

Abstract
Continuous cerebral blood flow is essential for neuronal survival, but whether vascular tone influences resting neuronal function is not known. Using a multidisciplinary approach in both rat and mice brain slices, we determined whether flow/pressure-evoked increases or decreases in parenchymal arteriole vascular tone, which result in arteriole constriction and dilation, respectively, altered resting cortical pyramidal neuron activity. We present evidence for intercellular communication in the brain involving a flow of information from vessel to astrocyte to neuron, a direction opposite to that of classic neurovascular coupling and referred to here as vasculo-neuronal coupling (VNC). Flow/pressure increases within parenchymal arterioles increased vascular tone and simultaneously decreased resting pyramidal neuron firing activity. On the other hand, flow/pressure decreases evoke parenchymal arteriole dilation and increased resting pyramidal neuron firing activity. In GLAST-CreERT2; R26-lsl-GCaMP3 mice, we demonstrate that increased parenchymal arteriole tone significantly increased intracellular calcium in perivascular astrocyte processes, the onset of astrocyte calcium changes preceded the inhibition of cortical pyramidal neuronal firing activity. During increases in parenchymal arteriole tone, the pyramidal neuron response was unaffected by blockers of nitric oxide, GABAA, glutamate, or ecto-ATPase. However, VNC was abrogated by TRPV4 channel, GABAB, as well as an adenosine A1 receptor blocker. Differently to pyramidal neuron responses, increases in flow/pressure within parenchymal arterioles increased the firing activity of a subtype of interneuron. Together, these data suggest that VNC is a complex constitutive active process that enables neurons to efficiently adjust their resting activity according to brain perfusion levels, thus safeguarding cellular homeostasis by preventing mismatches between energy supply and demand.
SIGNIFICANCE STATEMENT We present evidence for vessel-to-neuron communication in the brain slice defined here as vasculo-neuronal coupling. We showed that, in response to increases in parenchymal arteriole tone, astrocyte intracellular Ca2+ increased and cortical neuronal activity decreased. On the other hand, decreasing parenchymal arteriole tone increased resting cortical pyramidal neuron activity. Vasculo-neuronal coupling was partly mediated by TRPV4 channels as genetic ablation, or pharmacological blockade impaired increased flow/pressure-evoked neuronal inhibition. Increased flow/pressure-evoked neuronal inhibition was blocked in the presence of adenosine A1 receptor and GABAB receptor blockade. Results provide evidence for the concept of vasculo-neuronal coupling and highlight the importance of understanding the interplay between basal CBF and resting neuronal activity.
Keywords: adenosine, astrocytes, calcium, myogenic tone, neurovascular coupling, TRPV4 channels
Introduction
A question in neurobiology is how increases in neuronal activity evokes changes in cerebral blood flow (CBF). This process, termed neurovascular coupling (NVC), supports brain information processing, commonly assessed using techniques, such as fMRI. On the other hand, little is known about constitutive mechanisms that regulate basal CBF and resting neuronal activity. Given that a large proportion of the brain's energy is used under resting conditions (Raichle, 2009), gaining information on brain homeostatic processes is crucial. Basal CBF is maintained through dynamic processes and signaling interactions between blood vessels and the surrounding neuropil collectively referred to as cerebral autoregulation (Faraci et al., 1989; Paulson et al., 1990). A cellular mechanism governing cerebral autoregulation is the myogenic response, a process intrinsic to smooth muscle in which the contractile state (myogenic tone) of small, resistance arteries and arterioles dynamically responds to changes in intraluminal pressure (Cipolla, 2009). Thus, as pressure increases, the diameter of such vessels decrease; the opposite occurs when pressure decreases (Ngai and Winn, 1995; Golding et al., 1998; Knot and Nelson, 1998; Cipolla, 2009; Pires et al., 2013). This allows for constant CBF over a wide range of systemic pressures, protecting the brain from sudden blood pressure changes and preventing hyperperfusion or hypoperfusion, which can compromise brain function. In a previous study, we demonstrated that brain parenchymal arterioles (PAs) intrinsically respond to changes in luminal pressure (Kim et al., 2015). Importantly, we showed that the sustained application of a hemodynamic stimulus (e.g., increased flow/pressure) engaged perivascular astrocytes, which in turn signaled PAs to maintain vascular tone (Kim et al., 2015). Whether this dynamic, intercellular communication modality translates into changes in neuronal activity is not known.
Based on previous functional and structural observations (Kim et al., 2015), we hypothesized the neurovascular unit provides a route for bidirectional communication, such that vascular-derived signals and/or biomechanical changes may also influence neuronal activity (Paton et al., 2007; Moore and Cao, 2008; del Zoppo, 2010). If so, it would suggest that changes in vascular tone during basal CBF could modulate resting neuronal activity. Experimental evidence for vasculo-to-neuronal communication would support adjustments in resting neuronal activity according to the status of perfusion pressure to the brain providing a protective negative feedback mechanism that balances energy supply with demands while protecting the brain from hyperperfusion and hypoperfusion.
The brain is vulnerable to blood volume and pressure fluctuations. Thus, a negative feedback mechanism that regulates resting neuronal activity with vascular flow/pressure changes would be beneficial. TRPV4 channels (Ca2+-permeable, nonselective cation channels expressed in numerous tissues), including brain (Plant and Strotmann, 2007), serve as molecular detectors and transducers of hemodynamic changes (Hill-Eubanks et al., 2014; Kim et al., 2015). TRPV4 channels have been implicated in multiple functions, including mechanosensation and vascular tone (Vriens et al., 2004; Plant and Strotmann, 2007; Earley et al., 2009). In the cerebral cortex, TRPV4 channels are strategically localized to astrocytic membranes, including structural subdomains termed endfeet (Benfenati et al., 2007, 2011), where they can be activated by hemodynamic stimuli (pressure/flow) (Kim et al., 2015). In addition to the well-defined functional implications of the astrocyte tripartite synapse (Araque et al., 1999; Pérez-Alvarez and Araque, 2013), which enables astrocyte signaling to modulate neuronal synaptic transmission, vessels can also modify basal astrocyte function (Kim et al., 2015), raising the question of whether hemodynamic-evoked astrocyte Ca2+ alters resting neuronal activity.
Using an in vitro model of pressurized/perfused PAs with simultaneous monitoring of astrocyte and neuronal function (Kim and Filosa, 2012; Kim et al., 2015) we provide evidence for vessel-to-neuron communication (Moore and Cao, 2008), defined here as vasculo-neuronal coupling (VNC). We show that local hemodynamic stimuli are sensed by perivascular astrocytes and transduced into changes in neuronal activity. Our data support TRPV4-dependent increases in astrocytic Ca2+ as a key step preceding astrocyte-to-neuron communication and suggest adenosine as the glial-derived signal involved in translating PA constriction into pyramidal neuron inhibition. Supporting a complex intercellular dynamic process, PA constriction was associated with activation of a subtype of GABAergic interneurons.
Materials and Methods
Brain slice preparation.
Cortical brain slices were prepared from male juvenile (P21-P28) Wistar rats or 6- to 10-week-old mice, including TRPV4+/+, TRPV4−/− (see Fig. 8) bred on a C57BL6 background (kindly provided by Dr. Wolfgang Liedtke), GLAST-CreERT2; R26-lsl-GCaMP3 mice obtained from breeding Tg (Slc1a3-cre/ERT)1Nat/J (The Jackson Laboratory, stock #012586) with B6;129S-Gt (ROSA)26Sor<tm38 (CAG-GCaMP3)Hze>/J (The Jackson Laboratory, stock #014538) (Otsu et al., 2015) (see Fig. 2) and FVB-Tg (GadGFP)45704Swn/J, GFP (and somatostatin) positive interneuron mice (GIN) (The Jackson Laboratory, stock #003718) (see Fig. 6C,D) following protocols approved by the animal care and use committee of Augusta University. Of note, GIN mice label a subset of somatostatin GABAergic interneurons in layers 2/3 with some additional cells in layer 5 (Oliva et al., 2000; Ma et al., 2006). Before experimentation, animals were housed in a room maintained at 20°C–22°C with a 12 h:12 h light-dark cycle and given ad libitum access to food and water. Following anesthesia with sodium pentobarbital, the brain was removed and cut into 250- to 300-μm-thick coronal slices using a vibratome (Leica VT 1200S, Leica Microsystems) in cold aCSF containing the following (in mm): KCl 3, NaCl 120, MgCl2 1, NaHCO3 26, NaH2PO4 1.25, glucose 10, CaCl2 2, and 400 μm l-ascorbic acid, with osmolarity at 300–305 mOsm, equilibrated with 95% O2-5% CO2. Slices were kept at room temperature in aCSF equilibrated with 95% O2-5% CO2, pH 7.4, until transferred to the microscopy chamber.
