mapping neuronal activation using c-Fos immunohistochemistry
Having ascertained that PPARγ-mediated response in anxiety is regulated by neuronal mechanisms, we used c-Fos immunohistochemistry to map neuronal activation in WT and KO mice under basal conditions and in response to a mild stressful stimulus consisting of the exposure to the light side of the LD compartment. The results provided two important pieces of information: (1) basal c-Fos expression was generally higher in KO mice compared with WT mice (effect significant in the PVN) and (2) in WT mice, exposure to LD enhanced c-Fos expression in areas responsible for emotional control (i.e., AMY and HIPP) and in structures mediating the endocrine response to stress (i.e., PVN and PVA). Conversely, in KO mice, enhanced c-Fos expression after stress was only observed in endocrine-relevant areas and in particular in the PVN, which plays a key role in initiating the hypothalamic–pituitary–adrenal (HPA) response to stress (Cullinan et al., 1995; Herman et al., 2002; Herman et al., 2003; Smith and Vale, 2006). Based on c-Fos expression pattern, we anticipated no differences in HPA response to stress between KO and WT mice, but an altered emotional processing occurring in mice lacking PPARγ function in limbic areas such as the AMY or the HIPP.
Our prediction was confirmed by endocrinology data showing no difference between WT and KO mice in the basal corticosterone level or levels after stimulation by acute stress. This finding is consistent with previous work demonstrating that the protective effects of PPARγ ligands on stress were independent of systemic hormonal responses because no differences were observed in plasma corticosterone levels in rats treated with rosiglitazone or vehicle and exposed to restraint stress (García-Bueno et al., 2005).
AMY is the site of action of PPARγ-mediated anxiety responses
After the hypothesis of an altered emotional processing in response to stress, we next focused our attention on the limbic system and, in particular, on the AMY, in which marked c-Fos hyporesponsivity was observed in PPARγ KO mice compared with WT mice. The AMY is an important relay responsible for the modulation of anxiety and is activated in response to stress (Roozendaal et al., 2009). PPARγ in this area appears to modulate gene networks linked to GABA and glutamate neurotransmission (Ferguson et al., 2014), which are both important in the regulation of affective responses (Hartmann et al., 1993). Therefore, we monitored the effect of intra-AMY injection of pioglitazone on anxiety-like behavior in WT mice exposed to restraint stress. As predicted, the selective activation of PPARγ in the AMY abolished the anxiogenic-like effect of restraint stress. Next, considering that differential c-Fos activation between PPARγ KO and WT mice was detected also in the HIPP, we investigated the effect of pioglitazone injection in this region. Activation of PPARγ in this region was, however, unable to prevent the anxiogenic-like effect of restraint stress. This suggests that the AMY is a specific neuronal substrate for the anxiolytic effect of pioglitazone.
Subsequently, we studied the localization of PPARγ in these regions. In the AMY, results confirmed a relatively high expression of the receptor in cells positive for the GABAergic marker GAD2. Interestingly, recent findings from cross-species genome-wide approaches have identified GAD2 and PPARγ as intriguing candidate genes in anxiety disorders (Sokolowska and Hovatta, 2013). In the HIPP, lower receptor expression levels were detected even though colocalization with GAD2-positive cells was confirmed. These neuroanatomical data may reflect the possibility that PPARγ activation elicits anxiolytic action through modulation of the GABAergic transmission within the AMY.
One aspect of the study that remains elusive is why, in outbred mice, pioglitazone showed its anxiolytic effect only after exposure to stress, whereas in PPARγ KO mice, excessive anxiety also appeared under basal conditions, indicating a tonic control of this emotional state by PPARγ. One possibility is that the genetic deletion of the receptor sets the condition of reduced resilience to environmental changes, dampening the innate ability of the mouse to adapt to external stimuli that are then perceived as stressors. If moderately stressful stimuli, such as those experienced in the LD or EPM tests, are perceived as highly aversive, then they may also activate stress mechanisms under basal conditions. An aspect of the study that has remained unexplored is whether activation of PPARγ after exposure to stress is able to attenuate anxiety and restore normal affective condition
Another element of the study to be considered is that the results from the KO mice point to a neuronal-mediated mechanism for the modulation of anxiety by PPARγ. This finding is particularly important because recent clinical data have provided preliminary evidence for the efficacy of pioglitazone and rosiglitazone in attenuating depression and bipolar disorder in patients (Kemp et al., 2014; Zeinoddini et al., 2015). In these studies, the anti-inflammatory and glia inhibitory properties of PPARγ agonists were proposed as key factors for the effect of these two TDZs. Challenging this assumption, we provide here evidence that PPARγ activity is mediated by neuronal extrahypothalamic mechanisms involving the AMY. The exact cellular processes through which PPARγ modulates AMY transmission remain to be determined. Here, we have shown a high level of colocalization with GABA cells. Moreover, we have shown recently that PPARγ activation modulates presynaptic GABA transmission and attenuates opioid-induced stimulation of DA neurons in the VTA. Finally, in a recent gene expression study, it was shown that PPARγ stimulation results in marked changes in the levels of several transcripts linked to GABA and glutamate transmission in the AMY (Ferguson et al., 2014). It is well known that intrinsic and extrinsic circuitry formed by GABA and glutamate neurons in the BLA–CeA plays a critical role in the regulation of mood and affect (Veinante and Freund-Mercier, 1998). Therefore, it is possible that PPARγ, by regulating the connectivity of these circuitries through modulation of amino acid transmissions, may affect anxiety and possibly also other affective states. One limitation of our study is that we have not been able to determine the specific role of these two AMY subregions that, based on our ISH and c-Fos results, could be both involved in mediating the effects of PPARγ. In fact, due to the small size of the BLA and the CeA, it was not possible to target these neighboring regions separately through microinjection experiments in the mouse. Future studies, possibly using electrophysiology recordings, are needed to determine the exact role of PPARγ in the regulation of GABA and glutamate neurocircuitry in these different subregions of the AMY.
