

Abstract
Iron deficiency (ID), with and without anemia, affects an estimated 2 billion people worldwide. ID is particularly deleterious during early-life brain development, leading to long-term neurological impairments, including deficits in hippocampus-mediated learning and memory. Neonatal rats with fetal/neonatal ID anemia (IDA) have shorter hippocampal CA1 apical dendrites with disorganized branching. ID-induced dendritic structural abnormalities persist into adulthood despite normalization of iron status. However, the specific developmental effects of neuronal iron loss on hippocampal neuron dendrite growth and branching are unknown. Embryonic hippocampal neuron cultures were chronically treated with deferoxamine (DFO, an iron chelator) beginning at 3 days in vitro (DIV). Levels of mRNA for Tfr1 and Slc11a2, iron-responsive genes involved in iron uptake, were significantly elevated in DFO-treated cultures at 11DIV and 18DIV, indicating a similar degree of neuronal ID as seen in rodent ID models. DFO treatment decreased mRNA levels for genes indexing dendritic and synaptic development (i.e., BdnfVI, Camk2a, Vamp1, Psd95, Cfl1, Pfn1, Pfn2, and Gda) and mitochondrial function (i.e., Ucp2, Pink1, and Cox6a1). At 18DIV, DFO reduced key aspects of energy metabolism including basal respiration, maximal respiration, spare respiratory capacity, ATP production, and glycolytic rate, capacity, and reserve. Sholl analysis revealed a significant decrease in distal dendritic complexity in DFO-treated neurons at both 11DIV and 18DIV. At 11DIV, the length of primary dendrites and the number and length of branches in DFO-treated neurons was reduced. By 18DIV, a partial recovery of dendritic branch number in DFO-treated neurons was counteracted by a significant reduction in the number and length of primary dendrites and length of branches. Our findings suggest that early neuronal iron loss, at least partially driven through altered mitochondrial function and neuronal energy metabolism, is responsible for the effects of fetal/neonatal ID and IDA on hippocampal neuron dendritic and synaptic maturation. Impairments in these neurodevelopmental processes likely underlie the negative impact of early life ID and IDA on hippocampus-mediated learning and memory.
Keywords: Branching, anemia, developing brain, energy metabolism, metabolism, dendritogenesis, mitochondria, glycolysis
Introduction
Iron deficiency (ID) is the most common micronutrient deficiency, affecting an estimated 2 billion people and 40–50% of pregnant women and children, worldwide [1,2]. This is not just a problem in low- and middle-income countries because 18% of pregnant women and 14% of 1–2 year old children in the United States are iron-deficient [3,4]. These population groups are particularly important because the developing fetal and early postnatal brain has greater iron requirements than the mature brain [5]. In both humans and rodents, ID during fetal and early postnatal life impairs learning and memory [6]. Even more concerning is that learning and memory impairments persist into adolescence and adulthood despite prompt iron repletion during infancy, findings supported experimentally in rodent models of fetal/neonatal ID [6].
Declarative learning and memory processing and consolidation depends on proper development of the hippocampus [7]. Hippocampal development, especially neuronal maturation (i.e. axon/dendrite growth and branching and spine/synapse formation), is a metabolically demanding process requiring appropriate levels and intracellular trafficking of iron, ATP and other energetic substrates [8–11]. Iron provides the catalytic component for several mitochondrial enzymes [12]. Fetal/neonatal ID reduces mitochondrial cytochrome c oxidase activity in a brain-region specific manner, with the greatest loss of activity occurring in the hippocampus [13] during the early stage of rapid dendrite maturation.
Dendrite and synapse formation are dynamic processes requiring continuous remodeling of the cytoskeleton [14], a process facilitated by actin- and microtubule-associated proteins (e.g. cofilin, profilins, and guanine deaminase) [15–17]. This process also depends on extrinsic signaling from neurotrophins (e.g., brain derived neurotrophic factor, BDNF) and neurotransmitters [18], which is supported by a variety of synaptic proteins including calcium/calmodulin-dependent protein kinase II-alpha (CaMKIIα), synaptobrevin 1 (i.e., vesicle-associated membrane kinase 1, VAMP1), post synaptic density protein 95 (PSD95), and regulators of synaptic protein gene expression (e.g. early growth response protein 1, EGR1) [19–22].