Figure 8.

TRPV4 channels contribute to VNC. A, Representative trace showing inhibition of pyramidal neuron firing activity following a modest increase in flow rate (0.1 to <0.5 μl/min, ↑Q) in the absence (left) and presence (middle) of the TRPV4 blocker HC067047 (3 μm); summary data of neuronal firing activity responses (far right). B, Representative trace showing inhibition of pyramidal neuron firing activity following ↑Q in the absence (left) and presence (middle) of the TRPV4 blocker RN1734 (50 μm); summary data of neuronal firing activity responses (far right) (A,B, rat experiments). C, Representative trace showing inhibition of pyramidal neuron firing activity following ↑Q from TRPV4+/+ mice (left) and summary data of neuronal firing activity from TRPV4+/+ mice and TRPV4−/− mice (right). Error bars indicate SEM. *p < 0.05 (one-way repeated-measures ANOVA followed by Dunnett's test). **p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test).
Figure 2.

Flow/pressure-evoked Ca2+ oscillations in cortical astrocytes from GLAST-CreERT2; R26-lsl-GCaMP3 mice. A, Representative image with ROIs for larger process (black trace) and MD (gray trace). Scale bar, 10 μm. B, Representative traces for Ca2+ oscillations for processes and MD. C, Onset time for astrocyte process and MD in response to increases in parenchymal arteriole flow (↑Q) in the presence or absence of the Na+ channel blocker TTX. D, Summary data showing Ca2+ peak amplitude before, during, and 2 min following the ↑Q in the presence or absence of TTX. E, Summary data showing Ca2+ frequency before, during, and 2 min following the ↑Q in the presence or absence of TTX. F, Summary data showing AUC before, during, and 2 min following the ↑Q in the presence and absence of TTX. Control and TTX experiments were conducted in different sets of animals; n = 4 mice for each group. Error bars indicate SEM. **p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test).
Figure 6.

Increased flow/pressure-evoked changes in interneuron firing activity. A, Representative square pulse trace showing various subtypes of interneuron firing pattern. B, Left, Representative trace and summary data showing flow/pressure-evoked inhibition of interneuron firing activity in response to a modest increase in Q (0.1 to <0.5 μl/min) from rat cortical slices. Right, Representative trace and summary data showing flow/pressure-evoked increases in interneuron firing activity following ↑Q from rat cortical slices. C, Immunostaining corresponding to a paired recording from a pyramidal neuron (arrowhead) and a GFP+ somatostatin neuron (arrow) from GIN mouse. Scale bar, 20 μm. D, Paired recording in current clamp showing action potential firing activity for a pyramidal neuron (left) and a GFP+ and somatostatin+ neuron (right) in response to increases in flow (↑Q). Error bars indicate SEM. *p < 0.05 (one-way repeated-measures ANOVA followed by Dunnett's test). **p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test).
Vessel cannulation.
Parenchymal arterioles were visualized using a 60× Nikon objective (NIR Apo, 60×/1.0w, DIC N2, ∞/0 WD 2.8) equipped with infrared differential interference contrast (DIC) optics. Cannulas (ID 1.17 mm and OD 1.50 mm, G150TF-3, Warner Instruments) were pulled with a micropipette puller (P-97 puller, Sutter Instruments) and mounted onto a micromanipulator, averaged cannula tip size was ∼8 μm. Luminal flow was controlled with a syringe pump (PHD 2000, Harvard Apparatus). A pressure transducer was placed just before the cannula for constant pressure monitoring (Servo Pump PS/200, Living System Instrumentation), as previously described (Kim and Filosa, 2012). The internal cannula solution consisted of the following (in mm): KCl 3, NaCl 135, MgCl2 1, glucose 10, HEPES 10, CaCl2 2, and 1% albumin (Duling et al., 1981) with osmolarity at 300–305 mOsm and pH 7.4 adjusted with NaOH. The tip of the cannula was maneuvered toward the entrance of the arteriole and slowly introduced into the vessel lumen (Kim and Filosa, 2012). Following cannula insertion, flow rate (Q) was increased to ∼0.1 μl/min to induce myogenic tone.
Calcium imaging.
Experiments were conducted using the Andor Revolution system (Andor Technology). A Nikon microscope (Eclipse FN 1) was connected to a laser confocal spinning unit (CSU-X1, Yokogawa) attached to a Sutter filter wheel and an ultrasensitive EMCCD camera (iXonEM, Andor Technology) (Kim and Filosa, 2012). The microscope chamber was continuously perfused with aCSF using a peristaltic pump (Miniplus 3, Gilson) at a rate of 2–3 ml/min. Chamber temperature was maintained at 36 ± 1°C using a single line solution heater (SH-28B, Warner Instruments) connected to a DC power supply (1735A, BK Precision). Calcium imaging experiments were conducted using both GLAST-CreERT2; R26-lsl-GCaMP3 mice (see Fig. 2) and Wistar rats (see Fig. 7). Expression of the Ca2+ indicator GCaMP3 was induced following 5 consecutive daily tamoxifen in oil (∼80 mg/kg) injections to 4- to 7-week-old mice; the brain was used 15–18 d after injections. Rat cortical brain slices were incubated at room temperature in aCSF containing 10 μm Fluo-4 AM and pluronic acid (2.5 μg/ml). Following a 2 h incubation period, slices were placed in room temperature aCSF until needed. Fluorescence images were obtained using a krypton/argon laser (488 nm excitation and >495 nm emission). Images were acquired at 2 frames/s for ∼10 min. In a few cases, we used a 20× objective to increase the field of view and added Alexa-555 (10 μm) to the cannula solution to visualized the cannulated PA and downstream vascular network (see Fig. 4).
Figure 7.
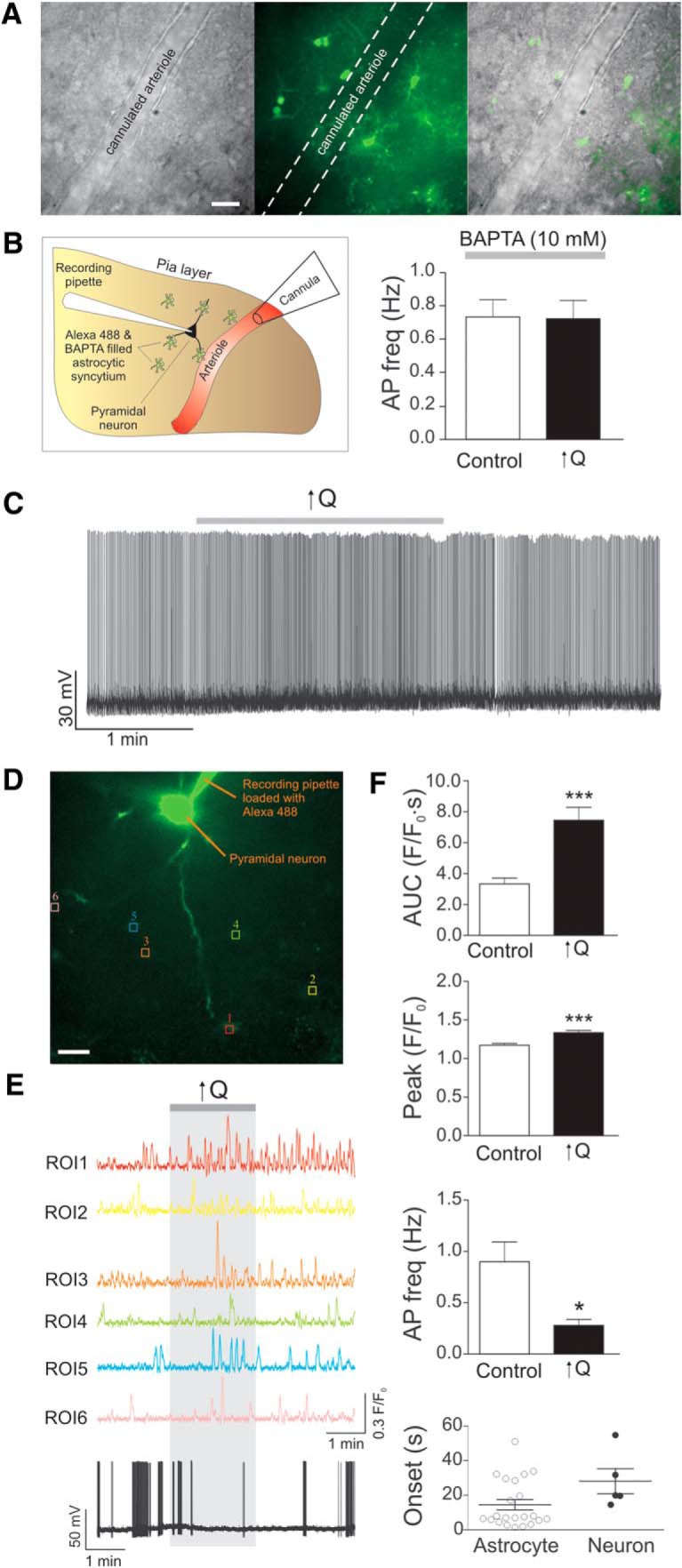
Astrocytes contribute to VNC. A, DIC image of a cannulated arteriole (left), fluorescence image of Alexa-488 diffused within the astrocytic syncytium (middle), and overlapped DIC/fluorescence image (right). Scale bar, 20 μm. B, Schematic representation (left), including cannulated arteriole, patched neuron, and astrocytes loaded with Alexa-488 and BAPTA (10 mm) and summary data showing changes in neuronal firing activity in response to ↑Q in the presence of BAPTA-loaded astrocytes. C, Representative trace showing neuronal firing activity in response to a modest increase in flow rate (0.1 to <0.5 μl/min, ↑Q) in the presence of BAPTA-loaded astrocytes. D, Confocal image of Fluo4-loaded astrocytes with corresponding ROIs and Alexa-488-loaded pyramidal neuron and recording pipette. Scale bar, 20 μm. E, Representative traces of astrocytic Ca2+ activity and neuronal firing activity in response to Q change. F, Summary data showing AUC, Ca2+ peak amplitude, neuronal firing activity, and onset times for neurons and astrocytes in response to ↑Q. Error bars indicate SEM. *p < 0.05 (paired t test). ***p < 0.0001 (paired t test).