In conclusion, our findings open new vistas on the role of PPARγ in the regulation of mood disorder, indicating that dampened transmission may contribute to exacerbate anxiety and the negative effects of stress. The results also suggest that the activation of PPARγ may be beneficial in the treatment of psychiatric conditions associated with stress and, in particular, anxiety disorders. We have also demonstrated previously marked efficacy of PPARγ agonists in preventing relapse to alcohol seeking by stress (Stopponi et al., 2011; Le Foll et al., 2013; Stopponi et al., 2013).
Footnotes
This work was supported by the Italian Society of Pharmacology (fellowship to E.D.), the Bundesministeriumfür Bildung und Forschung (e:Med program Grant FKZ: 01ZX1311A, Spanagel et al., 2013 and Grant RO1 AA017447 to M.R.). We thank Dr Kevin Niswender at Vanderbilt University for providing PPARγ-engineered mouse lines; Rina Righi and Mariangela Fiorelli for animal care; and Alfredo Fiorelli, Elisabeth Röbl, and Claudia Schäfer for technical support.
The authors declare no competing financial interests.
References
- Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Bernardi RE, Uhrig S, Spanagel R, Hansson AC. Transcriptional regulation of L-type calcium channel subtypes Cav1.2 and Cav1.3 by nicotine and their potential role in nicotine sensitization. Nicotine Tob Res. 2014;16:774–785. doi: 10.1093/ntr/ntt274. [DOI] [PubMed] [Google Scholar]
- Bourin M, Hascoët M. The mouse light/dark box test. Eur J Pharmacol. 2003;463:55–65. doi: 10.1016/S0014-2999(03)01274-3. [DOI] [PubMed] [Google Scholar]
- Breidert T, Callebert J, Heneka MT, Landreth G, Launay JM, Hirsch EC. Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson's disease. J Neurochem. 2002;82:615–624. doi: 10.1046/j.1471-4159.2002.00990.x. [DOI] [PubMed] [Google Scholar]
- Carta AR, Frau L, Pisanu A, Wardas J, Spiga S, Carboni E. Rosiglitazone decreases peroxisome proliferator receptor-gamma levels in microglia and inhibits TNF-alpha production: new evidences on neuroprotection in a progressive Parkinson's disease model. Neuroscience. 2011;194:250–261. doi: 10.1016/j.neuroscience.2011.07.046. [DOI] [PubMed] [Google Scholar]
- Chang F, Jaber LA, Berlie HD, O'Connell MB. Evolution of peroxisome proliferator-activated receptor agonists. Ann Pharmacother. 2007;41:973–983. doi: 10.1345/aph.1K013. [DOI] [PubMed] [Google Scholar]
- Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, Krishnan V, Reyes CM, Han MH, Ables JL, Eisch AJ, Dietz DM, Ferguson D, Neve RL, Greengard P, Kim Y, Morrison JH, Russo SJ. IkappaB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Battaglia DF, Akil H, Watson SJ. Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience. 1995;64:477–505. doi: 10.1016/0306-4522(94)00355-9. [DOI] [PubMed] [Google Scholar]
- de Guglielmo G, Kallupi M, Scuppa G, Stopponi S, Demopulos G, Gaitanaris G, Ciccocioppo R. Analgesic tolerance to morphine is regulated by PPARgamma. Br J Pharmacol. 2014;171:5407–5416. doi: 10.1111/bph.12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guglielmo G, Melis M, De Luca MA, Kallupi M, Li HW, Niswender K, Giordano A, Senzacqua M, Somaini L, Cippitelli A, Gaitanaris G, Demopulos G, Damadzic R, Tapocik J, Heilig M, Ciccocioppo R. PPARgamma activation attenuates opioid consumption and modulates mesolimbic dopamine transmission. Neuropsychopharmacology. 2015;40:927–937. doi: 10.1038/npp.2014.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein DL. Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes Technol Ther. 2003;5:67–73. doi: 10.1089/152091503763816481. [DOI] [PubMed] [Google Scholar]
- Ferguson LB, Most D, Blednov YA, Harris RA. PPAR agonists regulate brain gene expression: relationship to their effects on ethanol consumption. Neuropharmacology. 2014;86:397–407. doi: 10.1016/j.neuropharm.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- García-Bueno B, Madrigal JL, Lizasoain I, Moro MA, Lorenzo P, Leza JC. Peroxisome proliferator-activated receptor gamma activation decreases neuroinflammation in brain after stress in rats. Biol Psychiatry. 2005;57:885–894. doi: 10.1016/j.biopsych.2005.01.007. [DOI] [PubMed] [Google Scholar]