Gene and protein expression for TfR-1 and DMT-1, two iron-responsive proteins that regulate neuronal iron uptake, in the hippocampus increases between postnatal day (P)5 and P15 [23,24], just prior to the period of rapid hippocampal neuron maturation [25]. Dietary-induced fetal/neonatal IDA in the rat leads to aberrant apical and basal dendrite branching and thinner spine heads in hippocampal sub-region CA1 pyramidal neurons in early postnatal life [26,27]. CA1 pyramidal neurons from adult formerly IDA rats have shorter branches and a decrease in distal branching, despite neonatal iron repletion, indicating early-life IDA causes long-term impairments in neuronal structure [26]. This manifests electrophysiologically as lower CA1 long-term potentiation (LTP) expression [28] and behaviorally as impaired performance in spatial and recognition learning/memory tasks in formerly IDA rats [29,30], suggesting permanently impaired synaptic function and efficacy.
The rat dietary IDA model is characterized by several secondary pathophysiologies with potential neuro-toxic effects including anemia, tissue hypoxia, hypothyroidism, and divalent metal toxicity [6] making it impossible to determine whether the specific structural defects are due to neuronal iron loss. Genetic mouse models of early-life hippocampal neuron-specific ID, without these potential confounders, are characterized by abnormal CA1 pyramidal neuron structure, accompanied by reduced LTP and spatial learning/memory deficits in adulthood [8,31,32], indicating that the long-term structural and functional deficits seen in IDA rats are specifically due to reduced iron in CA1 pyramidal neurons. However, the specific molecular and cellular effects of hippocampal neuron-specific ID on dendrite growth and branching during the period of rapid hippocampal neuron development are still unknown. Determining how neuronal iron loss during this early-life period specifically affects dendrite maturation is necessary to reveal the developmental origins of the long-term structural deficits.
We developed a novel culture model of chronic hippocampal neuron ID during peak dendrite development, which generates a similar degree of ID documented in rodent ID models and human iron-deficient newborn infants. Neuronal ID impaired mRNA expression of genes involved in dendrite and synapse development, as well as mitochondrial function. Both mitochondrial respiration and glycolysis were impaired in iron-deficient neurons. Ultimately, neuronal ID caused an overall reduction in dendritic arborization with age-dependent differences in the relative contributions of branches and primary dendrites to the total arbor. These data suggest intrinsic (e.g., energy metabolism) or extrinsic (e.g., synaptic activity) mechanisms may promote dendrite branching in the face of ID and/or iron prioritization to dendrite maturation processes.
Materials and Methods
Animals
Mice were given free access to food and drinking water and were housed at constant temperature and humidity on a 12h light:dark cycle. All animal procedures were conducted in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals. The local Institutional Animal Care and Use Committee approved these procedures.
Cell culture
Primary hippocampal neuron cultures were prepared as previously described [33]. Timed-pregnant CD1 mice were ordered from Charles River Laboratories. The hippocampus was removed from embryonic day 16 (E16) embryos. Pooled hippocampi were incubated at 37°C for 15 minutes in 0.25% trypsin-EDTA (Sigma-Aldrich, T4174), rinsed three times for five minutes in calcium- and magnesium-free Hanks balanced salt solution (CMF-HBSS) and resuspended in neuronal plating media (10 mM HEPES, 10 mM sodium pyruvate, 0.5 mM glutamine, 12.5 μM glutamate, 10% fetal bovine serum (FBS), 0.6% glucose in minimal essential media plus Earl’s salt (EMEM)). Pooled hippocampi were dissociated by trituration with a fire-polished pipette and then viable cells were counted using trypan blue and a hemocytometer. Cells were plated in neuronal plating media at 200 cells/mm2 in 35 mm dishes containing five 12 mm acid-washed German glass coverslips coated with 100 μg/ml poly-d-lysine and 4 μg/ml laminin. After the cells adhered (1–3 hours after plating), plating media was replaced with glia-conditioned neural growth media (Neurobasal, 1X B27 (Invitrogen), 0.5 mM glutamine, 1X penicillin/streptomycin). Dishes were maintained at 37 °C, 5% CO2.