Figure 4.

Flow/pressure-evoked changes in neuronal network activity. A, Representative DIC and fluorescence images showing the position of the patched neuron relative to the cannulated arteriole. Top row, Patched neuron ∼150 μm away from the cannulated arteriole. Bottom row, Patched neuron ∼300 μm away from the cannulated arteriole. Scale bar, 25 μm. B, Summary data of changes in neuronal firing rate induced by a modest increase in flow rate (0.1 to <0.5 μl/min, ↑Q) grouped according to proximity to the cannulated arteriole. C, Representative traces of diameter changes (top) in a cannulated arteriole and sEPSC (middle). Bottom, Expanded traces showing sEPSC before (left), during (middle), and after (right) ↑Q. D, Cumulative probability (Prob.) of sEPSC interevent interval (left) before (control), during (↑Q), and after (recovery) the flow/pressure stimulus and summary data of mean sEPSC frequency. E, Cumulative probability plot of amplitude (left) before (control), during (↑Q), and after (recovery) the flow/pressure stimulus and summary of mean sEPSC amplitude (right) Error bars indicate SEM. *p < 0.05 (paired t test). **p < 0.01 (paired t test). ***p < 0.0001 (paired t test).
Electrophysiology.
Cortical neurons and astrocytes in close proximity (<150 μm) to a PA within cortical layers III-IV were identified with a 60× water-immersion objective, and whole-cell current recordings obtained using an Axopatch 200B amplifier (Axon Instruments). Patch pipettes were made from thin-walled borosilicate glass (outer diameter 1.5 mm, internal diameter 0.86 mm; BF150–86-7.5, Sutter Instruments) and pulled (P-97 puller, Sutter Instruments) to resistances between 4 and 6 mΩ for neurons and 6–8 mΩ for astrocytes. The internal solution for whole-cell neuronal recordings consisted of the following (in mm): K+ gluconate 135, HEPES 10, EGTA 0.2, KCl 10, MgCl2 0.9, Mg2ATP 4, Na2GTP 0.3, phosphocreatine 20, with osmolarity 291–295 mOsm and pH adjusted to 7.2 with KOH. For Figures 5 and 6C, D experiments, we performed paired whole-cell recordings from two neurons with somas <100 μm from each other. For Figure 7A–C experiments, the internal solution for astrocytes recordings consisted of the following (in mm): K+ gluconate 130, HEPES 10, BAPTA 10, KCl 10, MgCl2 0.9, Mg2ATP 4, Na2GTP 0.3, phosphocreatine 20, added 100 μm Alexa-488, with osmolarity 291–295 mOsm and pH adjusted to 7.2 with KOH. For voltage-clamp recordings (see Fig. 4), to isolate spontaneous postsynaptic currents, we used an internal solution consisting of the following (in mm): Cs+ methanesulfonate 135, HEPES 10, MgCl2 1, EGTA 0.2, Mg2ATP 4, Na2GTP 0.3, phosphocreatine 20, QX-314 5, with osmolarity 291–295 mOsm and pH adjusted to 7.2 with KOH. To better understand and isolate spontaneous EPSCs, we bath applied the GABAA receptor antagonist picrotoxin (300 μm) and held membrane potential at −80 mV. EPSC peak amplitude and frequency were obtained and results plotted either as running averages (10 s bins) or as cumulative probability histograms. Current signals were filtered at 1 kHz low pass filter and digitized with a Digidata 1320 board (Axon Instruments). pClamp 10.2 (Axon Instruments) was used for data acquisition and storage. Pyramidal neurons were identified based on having a morphological triangular shape soma and presence of regular spiking activity with frequency adaptation following a depolarizing square pulse (van Aerde et al., 2015) (see, e.g.; Fig. 3C). Interneurons, on the other hand, were mainly determined based on having a nontriangular shape soma and a firing pattern response to a depolarizing/hyperpolarizing square pulse characteristic of that of an interneuron subtype (e.g., fast spiking, low threshold spike, inward rectification with hyperpolarization; see, e.g.; Fig. 6A) (Cauli et al., 2004; Sosulina et al., 2010; Lecrux et al., 2011). To further corroborate the identity of the interneuron subtype “activated” during flow/pressure responses, at the end of the electrophysiological recording, the slice was fixed and processed for immunohistochemical staining (see below). In addition, interneuron responses were further corroborated in GIN. In current-clamp mode, to induce action potential firing, neurons were slowly depolarized with DC current injection until threshold was reached and a stable firing activity was established. On average, the resting Vm of all rat pyramidal neurons recorded in current-clamp mode was −70.7 ± 0.5 mV and input resistance 110.1 ± 4 mΩ (n = 120). The averaged resting Vm for rat interneurons activated during flow/pressure increases was −69.3 ± 1.4 mV and input resistance 163.6 ± 8.4 mΩ (n = 10). The averaged resting Vm for rat interneurons inhibited during flow/pressure increases was −70 ± 2.3 mV and input resistance 226.7 ± 37.5 mΩ (n = 6). For mice, the averaged resting Vm of all pyramidal neurons recorded in current-clamp mode was −71.9 ± 1 mV and input resistance 150.2 ± 9.8 mΩ (n = 14). The averaged resting Vm for somatostatin-positive interneurons was −63.1 ± 0.9 mV and input resistance 417.5 ± 19.6 mΩ (n = 7). Values were not corrected for junction potential offset.
Figure 5.

Paired pyramidal neuron recordings in response to increased PA flow/pressure. A, Schematic representation of the dual recording setup in addition to arteriole cannulation. B, Summary data showing pyramidal neuron 1 and neuron 2 firing activity in response to increased flow/pressure within a PA under basal conditions (control), during the stimulus (↑Q), and 2 min after the stimulus. C, Representative trace showing simultaneous pyramidal neuron 1 and neuron 2 firing activity in response to increased flow rate (↑Q). Error bars indicate SEM. *p < 0.05 (one-way repeated-measures ANOVA followed by Dunnett's test). **p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test).
Figure 3.

Increased flow/pressure-evoked inhibition of cortical pyramidal neuron activity. A, Representative DIC images of a cannulated arteriole (a), a patched neuron next to a cannulated arteriole (different slice from a) (b), and the corresponding confocal fluorescence image of the neuron loaded with Alexa-488 and the arteriole perfused with Alexa-555 (c); the same slice was also fixed and imaged after the experiment to better show the morphology of the neuron (d). Scale bar, 25 μm. **Cannula. *Recording pipette. B, Left, Schematic representation corresponding to Aa–Ad. Right, Representative trace of arteriolar diameter changes before and after changes in Q from 0.1 to 0.3 μl/min (right). a, Time at which the cannula is introduced into the arteriole lumen. b, Diameter difference corresponding to the development of myogenic tone. c, Diameter difference corresponding to flow/pressure-induced constriction. C, Representative trace showing pyramidal neuron firing pattern in response to a square pulse depolarization (far left) and changes in action potential frequency in response to a modest increase in flow rate (0.1 to <0.5 μl/min, ↑Q) (middle left). Summary data showing flow/pressure-induced changes in action potential frequency following modest ↑Q (middle right) and higher (0.1 to >0.5 μl/min, ↑↑↑Q) Q increases (far right). D, Representative trace of repetitive ↑↑↑Q-induced inhibition of firing activity in a pyramidal neuron. Error bars indicate SEM. *p < 0.05 (one-way repeated-measures ANOVA followed by Dunnett's test). **p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test).