- García-Bueno B, Caso JR, Leza JC. Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci Biobehav Rev. 2008a;32:1136–1151. doi: 10.1016/j.neubiorev.2008.04.001. [DOI] [PubMed] [Google Scholar]
- García-Bueno B, Madrigal JL, Pérez-Nievas BG, Leza JC. Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology. 2008b;149:1969–1978. doi: 10.1210/en.2007-0482. [DOI] [PubMed] [Google Scholar]
- Gofflot F, Chartoire N, Vasseur L, Heikkinen S, Dembele D, Le Merrer J, Auwerx J. Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell. 2007;131:405–418. doi: 10.1016/j.cell.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Björk K, Soverchia L, Terasmaa A, Massi M, Heilig M, Ciccocioppo R. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci U S A. 2006;103:15236–15241. doi: 10.1073/pnas.0604419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Künig G, Riederer P. Involvement of transmitter systems in neuropsychiatric diseases. Acta Neurol Scand Suppl. 1993;146:18–21. [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Ziegler DR, Tasker JG. Role of the paraventricular nucleus microenvironment in stress integration. Eur J Neurosci. 2002;16:381–385. doi: 10.1046/j.1460-9568.2002.02133.x. [DOI] [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Hou R, Tang Z, Baldwin DS. Potential neuroimmunological targets in the treatment of anxiety disorders. Mod Trends Pharmacopsychiatri. 2013;29:67–84. doi: 10.1159/000351965. [DOI] [PubMed] [Google Scholar]
- Jones JR, Shelton KD, Guan Y, Breyer MD, Magnuson MA. Generation and functional confirmation of a conditional null PPARgamma allele in mice. Genesis. 2002;32:134–137. doi: 10.1002/gene.10042. [DOI] [PubMed] [Google Scholar]
- Kemp DE, Schinagle M, Gao K, Conroy C, Ganocy SJ, Ismail-Beigi F, Calabrese JR. PPAR-gamma agonism as a modulator of mood: proof-of-concept for pioglitazone in bipolar depression. CNS Drugs. 2014;28:571–581. doi: 10.1007/s40263-014-0158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107:2669–2674. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, Plunket KD, Shenk JL, Stimmel JB, Therapontos C, Willson TM, Blanchard SG. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41:6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Di Ciano P, Panlilio LV, Goldberg SR, Ciccocioppo R. Peroxisome proliferator-activated receptor (PPAR) agonists as promising new medications for drug addiction: preclinical evidence. Curr Drug Targets. 2013;14:768–776. doi: 10.2174/1389450111314070006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Leonard BE, Myint A. The psychoneuroimmunology of depression. Hum Psychopharmacol. 2009;24:165–175. doi: 10.1002/hup.1011. [DOI] [PubMed] [Google Scholar]
- Li X, Rubio FJ, Zeric T, Bossert JM, Kambhampati S, Cates HM, Kennedy PJ, Liu QR, Cimbro R, Hope BT, Nestler EJ, Shaham Y. Incubation of methamphetamine craving is associated with selective increases in expression of Bdnf and trkb, glutamate receptors, and epigenetic enzymes in cue-activated fos-expressing dorsal striatal neurons. J Neurosci. 2015;35:8232–8244. doi: 10.1523/JNEUROSCI.1022-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O'Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- Moreno S, Farioli-Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience. 2004;123:131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- Morgenweck J, Abdel-Aleem OS, McNamara KC, Donahue RR, Badr MZ, Taylor BK. Activation of peroxisome proliferator-activated receptor gamma in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology. 2010;58:337–345. doi: 10.1016/j.neuropharm.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozhui K, Karlsson RM, Kash TL, Ihne J, Norcross M, Patel S, Farrell MR, Hill EE, Graybeal C, Martin KP, Camp M, Fitzgerald PJ, Ciobanu DC, Sprengel R, Mishina M, Wellman CL, Winder DG, Williams RW, Holmes A. Strain differences in stress responsivity are associated with divergent amygdala gene expression and glutamate-mediated neuronal excitability. J Neurosci. 2010;30:5357–5367. doi: 10.1523/JNEUROSCI.5017-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2003. [Google Scholar]
- Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci. 2009;10:423–433. doi: 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- Sadaghiani MS, Javadi-Paydar M, Gharedaghi MH, Fard YY, Dehpour AR. Antidepressant-like effect of pioglitazone in the forced swimming test in mice: the role of PPAR-gamma receptor and nitric oxide pathway. Behav Brain Res. 2011;224:336–343. doi: 10.1016/j.bbr.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Sarruf DA, Yu F, Nguyen HT, Williams DL, Printz RL, Niswender KD, Schwartz MW. Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology. 2009;150:707–712. doi: 10.1210/en.2008-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnegg CI, Robbins ME. Neuroprotective mechanisms of PPAR delta: modulation of oxidative stress and inflammatory processes. PPAR Res. 2011;2011:373560. doi: 10.1155/2011/373560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searcy JL, Phelps JT, Pancani T, Kadish I, Popovic J, Anderson KL, Beckett TL, Murphy MP, Chen KC, Blalock EM, Landfield PW, Porter NM, Thibault O. Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2012;30:943–961. doi: 10.3233/JAD-2012-111661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. 2006;8:383–395. doi: 10.31887/DCNS.2006.8.4/ssmith. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowska E, Hovatta I. Anxiety genetics: findings from cross-species genome-wide approaches. Biol Mood Anxiety Disord. 2013;3:9. doi: 10.1186/2045-5380-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer WH, Costa RM, Hansson AC. Dopamine systems adaptation during acquisition and consolidation of a skill. Front Integr Neurosci. 2014;8:87. doi: 10.3389/fnint.2014.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, et al. A systems medicine research approach for studying alcohol addiction. Addict Biol. 2013;18:883–896. doi: 10.1111/adb.12109. [DOI] [PubMed] [Google Scholar]
- Stopponi S, Somaini L, Cippitelli A, Cannella N, Braconi S, Kallupi M, Ruggeri B, Heilig M, Demopulos G, Gaitanaris G, Massi M, Ciccocioppo R. Activation of nuclear PPARgamma receptors by the antidiabetic agent pioglitazone suppresses alcohol drinking and relapse to alcohol seeking. Biol Psychiatry. 2011;69:642–649. doi: 10.1016/j.biopsych.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Stopponi S, de Guglielmo G, Somaini L, Cippitelli A, Cannella N, Kallupi M, Ubaldi M, Heilig M, Demopulos G, Gaitanaris G, Ciccocioppo R. Activation of PPARgamma by pioglitazone potentiates the effects of naltrexone on alcohol drinking and relapse in msP rats. Alcohol Clin Exp Res. 2013;37:1351–1360. doi: 10.1111/acer.12091. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Furuyashiki T, Kitaoka S, Senzai Y, Imoto Y, Segi-Nishida E, Deguchi Y, Breyer RM, Breyer MD, Narumiya S. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J Neurosci. 2012;32:4319–4329. doi: 10.1523/JNEUROSCI.5952-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- Veinante P, Freund-Mercier MJ. Intrinsic and extrinsic connections of the rat central extended amygdala: an in vivo electrophysiological study of the central amygdaloid nucleus. Brain Res. 1998;794:188–198. doi: 10.1016/S0006-8993(98)00228-5. [DOI] [PubMed] [Google Scholar]
- Woster AP, Combs CK. Differential ability of a thiazolidinedione PPARgamma agonist to attenuate cytokine secretion in primary microglia and macrophage-like cells. J Neurochem. 2007;103:67–76. doi: 10.1111/j.1471-4159.2007.04706.x. [DOI] [PubMed] [Google Scholar]
- Yuan G, Chen X, Li D. Modulation of peroxisome proliferator-activated receptor gamma (PPAR gamma) by conjugated fatty acid in obesity and inflammatory bowel disease. J Agric Food Chem. 2015;63:1883–1895. doi: 10.1021/jf505050c. [DOI] [PubMed] [Google Scholar]
- Zeinoddini A, Sorayani M, Hassanzadeh E, Arbabi M, Farokhnia M, Salimi S, Ghaleiha A, Akhondzadeh S. Pioglitazone adjunctive therapy for depressive episode of bipolar disorder: a randomized, double-blind, placebo-controlled trial. Depress Anxiety. 2015;32:167–173. doi: 10.1002/da.22340. [DOI] [PubMed] [Google Scholar]