At 3DIV, cultures were treated with 67.5 μM 5-fluoro-2′-deoxyuridine (Sigma-Aldrich #F0503)/136 μM uridine (Sigma-Aldrich #U6381) (5-FU) to inhibit glia proliferation, allowing a nearly pure neuronal culture. To create chronic ID that extended through the major time period of dendrite development, hippocampal cultures were treated with 10 μM deferoxamine (DFO, Cayman Chemicals #14595), an iron chelator, beginning at 3DIV. Each week, beginning at 7DIV, half of the media was removed and replaced with fresh glia-conditioned neuronal growth media containing 1X 5-FU and 1X DFO. To compensate for DFO breakdown, media was supplemented with fresh DFO (assuming 50% breakdown) each week beginning at 10DIV. Cultures were analyzed just after the beginning of dendritic branching (i.e., 11DIV) and during peak branching and synapse formation (i.e., 18DIV) [34].
Postnatal glia cultures were prepared as previously described [35]. Glia media (10% FBS, 2 mM glutamine, 1X penicillin/streptomycin in EMEM) was changed each week. Glia-conditioned media was prepared by replacing glia media with neural growth media on confluent glial plates for 24 hr. Once the conditioning period was complete, conditioned media was removed and replaced with fresh glia media.
mRNA expression analysis
Total RNA was extracted from hippocampal cultures at 11DIV and 18DIV using a Quick-RNA MicroPrep kit (Zymo Research) according to the manufacturer’s protocol. Cells were lysed directly in the tissue culture dishes. To eliminate potential genomic DNA contamination, the optional on-column digestion of DNA with DNase was performed. RNA integrity and purity was established spectrophotometrically using a Nanodrop spectrophotometer. cDNA was synthesized from 250 or 500 ng total RNA using a High Capacity RNA-to-cDNA Kit (Applied Biosystems) in a 20 μL reaction volume. Quantitative real-time polymerase chain reaction (qPCR) was performed using a 2x FastStart Universal Probe Master (Rox) kit (Roche Applied Science) and a Stratagene MX3000P qPCR machine. PCR reactions were performed on cDNA equivalent to 10 ng of total RNA according to the manufacturer’s protocol except that a final volume of 10 μl was used. TaqMan qPCR probes for the genes assessed are described in Table 1. Quantification cycle (CQ) values were determined in the log-linear amplification phase using the qPCR machine’s software. Relative mRNA levels for the genes of interest were calculated relative to an internal calibrator cDNA sample and were normalized to a reference gene, TATA box binding protein, Tbp.
Table 1.
TaqMan qPCR probes
| Gene Name | GenBank mRNA Accession # | TaqMan Probe Assay ID | Functional Category |
|---|---|---|---|
| BdnfVI | EF125674.1 | Mm01334042_m1 | Growth Factor, Synaptic Plasticity [18] |
| Camk2a | NM_001286809.1 | Mm00437967_m1 | |
| NM_009792.3 | Synaptic Calcium Signaling [21] | ||
| NM_177407.4 | |||
| Cfl1 | NM_007687.5 | Mm03057591_g1 | Actin Dynamics [17] |
| Cox6a1 | NM_007748.3 | Mm01612194_m1 | Mitochondrial Electron Transport [50] |
| Egr1 | NM_007913.5 | Mm00656724_m1 | Transcriptional Regulation [19] |
| Gda | NM_010266.2 | Mm00515820_m1 | Microtubule Dynamics [15] |
| Pfn1 | NM_011072.4 | Mm01245693_g1 | Actin Dynamics [16] |
| Pfn2 | NM_019410.3 | Mm01289572_m1 | Actin Dynamics [16] |
| Pink1 | NM_026880.2 | Mm00550827_m1 | Mitophagy Regulation [49] |
| Psd95 | NM_001109752.1 | Mm00492193_m1 | Synaptic Structure/Plasticity [22] |
| NM_007864.3 | |||