Immunofluorescence.
Whenever possible, immunofluorescence was used for further identification of the recorded interneuron (Cauli et al., 2004). The internal solution for electrophysiological recordings included 2% biocytin. At the end of the experiment, the cortical slice was fixed in 4% PFA PBS (4% PFD) for 24 h at 4°C. Following fixation, slices were incubated in 0.3% Triton X-100, 0.04% NaN3, and 10% normal horse serum for 2 h. For immunofluorescence reactions, slices were incubated for 48 h in goat polyclonal anti-somatostatin (D-20) (1:50, SC-7819, Santa Cruz Biotechnology), rabbit polyclonal anti-GFP (1:1000, A6455, Molecular Probes). Reactions with primary antibodies were followed by a 4 h incubation with secondary antibodies (1:50 anti-goat Alexa-647 and 1:250 anti-rabbit Alexa-488, Jackson ImmunoResearch Laboratories) and 1 μg/ml streptavidin-Cy3 (016-160-084, Sigma) in PBS containing 0.3% Triton X-100 and 0.04% NaN3. Mounted slices were imaged using a LSM510 confocal microscope (Zeiss Microimaging).
Drugs and chemicals.
All drugs, with the exception of Fluo 4 AM, Alexa-488, Alexa-555 (Invitrogen), RN1734, HC067047, gabazine, DPCPX, l-NAME, CGP52432 and CGP55845 (Tocris Bioscience), 4-α-PDD (LC Laboratories), TTX (Abcam), BSA (Fisher Scientific), U46619 (Cayman Chemical), and carboxy-PTIO (Enzo Life Sciences) were purchased from Sigma. Drugs were bath applied, unless otherwise specified.
Data analysis.
Analysis for electrophysiological recordings was performed using Clampfit 10.2, pClamp version 10.2 (Axon Instruments) and Mini analysis software (Synaptosoft, version 6.0.7). Ca2+ imaging in bulk-loaded Wistar brain slices was analyzed using Sparkan software (Adrian Bonev, University of Vermont). Fractional fluorescence (F/Fo) was determined by dividing the fluorescence intensity (F) within a region of interest (ROI) by a baseline fluorescence value (Fo) determined from ∼20 images showing no activity. The frequency of Ca2+ oscillations was determined within a ROI (10 × 10 pixels or 2.5 × 2.5 μm) on a cell exhibiting Ca2+ oscillations. To detect Ca2+ oscillations in brain slices from GLAST-CreERT2; R26-lsl-GCaMP3 mice (see Fig. 2), we used GECIquant software developed by the Khakh laboratory (Srinivasan et al., 2015). Approximately 35% of cortical astrocytes express GCamP3 in these mice (Paukert et al., 2014). To better understand Ca2+ events in astrocytic processes in response to flow/pressure changes, we measured events in larger processes comprising a defined area range between 5 and 30 μm2 and in microdomains comprising a defined area range from 0.5–4 μm2) as previously described (Srinivasan et al., 2015) (see Fig. 2). GECIquant automatically detects ROIs. For each ROI, basal F was determined during 30 s periods having no Ca2+ oscillations.
GraphPad software was used for all statistical analyses. All values are expressed as mean ± SEM. Experiments were conducted a minimum of four times in at least three different rats/mice, except for GIN mice where 2 mice were used. Differences between two means within groups were determined using Student's paired t test. Multiple within-group comparisons with control were evaluated using repeated-measures, one-way ANOVA with Dunnet's multiple-comparison post test (see Figs. 2; Fig. 5). Statistical significance was tested at 95% (p < 0.05) confidence level.
Results
Hemodynamic-evoked Ca2+ increases in astrocyte processes
We previously reported that increases in flow (and pressure) within PAs elevate Ca2+ in astrocyte somas (Kim et al., 2015). Here, using GLAST-CreERT2; R26-lsl-GCaMP3 mice, we found that increased PA tone caused an increase in Ca2+ dynamics in astrocyte processes. Vascular tone was increased by increasing flow rate (Q), and thus pressure, within perfused and pressurized PAs (Kim and Filosa, 2012) (Fig. 1). Specifically, increasing Q from 0.1 to 0.5 μl/min increased Ca2+ peak amplitude and frequency within both larger processes and fine structures, defined here as microdomains (MDs) (Fig. 2). In a previous study (Kim et al., 2015), we reported flow/pressure-evoked Ca2+ changes in astrocyte soma. Here we focused on the flow/pressure-evoked Ca2+ response of astrocytic processes. In larger processes, Ca2+ peak amplitude and frequency increased significantly during the stimulus (amplitude, p < 0.01; frequency, p < 0.01, n = 57). In MDs, the increases in Ca2+ peak amplitude during the stimulus were even larger, as were the corresponding increases in frequency (amplitude, p < 0.01; frequency p < 0.01, n = 148) (Fig. 2D,E). Consistent with the observed changes in Ca2+ peak amplitude and frequency, increasing Q (and pressure) also increased the area under the curve (AUC) in both larger processes (p < 0.01, n = 57) and within MDs (p < 0.01, n = 148) (Fig. 2F). The average response-onset time following the change in Q was 17.9 ± 1.7 s (n = 57) for larger processes and 18.43 ± 1.03 s (n = 148) for MDs (Fig. 2C). To evaluate whether flow/pressure-evoked increases in astrocyte Ca2+ were action potential, dependent experiments were repeated in the presence of TTX (1 μm). We observed comparable responses to those observed in the absence of TTX. In larger processes, Ca2+ peak amplitude, frequency, and AUC increased significantly during the stimulus (amplitude, p < 0.01; frequency, p < 0.01; AUC, p < 0.01, n = 46). In MDs, increased Ca2+ peak amplitude, frequency, and AUC during the stimulus were also significantly increased (amplitude, p < 0.01; frequency, p < 0.01; AUC, p < 0.01, n = 137) (Fig. 2D–F). The averaged response onset was also comparable: 18.8 ± 2.3 s for larger processes (n = 46) and 18.3 ± 0.9 for MDs (n = 137). Interestingly, under control and in the presence of TTX, Ca2+ increases persisted even 2 min following the increased flow/pressure stimulus. Together, these data support vessel-to-astrocyte signaling.
Figure 1.

Parenchymal arteriole myogenic tone. A, Representative arteriolar diameter (solid), shear stress (dashed), and luminal pressure changes (bottom) from a cannulated parenchymal arteriole in response to increases in flow rate (Q), and pressure, in the absence and then presence of TTX (1 μm). B, Summary data of flow/pressure-evoked changes in myogenic tone in the absence and presence of TTX. Error bars indicate SEM.
Flow/pressure evokes changes in cortical neuronal activity
Our previous data indicate that increases in flow/pressure within PAs elevate astrocyte intracellular Ca2+ (Kim et al., 2015). It is unclear, however, whether this response extends to changes in neuronal activity. Using a cannulated-arteriole brain slice preparation developed in our laboratory, we assessed cortical neuronal activity in response to increases in luminal flow/pressure in Wistar rats. Myogenic tone persisted in the presence of the sodium channel blocker TTX (1 μm), indicating that flow/pressure-evoked increases in vascular tone were independent of neuronal activity (Fig. 1). Following an equilibration period to establish myogenic tone, we patched a pyramidal neuron near the arteriole and monitored its membrane potential (Vm) and firing activity over time. Pyramidal neurons were identified based on their electrophysiological properties (Fig. 3C) and morphology, characterized by a long ascending apical dendrite and a triangular-shaped soma (Fig. 3A).
In response to a modest increase in Q (from 0.1 to ≤0.5 μl/min), which increases vascular tone (Kim and Filosa, 2012; Iddings et al., 2015; Kim et al., 2015), neuronal firing rate significantly decreased (p < 0.01, n = 12) (Fig. 3C), responses persisted following the stimulus. Similar responses were observed with larger increases in Q (from 0.1 to > 0.5–0.8 μl/min; p < 0.01, n = 7) (Fig. 3C,D). As shown in Fig. 3D, flow/pressure-evoked neuronal responses were repeatable and reproducible. As noted in Figure 3 C, D, increased Q responses persisted beyond the stimulus time.
To gain insight into the spatial extent of VNC, we recorded from neurons located at different distances (∼150 and 300 μm) from the cannulated PA. Neurons recorded ∼150 μm from the cannulated PA were always surrounded by perfused vessels (primarily capillaries), as determined by the presence of Alexa-555, added to the arteriole lumen solution (Fig. 4A). In contrast, perfusion was not evident in vessels surrounding neurons recorded ∼300 μm from the cannulated PA (Fig. 4A). Consistent with this difference in perfusion, flow/pressure-induced inhibition of pyramidal neuronal firing activity was consistently observed in neurons located ∼150 μm from the cannulated PA (Fig. 4A, top row, B) but absent in neurons recorded ∼300 μm away from the cannulated vessel (Fig. 4A, bottom row, B). These data are consistent with the idea that flow/pressure-evoked pyramidal neuron responses are dependent upon the presence of a perfused vessel in the proximity of the recorded neuron.