| Slc11a2 (Dmt1) | NM_001146161.1 | Mm00435356_g1 | Neuronal Iron Uptake [6] |
| NM_008732.2 | |||
| Tbp | NM_013684.3 | Mm00446971_m1 | Basal Transcription Factor (Reference Gene) [60] |
| Tfr1 | NM_011638.4 | Mm00441941_m1 | Neuronal Iron Uptake [6] |
| Ucp2 | NM_011671.5 | Mm00627599_m1 | Oxidative Stress [48] |
| Vamp1 | NM_009496.3 | Mm00772309_g1 | Synaptic Vesicle Trafficking [20] |
Genes: Brain derived neurotrophic factor transcript variant VI, BdnfVI; calcium/calmodulin-dependent protein kinase II alpha, Camk2a; Cofilin 1, Cfl1; Cytochrome c oxidase subunit VIa polypeptide 1, Cox6a1; Early growth response factor 1, Egr1; Guanine deaminase, Gda; Profilin 1, Pfn1; Profilin 2, Pfn2; PTEN induced putative kinase 1, Pink1; Post-synaptic density 95, Psd95; Solute carrier family 11 member 2, Slc11a2; TATA box binding protein, Tbp; Transferrin receptor 1, Tfr1; Uncoupling protein 2, Ucp2; Vesicle-associated membrane protein, Vamp1.
Neuronal bioenergetics
8,000 hippocampal cells were plated in each well of an XFe24 cell culture microplate (Seahorse Biosciences) designed for subsequent bioenergetic analysis. Iron-deficient and -sufficient hippocampal neuron cultures were prepared as described above. For these experiments, the DFO concentration was reduced to 6 μM due to increased evaporation in the Seahorse microplate wells compared to 35 mm dishes. At 18DIV, the growth media was exchanged for Assay Media (unbuffered DMEM plus 25 mM glucose, 1 mM sodium pyruvate, 31 mM NaCl, 2 mM GlutaMax, pH 7.4 at 37°C) and incubated at 37°C (non-CO2 incubator) for ~30 min prior to bioenergetic analyses [36]. Real-time oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were simultaneously measured using a Seahorse XFe24 Extracellular Flux Analyzer, at baseline and following treatment with 1 μM oligomycin (ATP synthase inhibitor), 4 μM FCCP (uncouples oxygen consumption from ATP production), and 1 μM antimycin A/rotenone (ETC complex III and I inhibitors, respectively). The assay protocol is outlined in Supplemental Table 1. Mitochondrial oxidative phosphorylation is the main determinant of OCR (31). ECAR is predominantly controlled by lactic acid formation and thus is a specific read-out of glycolytic energy metabolism (31). Key indicators of mitochondrial respiration and glycolysis were calculated including basal respiration (last baseline OCR–non-mitochondrial respiration (i.e., minimum OCR after antimycin A/rotenone)), maximal respiration (maximum OCR after FCCP–non-mitochondrial respiration), ATP production (last baseline OCR–minimum OCR after oligomycin), spare respiratory capacity (maximal respiration–basal respiration), glycolytic rate (maximum baseline ECAR), glycolytic capacity (maximum ECAR after oligomycin) and glycolytic reserve (glycolytic capacity – glycolytic rate).
Plasmids and neuronal transfection
For neuronal tracing experiments hrGFP (A “humanized” recombinant green fluorescent protein cDNA from R. reniformis adapted for high-level expression in mammalian cells) was expressed to enable visualization of individual neurons [37]. Briefly, a mammalian expression vector utilizing a CMV enhanced chicken B-actin (CBA) promoter to drive hrGFP expression was electroporated into hippocampal cells prior to plating using a Lonza Nucleofector II system and the Amaxa Mouse Neuron Nucleofector Kit (Lonza #VAPG-1001) as previously described [35]. Electroporated cells were then mixed 1:1 with non-electroporated cells before plating.