Because our approach is limited to one neuronal recording per cannulated PA (and its downstream vascular network), we monitored the degree of spontaneous EPSCs (sEPSCs) in patched pyramidal neurons as an indirect index of the degree of local neuronal network activity. Increased flow/pressure-evoked arteriole constriction (Fig. 4C, top) resulted in a small and transient inhibition of sEPSC frequency (ΔHz = −0.81 ± 0.15, p < 0.001, n = 8; Fig. 4C,D) and amplitude (ΔpA = −0.67 ± 0.18, p < 0.01, n = 8; Fig. 4C,E). Importantly, these voltage-clamp studies revealed that flow/pressure-induced constrictions resulted in a transient outward shift in the holding current (Δ13.2 ± 2.33 pA, p < 0.001, n = 8). Based on the pyramidal neuron input resistance from current-clamp recordings (110.1 ± 4 ΜΩ, n = 120), this outward shift likely contributes to the membrane hyperpolarization observed in current-clamp mode. Together, our results suggest that flow/pressure-evoked increases in vascular tone decrease local neuronal network activity. To provide further support for a neuronal network response to increases in vascular tone, we determined flow/pressure-evoked neuronal responses using paired whole-cell recordings (Fig. 5). Increasing flow/pressure within the PA inhibited pyramidal neuron firing activity (neuron 1, Δ firing rate = −0.32 ± 0.09 Hz, p < 0.05; neuron 2, Δ firing rate = −0.56 ± 0.18 Hz, p < 0.05; n = 4 experiments (Fig. 5B,C).
The intercommunication between blood vessels and pyramidal neurons could arise from direct vessel-to-pyramidal neuron signaling and/or indirectly via the action of interneurons and/or astrocytes, the latter enwrap blood vessels and contact synapses (Bushong et al., 2002). Thus, we first assessed the effects of flow/pressure-evoked vascular responses on interneuron activity. Interneurons were identified based on their lack of a triangular shape soma and electrophysiological properties following a depolarization/hyperpolarization square pulse (Fig. 6A). In contrast to pyramidal neurons, interneurons exhibited mixed responses to flow/pressure increases, with some showing inhibition of firing activity (p < 0.05, n = 6; Fig. 6B), and the remainder showing activation (p < 0.01, n = 10; Fig. 6B,D). Because fast spiking interneurons were mainly associated with flow/pressure-evoked inhibition, we expanded these studies by measuring interneuron responses from somatostatin GFP+ mice (GIN) (Fig. 6C). All SOM+ neurons responded to flow/pressure increases with an increase in firing activity (from 0.86 ± 0.2 to 1.34 ± 0.3 Hz, n = 7) (Fig. 6D), significance was however delayed (1.65 ± 0.5 Hz, p < 0.5, averaged value 2 min following the stimulus, n = 7). The data support somatostatin neurons as contributors of VNC.
To determine whether astrocytes contribute to vascular-to-neuronal signaling, we next tested whether introduction of the fast Ca2+ chelator BAPTA (10 mmol/L) and Alexa-488 (100 mol/L) into the astrocytic syncytium via a patch pipette altered vascular-to-neuronal coupling (Fig. 7A). We found that blunting Ca2+-dependent signaling in cortical astrocytes completely abrogated increased flow/pressure-induced inhibition of neuronal firing activity (n = 5, p = 0.68; Fig. 7B,C). Simultaneous measurements of astrocyte Ca2+ and neuronal activity during increases in PA flow/pressure revealed first an increase in the Ca2+ astrocyte response (14.54 ± 2.95 s, n = 22) followed by inhibition of pyramidal neuron activity (28.15 ± 7.23 s, n = 5) (Fig. 7F). The increase in astrocyte Ca2+ peak amplitude and AUC (Fig. 7E,F) was followed by a significant decrease in neuronal firing activity (n = 5, p = 0.013; Fig. 7E,F).
TRPV4 channels are key mediators of VNC
We previously showed that flow/pressure-evoked astrocyte Ca2+ increases are partially mediated by TRPV4 channels (Kim et al., 2015). Thus, we next sought to determine whether TRPV4 channels also contribute to flow/pressure-evoked changes in rat pyramidal neuron firing activity. Flow/pressure-induced neuronal responses were tested in the same neuron in the absence and then presence of the TRPV4 channel blocker HC067047 (3 μm) or RN1734 (50 μm). Flow/pressure-evoked decreases in neuronal firing activity were suppressed by TRPV4 channel blockade with HC067047 (n = 5) (Fig. 8A) and RN1734 (n = 6) (Fig. 8B).
To corroborate findings from rats, we also measured pyramidal neuron activity in response to increases in Q using TRPV4+/+ and TRPV4−/− mice. Increases in Q inhibited pyramidal neuron firing activity (p < 0.01, n = 6) in TRPV4+/+ mice but had no significant effect in TRPV4−/− mice (Fig. 8C). Together, these experiments support the critical involvement of TRPV4 channels in mediating VNC in the cortex.
Adenosine mediates astrocyte to neuronal signaling during VNC
Suppression of neuronal activity in response to increases in vascular tone suggests the release of an inhibitory signal. To explore the identity of such a signal, we perfused slices with blockers of four of the most likely candidates: nitric oxide (NO), released from the endothelium in response to increases in shear stress; GABA, released from interneurons; glutamate, released from pyramidal neurons and/or astrocytes; and ATP/adenosine, released from astrocytes. Using rat brain slices, whenever possible, neuronal responses to increases in PA flow/pressure were tested in the same neuron before and after the application of blockers. The NOS blocker l-NAME (100 μm) and NO scavenger carboxy-PTIO (100 μm) had no effect on neuronal responses (n = 9) (Fig. 9A). Similarly, flow/pressure-induced neuronal responses persisted in the presence of the glutamate receptor blocker kynurenate (2 mm) (n = 6) and the GABAA receptor blocker gabazine (10 μm) (n = 6) (Fig. 9B,C). Responses were, however, significantly blocked (p > 0.05, n = 6) by the presence of GABAB receptor blockers (CGP52432 and CGP55845, 5 μm) (Fig. 9D). Consistent with the possibility of interneuron-pyramidal neuron signaling, these result points to GABA as a contributing signal in VNC.
Figure 9.

Possible signals contributing to VNC. A, Representative trace showing changes in neuronal firing activity in response to a modest increase in flow rate (0.1 to <0.5 μl/min, ↑Q) before (left) and after (middle) application of the NO synthase inhibitor l-NAME (100 μm) and NO scavenger carboxy-PTIO (100 μm). Summary data showing changes in neuronal firing frequency in response to ↑Q in the absence (aCSF) or presence of l-NAME and c-PTIO (right); responses were measured before and during the stimulus. B, Representative trace showing changes in neuronal firing activity in response to ↑Q in the presence of glutamate receptor antagonist, kynurenate (2 mm) (left). Summary data showing changes in neuronal firing frequency in response to ↑Q in the presence of kynurenate (right). C, Representative trace showing changes in neuronal firing activity in response to ↑Q before (left) and after (middle) application of the GABAA receptor antagonist gabazine (10 μm). Summary data showing changes in neuronal firing frequency in response to ↑Q in the absence (aCSF) or presence of gabazine (right). D, Representative trace showing changes in neuronal firing activity in response to ↑Q before (left) and after (middle) application of GABAB receptor antagonists CGP 52432 and CGP 55845 (5 μm). Summary data showing changes in neuronal firing frequency in response to ↑Q in the absence (aCSF) or presence of gabazine (right). Error bars indicate SEM. *p < 0.05 (one-way repeated-measures ANOVA followed by Dunnett's test). **p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test).
Given our previous demonstration that flow/pressure-evoked astrocytic responses and subsequent changes in vascular tone may result from the release of a purinergic signal (Kim et al., 2015), we also measured flow/pressure-evoked neuronal responses in the presence of the ecto-ATPase inhibitor/ATP analog, ARL67156 (50 μm), and the adenosine A1 receptor blocker DPCPX (200 nm). Flow/pressure-evoked neuronal responses were unaffected by ARL67156 (n = 6) (Fig. 10A). However, flow/pressure-evoked neuronal responses were significantly blunted by the adenosine A1 receptor blocker DPCPX (n = 7) (Fig. 10B). These results support the involvement of adenosine, and not ATP, in astrocyte-to-neuronal signaling during VNC.
Figure 10.

Adenosine mediates astrocyte-to-neuron communication during VNC. A, Representative trace showing changes in neuronal firing activity in response to ↑Q in the absence (left) or presence (middle) of the ecto-ATPase inhibitor/ATP analog ARL67156 (50 μm). Summary data showing changes in neuronal firing frequency in response to ↑Q in the presence of ARL67156 (right). B, Representative trace showing changes in neuronal firing activity in response to ↑Q before (left) and after (middle) application of the A1 adenosine receptor antagonist DPCPX (200 nm). Summary data showing changes in neuronal firing frequency in response to ↑Q in the absence (aCSF) or presence of DPCPX (right). Error bars indicate SEM. *p < 0.05 (one-way repeated-measures ANOVA followed by Dunnett's test). **p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test).
The above experiments would suggest that increased flow/pressure-evoked PA constriction inhibits pyramidal neuron firing activity. Considering the importance of this mechanism as a homeostatic process, we then determined whether reversing the polarity of the vascular stimulus, decreasing flow/pressure within PAs resulting in dilations, altered resting pyramidal neuron firing activity. Following an equilibration period to establish myogenic tone, flow/pressure within the PA was decreased (from ∼0.3 to 0.1 μl/min), simultaneous pyramidal neuron recordings were conducted throughout the experiment. Decreasing flow within PAs significantly dilated arterioles (p < 0.001, n = 4) and increased pyramidal neuron firing activity (n = 4) (Fig. 11). Opposite neuronal responses (activation vs inhibition) upon reversal of the vascular response (dilation vs constriction) support the presence of a negative feedback pathway establishing pyramidal neuron resting activity relative to PA vascular tone.
Figure 11.

Decreased flow/pressure-evoked activation of cortical pyramidal neuron firing activity. A, Left, Representative trace showing pyramidal neuron action potential frequency in response to a decrease in flow rate (0.3 to <0.1 μl/min, ↓Q) within a cannulated parenchymal arteriole. Bottom, Corresponding arteriole diameter changes. Right, Summary data showing decreased flow/pressure-induced increases in action potential frequency following ↓Q. Error bars indicate SEM. *p < 0.05 (one-way repeated-measures ANOVA followed by Dunnett's test). ***p < 0.01 (one-way repeated-measures ANOVA followed by Dunnett's test). B, Averaged Δtone (percentage) in response to flow/pressure decreases within parenchymal arterioles. Error bars indicate SEM. **p < 0.01 (paired t test).
Discussion
The present study provides supportive evidence for vessel-to-neuron signaling in the neurovascular unit as proposed by Moore and Cao (2008). Moreover, we demonstrate in brain slices that changes in vascular tone altered astrocyte and neuronal activity, a process referred to here as VNC (Fig. 12). Specifically, we show that increased flow/pressure within perfused PAs increased astrocytic Ca2+ and inhibited cortical pyramidal neuron activity. On the other hand, decreases in PA tone increased cortical pyramidal neuron activity. Experiments addressing the time course of VNC (Fig. 7) support sequential vessel-to-astrocyte-to-neuron signaling. Corroborating previous findings (Kim et al., 2015), we identified astrocytic mechanosensitive TRPV4 channels as important molecular players in VNC. The present study was mainly focused on increased PA flow/pressure-evoked astrocyte and neuronal responses. Future studies addressing the cellular mechanisms underlying decreased flow/pressure-evoked PA dilation and concomitant increases in pyramidal neuronal activity are needed.
Figure 12.
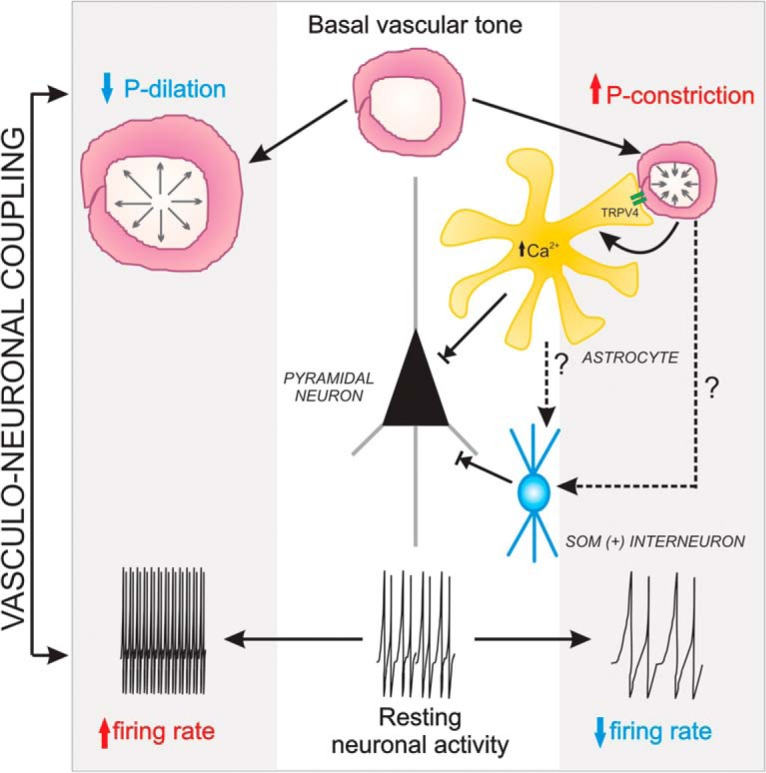
VNC working model. Middle, Resting conditions for both pyramidal neuron activity and parenchymal arteriole (steady-state myogenic tone). Increased luminal pressure within PAs increases PA vascular tone (constriction), which evokes an increase in astrocyte Ca2+ via TRPV4 channels. As previously demonstrated (Kim et al., 2015), the rise in astrocyte Ca2+ contributes to the maintenance of vascular tone during the myogenic response. In addition, astrocytes release ATP/adenosine via a Ca2+-dependent mechanism contributing to inhibition of pyramidal neuron firing activity. According to BAPTA experiments shown in Figure 7, we anticipate pressure-evoked PA constrictions to evoke astrocytes-to-interneuron signaling (putatively somatostatin interneurons), further contributing to the modulation of pyramidal neuron resting activity. To the left, we show the expected arteriole dilation resulting from a decrease in flow/pressure. However, under these conditions, pyramidal neuron firing activity is increased and the cellular signaling mechanisms underlying this response was still to be determined.
In 2008, Moore and Cao (Moore and Cao, 2008; Cao et al., 2009) proposed the hemo-neuronal hypothesis, which posits that signals derived from the microcirculation can modify astrocyte and neuronal function contributing to information processing in the brain. The idea that signals from blood vessels can alter neuronal activity is supported by studies showing that stretching of the vascular wall induces astrocyte GABA release (Kozlov et al., 2006) and that angiotensin II-induced, endothelium-derived NO alters brainstem neuronal activity (Waki et al., 2006). In vivo data from our previous study (Kim et al., 2015) showed that systemic administration of phenylephrine, which increases arterial blood pressure and evokes cerebral autoregulation (i.e., PA vasoconstriction) (Niwa et al., 2002; Ayata et al., 2004), increased astrocytic Ca2+ to levels comparable with those observed in vitro. Data from this study, together with our previous findings (Kim et al., 2015), thus support reverse flow of information at the neurovascular unit (from vessel-to-astrocyte/neuron). However, whether VNC is involved in brain information processing (Moore and Cao, 2008) is still to be determined as NVC-mediated responses were not assessed in this study. Rather, we suggest VNC as a constitutive mechanism contributing to the modulation of resting neuronal activity according to the levels of PA vascular tone. We anticipate that, in the healthy brain, VNC is overridden during increases in neuronal activity, such as those evoked during NVC.
Our findings indicate that changes in baseline vascular tone rapidly (i.e., seconds) signal the state of perfusion to pyramidal neurons, which in turn respond with a change in firing activity. In the specific setting of flow/pressure-evoked constriction, this results in inhibition of pyramidal neuron firing activity and sEPSCs. Our data support a role for astrocytes as key intermediaries in vessel-to-neuronal signaling. Astrocytes transduced changes in intravascular pressure into increased Ca2+ activity both in the current study and our previous published data (Kim et al., 2015), an increase that preceded neuronal responses. Importantly, flow/pressure-evoked neuronal responses were blunted by loading the astrocytic syncytium with BAPTA, supporting the conclusion that flow/pressure-evoked inhibition of pyramidal neuron firing activity is dependent on astrocytic Ca2+ signaling. Consistent with our previous demonstration (Kim et al., 2015), we found that pharmacological blockade of TRPV4 channels blunted flow/pressure-induced neuronal inhibition; responses were also significantly reduced in TRPV4−/− mice compared with TRPV4+/+ mice. Interestingly, decreases in PA flow/pressure did not seem to evoke an increase in astrocyte Ca2+ (data not shown), suggesting that a different mechanism may operate during flow/pressure-evoked PA dilation and increases of pyramidal neuron resting activity.
Suppression of pyramidal neuron activity following increased flow/pressure-mediated astrocyte activation suggests that a gliotransmitter is released during VNC. In agreement with the suggestion that adenosine serves as a neuroprotective gliotransmitter (Carmignoto and Haydon, 2012) and modulator of excitatory synaptic transmission (Zhang et al., 2003; Pascual et al., 2005; Serrano et al., 2006; Panatier et al., 2011; Zorec et al., 2012), we provide evidence that flow/pressure-induced neuronal inhibition involves the activation of A1R. A perplexing finding from our study, however, is that while the main adenosine neuronal effect is inhibitory (van Aerde et al., 2015), on the vasculature adenosine acts as a potent vasodilator (Pelligrino et al., 2011). It is possible, however, that the adenosine-evoked suppression of pyramidal neuron activity requires concentrations below those needed to elicit arteriole dilations and override ongoing myogenic mechanisms contributing to PA constrictions. In addition, the polarity of the adenosine-evoked vascular response (dilation vs constriction) may also depend on the adenosine receptor subtype (A1 vs A2) targeted during this process (Guan et al., 2014). In contrast to the results of Panatier et al. (2011), who reported that astrocyte-derived adenosine mediates upregulation of synaptic transmission via A2 adenosine receptor activation (Panatier et al., 2011), flow/pressure-induced neuronal inhibition was dependent on A1 receptor activation, suggesting that the stimulus type targeting astrocytes may dictate the overall neuronal response outcome as well as the possibility that distinct purinergic signaling mechanisms may operate at the endfoot versus synapse side of the astrocyte.
Increased PA flow/pressure evoked an outward current in pyramidal neurons. These results are in agreement with findings from Newman (2003) showing that astrocyte-derived adenosine inhibits retinal neurons via activation of Kir channels. Although our results are consistent with the interpretation that adenosine acts as a primary signal during flow/pressure-evoked changes in pyramidal neuron inhibition, other signaling candidates should not be ruled out, as the hemodynamic-induced stimulus is complex and likely involves multiple glial, vascular, and/or neuronal signals (e.g., K+, arachidonic acid metabolites, polyamines, and endocanabinoids). To this end, a number of endothelium-mediated signaling pathways could be activated during PA adjustments in vascular tone, resulting in the release of signals that can readily cross the blood–brain barrier and/or alter the activity of adjacent cells via ion channels and/or myoendothelial junctions (Andresen et al., 2006; Longden et al., 2011; De Bock et al., 2013). The observation that pyramidal neuron inhibition persisted in the presence of gabazine and kynurenate suggests that synaptic transmission is not essential for this form of communication. However, because a subpopulation of GABAergic interneurons was activated during PA constrictions, a modulatory role by these cells is likely. To this end, it is unclear whether changes in vascular reactivity directly activate GABAergic interneurons or whether these receive signals from astrocytes. Moreover, it is also unclear whether the subpopulation of activated GABAergic interneurons during PA constrictions (i.e., somatostatin) contribute to the maintenance of the vascular response by increasing arteriole tone (Long et al., 1992; Cauli et al., 2004) and/or signal pyramidal neurons to decrease firing activity. Answers to these questions are needed to better understand how interneurons shape the VNC response.
The idea that increased vascular tone can alter resting neuronal activity becomes important in the context of diseases known to impair cerebral autoregulation (e.g., hypertension) (de la Torre, 2012; Dunn and Nelson, 2014) as these could influence the state of baseline perfusion and resting neuronal activity compromising important brain homeostatic processes. As previously eluted, we did not evaluated whether VNC alters the NVC response following neuronal stimulation. Interestingly however, a study by Dunn et al. (2013) showed that disruption of a key player in VNC, TRPV4 channels, altered NVC-mediated responses. The authors proposed a bidirectional synergy between IP3Rs and TRPV4 channels, which enhanced astrocyte endfoot Ca2+ responses, contributing to the magnitude of the NVC response (Dunn et al., 2013). It is thus possible that changes in TRPV4 channel function may alter constitutive VNC signaling, which in turn affect the NVC response. Along these lines, Zhang et al. (2013) demonstrated impaired TRPV4-mediated acetylcholine-induced dilations in cerebral vessels from Alzheimer's disease mice where cerebral hypoperfusion and impaired vessel-to-neuronal signaling (VNC) are at play (Zlokovic, 2005; Nicolakakis and Hamel, 2011; de la Torre, 2012; Hébert et al., 2013; Iadecola, 2013; Rosenberg et al., 2016; Yang et al., 2016). While these two studies focused on the role TRPV4 channels have on vasodilation, it becomes apparent that impairments in TRVP4 channel function could compromise flow of information at the neurovascular unit impairing CBF regulation. Future studies addressing how alterations in the balance between steady-state perfusion to the brain and resting neuronal activity impact NVC and may contribute to cognitive dysfunction are needed.
The functional significance of VNC is currently unclear. However, it raises the question of whether the intrinsic properties of the brain are further modulated by minute-to-minute systemic changes in blood pressure that define arteriole reactivity. We propose that VNC serves as a constitutive protective negative feedback mechanism that balances energy supply with demands while protecting the brain against hyperperfusion, blood–brain barrier breakdown, and edema formation on the one hand, and hypoperfusion and ischemia on the other. Under physiological conditions, where cerebral autoregulation mechanisms are operating efficiently, graded adjustments in neuronal activity triggered by the release of adenosine may serve as a signal encoding the various degrees of vascular tone, thus providing a mechanism that enables the cortex to monitor changes in brain perfusion pressures. This mechanism may be compromised in cases where cerebral autoregulation is impaired, such as ischemic stroke, traumatic brain injury, and hypertension, thereby increasing brain vulnerability to the deleterious effects of high/low perfusion pressures. Thus, understanding the consequences that changes in perfusion/pressure have on glial and neuronal function is essential as these may constitute unexplored parameters that contribute to the resetting of resting brain activity.
In conclusion, we show that increased flow/pressure-induced PA vasoconstriction (Kim and Filosa, 2012) augmented Ca2+ signaling in cortical astrocytes and decreased cortical pyramidal neuron activity. Conversely, decreased flow/pressure-evoked PA dilation increased resting pyramidal neuron activity. In addition, we present evidence that, during flow/pressure-evoked PA vasoconstrictions, astrocyte TRPV4 channels and adenosine are key molecular players in VNC. Together, our results support the existence of a physiological process by which the reverse flow of information, from vascular elements to neurons, occurs within the neurovascular unit. We propose that this process serves as a neuroprotective function to dynamically modulate resting neuronal function according to the status of vascular tone in penetrating cerebral arterioles.
Footnotes
This work was supported by National Heart, Lung, and Blood Institute, National Institutes of Health Grant R01 HL089067-02 to J.A.F. and American Heart Association predoctoral fellowship 11PRE7400037 to J.A.I. We thank Dr. Javier Stern for help with electrophysiological experiments.
The authors declare no competing financial interests.
References
- Andresen J, Shafi NI, Bryan RM., Jr Endothelial influences on cerebrovascular tone. J Appl Physiol. 2006;100:318–327. doi: 10.1152/japplphysiol.00937.2005. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/S0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy-OZdemir Y, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Benfenati V, Amiry-Moghaddam M, Caprini M, Mylonakou MN, Rapisarda C, Ottersen OP, Ferroni S. Expression and functional characterization of transient receptor potential vanilloid-related channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience. 2007;148:876–892. doi: 10.1016/j.neuroscience.2007.06.039. [DOI] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A. 2011;108:2563–2568. doi: 10.1073/pnas.1012867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Higashikubo BT, Cardin J, Knoblich U, Ramos R, Nelson MT, Moore CI, Brumberg JC. Pinacidil induces vascular dilation and hyperemia in vivo and does not impact biophysical properties of neurons and astrocytes in vitro. Cleve Clin J Med. 2009;76(Suppl 2):S80–S85. doi: 10.3949/ccjm.76.s2.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Haydon PG. Astrocyte calcium signaling and epilepsy. Glia. 2012;60:1227–1233. doi: 10.1002/glia.22318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J, Hamel E. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci. 2004;24:8940–8949. doi: 10.1523/JNEUROSCI.3065-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ. The cerebral circulation. San Rafael, CA: Morgan and Claypool Life Sciences; 2009. [PubMed] [Google Scholar]
- de la Torre JC. Cardiovascular risk factors promote brain hypoperfusion leading to cognitive decline and dementia. Cardiovasc Psychiatry Neurol. 2012;2012:367516. doi: 10.1155/2012/367516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock M, Wang N, Decrock E, Bol M, Gadicherla AK, Culot M, Cecchelli R, Bultynck G, Leybaert L. Endothelial calcium dynamics, connexin channels and blood–brain barrier function. Prog Neurobiol. 2013;108:1–20. doi: 10.1016/j.pneurobio.2013.06.001. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267:156–171. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duling BR, Gore RW, Dacey RG, Jr, Damon DN. Methods for isolation, cannulation, and in vitro study of single microvessels. Am J Physiol. 1981;241:H108–H116. doi: 10.1152/ajpheart.1981.241.1.H108. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Nelson MT. Neurovascular signaling in the brain and the pathological consequences of hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H1–H14. doi: 10.1152/ajpheart.00364.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A. 2013;110:6157–6162. doi: 10.1073/pnas.1216514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol. 2009;297:H1096–H1102. doi: 10.1152/ajpheart.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM, Baumbach GL, Heistad DD. Myogenic mechanisms in the cerebral circulation. J Hypertens Suppl. 1989;7:S61–S64. discussion S65. [PubMed] [Google Scholar]
- Golding EM, Robertson CS, Bryan RM., Jr Comparison of the myogenic response in rat cerebral arteries of different calibers. Brain Res. 1998;785:293–298. doi: 10.1016/S0006-8993(97)01419-4. [DOI] [PubMed] [Google Scholar]
- Guan Z, Fellner RC, Van Beusecum J, Inscho EW. P2 receptors in renal autoregulation. Curr Vasc Pharmacol. 2014;12:818–828. doi: 10.2174/15701611113116660152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert F, Grand'maison M, Ho MK, Lerch JP, Hamel E, Bedell BJ. Cortical atrophy and hypoperfusion in a transgenic mouse model of Alzheimer's disease. Neurobiol Aging. 2013;34:1644–1652. doi: 10.1016/j.neurobiolaging.2012.11.022. [DOI] [PubMed] [Google Scholar]
- Hill-Eubanks DC, Gonzales AL, Sonkusare SK, Nelson MT. Vascular TRP channels: performing under pressure and going with the flow. Physiology. 2014;29:343–360. doi: 10.1152/physiol.00009.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iddings JA, Kim KJ, Zhou Y, Higashimori H, Filosa JA. Enhanced parenchymal arteriole tone and astrocyte signaling protect neurovascular coupling mediated parenchymal arteriole vasodilation in the spontaneously hypertensive rat. J Cereb Blood Flow Metab. 2015;35:1127–1136. doi: 10.1038/jcbfm.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Filosa JA. Advanced in vitro approach to study neurovascular coupling mechanisms in the brain microcirculation. J Physiol. 2012;590:1757–1770. doi: 10.1113/jphysiol.2011.222778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Iddings JA, Stern JE, Blanco VM, Croom D, Kirov SA, Filosa JA. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J Neurosci. 2015;35:8245–8257. doi: 10.1523/JNEUROSCI.4486-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci U S A. 2006;103:10058–10063. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrux C, Toussay X, Kocharyan A, Fernandes P, Neupane S, Lévesque M, Plaisier F, Shmuel A, Cauli B, Hamel E. Pyramidal neurons are “neurogenic hubs” in the neurovascular coupling response to whisker stimulation. J Neurosci. 2011;31:9836–9847. doi: 10.1523/JNEUROSCI.4943-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JB, Rigamonti DD, Dosaka K, Kraimer JM, Martinez-Arizala A. Somatostatin causes vasoconstriction, reduces blood flow and increases vascular permeability in the rat central nervous system. J Pharmacol Exp Ther. 1992;260:1425–1432. [PubMed] [Google Scholar]
- Longden TA, Dunn KM, Draheim HJ, Nelson MT, Weston AH, Edwards G. Intermediate-conductance calcium-activated potassium channels participate in neurovascular coupling. Br J Pharmacol. 2011;164:922–933. doi: 10.1111/j.1476-5381.2011.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hu H, Berrebi AS, Mathers PH, Agmon A. Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice. J Neurosci. 2006;26:5069–5082. doi: 10.1523/JNEUROSCI.0661-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CI, Cao R. The hemo-neural hypothesis: on the role of blood flow in information processing. J Neurophysiol. 2008;99:2035–2047. doi: 10.1152/jn.01366.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. 2003;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngai AC, Winn HR. Modulation of cerebral arteriolar diameter by intraluminal flow and pressure. Circ Res. 1995;77:832–840. doi: 10.1161/01.RES.77.4.832. [DOI] [PubMed] [Google Scholar]
- Nicolakakis N, Hamel E. Neurovascular function in Alzheimer's disease patients and experimental models. J Cereb Blood Flow Metab. 2011;31:1354–1370. doi: 10.1038/jcbfm.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol. 2002;283:H315–H323. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20:3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE, Charpak S. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci. 2015;18:210–218. doi: 10.1038/nn.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Valleé J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–798. doi: 10.1016/j.cell.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Paton JF, Waki H, Abdala AP, Dickinson J, Kasparov S. Vascular-brain signaling in hypertension: role of angiotensin II and nitric oxide. Curr Hypertens Rep. 2007;9:242–247. doi: 10.1007/s11906-007-0043-1. [DOI] [PubMed] [Google Scholar]
- Paukert M, Agarwal A, Cha J, Doze VA, Kang JU, Bergles DE. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron. 2014;82:1263–1270. doi: 10.1016/j.neuron.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev. 1990;2:161–192. [PubMed] [Google Scholar]
- Pelligrino DA, Vetri F, Xu HL. Purinergic mechanisms in gliovascular coupling. Semin Cell Dev Biol. 2011;22:229–236. doi: 10.1016/j.semcdb.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Alvarez A, Araque A. Astrocyte-neuron interaction at tripartite synapses. Curr Drug Targets. 2013;14:1220–1224. doi: 10.2174/13894501113149990203. [DOI] [PubMed] [Google Scholar]
- Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol. 2013;304:H1598–H1614. doi: 10.1152/ajpheart.00490.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant TD, Strotmann R. TRPV4. Handb Exp Pharmacol. 2007;179:189–205. doi: 10.1007/978-3-540-34891-7_11. [DOI] [PubMed] [Google Scholar]
- Raichle ME. A paradigm shift in functional brain imaging. J Neurosci. 2009;29:12729–12734. doi: 10.1523/JNEUROSCI.4366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Wallin A, Wardlaw JM, Markus HS, Montaner J, Wolfson L, Iadecola C, Zlokovic BV, Joutel A, Dichgans M, Duering M, Schmidt R, Korczyn AD, Grinberg LT, Chui HC, Hachinski V. Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab. 2016;36:6–25. doi: 10.1038/jcbfm.2015.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosulina L, Graebenitz S, Pape HC. GABAergic interneurons in the mouse lateral amygdala: a classification study. J Neurophysiol. 2010;104:617–626. doi: 10.1152/jn.00207.2010. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS. Ca (2+) signaling in astrocytes from Ip3r2 (−/−) mice in brain slices and during startle responses in vivo. Nat Neurosci. 2015;18:708–717. doi: 10.1038/nn.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aerde KI, Qi G, Feldmeyer D. Cell type-specific effects of adenosine on cortical neurons. Cereb Cortex. 2015;25:772–787. doi: 10.1093/cercor/bht274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Janssens A, Prenen J, Nilius B, Wondergem R. TRPV channels and modulation by hepatocyte growth factor/scatter factor in human hepatoblastoma (HepG2) cells. Cell Calcium. 2004;36:19–28. doi: 10.1016/j.ceca.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Waki H, Murphy D, Yao ST, Kasparov S, Paton JF. Endothelial NO synthase activity in nucleus tractus solitarii contributes to hypertension in spontaneously hypertensive rats. Hypertension. 2006;48:644–650. doi: 10.1161/01.HYP.0000238200.46085.c6. [DOI] [PubMed] [Google Scholar]
- Yang Y, Kimura-Ohba S, Thompson J, Rosenberg GA. Rodent models of vascular cognitive impairment. Transl Stroke Res. 2016;7:407–414. doi: 10.1007/s12975-016-0486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Papadopoulos P, Hamel E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: impairment and recovery in cerebrovascular pathologies related to Alzheimer disease. Br J Pharmacol. 2013;170:661–670. doi: 10.1111/bph.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Han H, Wang J, Wang H, Yang B, Wang Z. Impairment of human ether-a-go-go-related gene (HERG) K+ channel function by hypoglycemia and hyperglycemia: similar phenotypes but different mechanisms. J Biol Chem. 2003;278:10417–10426. doi: 10.1074/jbc.M211044200. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Zorec R, Araque A, Carmignoto G, Haydon PG, Verkhratsky A, Parpura V. Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro. 2012;4:e00080. doi: 10.1042/AN20110061. [DOI] [PMC free article] [PubMed] [Google Scholar]