Immunocytochemistry
At 11DIV and 18DIV, hippocampal cultures on coverslips were fixed at 37°C with 4%paraformaldehyde/PHEM (60 mM PIPES pH 7.0, 25 mM K-HEPES pH 7.0, 10 mM EGTA, 2 mM MgCl2)/0.12 M sucrose-buffered fixative for 30 minutes. Coverslips were rinsed two times (five minutes each) in PBS and then stored in 3% bovine serum albumin (BSA)/PBS at 4°C. Cells were permeabilized using 0.2% Triton X-100 in PBS for 10 min at room temperature, rinsed in PBS for five minutes, and blocked for 30 minutes in 3% BSA/PBS. The blocking reagent was removed and cells were incubated in rabbit anti-hrGFP (Agilent Technologies, #240142, 1:1000) and mouse anti-MAP2 (Abcam, #ab11268, 1:1000) antibodies overnight at 4°C in a humidified chamber. Coverslips were rinsed in PBS for five minutes and incubated in fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG (Jackson Immunoresearch, #711-095-152, 1:200) and Alexa Fluor 594-conjugated donkey anti-mouse IgG (Jackson Immunoresearch, #715-585-151, 1:200) for 1 hour at room temperature in the dark. All antibody mixtures were prepared in 1% BSA/PBS. Coverslips were rinsed for five minutes in PBS, incubated for 30 seconds in DAPI, and mounted on glass slides with a glycerol-based mounting media (80% glycerol, 2.5% 1,4-Diazabicyclo-[2.2.2]Octane, 150 mM Tris pH 8.0). Three-channel (hrGFP, MAP2, and DAPI) images of dendritic arbors for individual hrGFP-expressing neurons were collected with a Zeiss Axiovert 200M microscope with 20x plan-apo objective and Openlab software.
Morphological analyses
ImageJ or FIJI was used for all image processing and morphological analyses [38,39]. For dendrite tracing, MAP2 and hrGFP images for each individual neuron were merged and converted to 8-bit color images. Dendrites were traced using the NeuronJ plugin (http://www.imagescience.org/meijering/software/neuronj/) and quantified using XL-Calculation as previously described [35,40]. Semi-automated Sholl analysis [41] was performed on the NeuronJ tracings using the Sholl Analysis plugin (http://fiji.sc/Sholl_Analysis). It is estimated that 85–90% of neurons in the E16 hippocampus are pyramidal neurons [42]. Non-pyramidal neurons were excluded, based on morphology, from dendrite tracing and Sholl analyses.
To determine neuronal density DAPI and MAP2 images were merged and the number of neuronal nuclei per mm2 was calculated. Cell health was qualitatively assessed by visually examining nuclear morphology [43]. Healthy cells have a relatively equal distribution of chromatin throughout the nuclei (smooth, round nuclei), whereas unhealthy or apoptotic cells undergo nuclear chromatin condensation (crenellated nuclei). In addition, dendrites (MAP2 staining) were visually inspected for signs of poor health such as fasciculation, focal swelling (varicosity), and fragmentation.
Statistical Analysis
Statistical analyses and data graphing were carried out using Prism (GraphPad Software) software. The data from two or three independent cultures were pooled and Student’s t-tests used to determine differences between experimental groups for each parameter. When variances were unequal, ln transformation was used prior to statistical analysis. When variances were unequal and not normalized by ln transformation, the Mann-Whitney U test was utilized. All data are presented as mean ± standard error of the mean (SEM). An α = 0.05 was chosen to define significant differences.
Results
Cell health and neuronal density
MAP2 immunocytochemistry demonstrated healthy neurons with minimal fasciculation, fragmentation, or dendrite swelling throughout both untreated and DFO-treated cultures (Figure 1A shows representative healthy neurons). Based on nuclear morphology, DFO treatment did not alter the percentage of healthy cells (i.e., cells without nuclear chromatin condensation) in 11DIV or 18DIV hippocampal cultures (Figure 1A insets). 11DIV neuron density was not altered by DFO treatment (Figure 1B). However, by 18DIV, there was a 29% reduction in neuron density in DFO-treated hippocampal cultures (Figure 1B), suggesting that some neurons died and detached from the coverslip.
Figure 1. The effect of DFO treatment on neuronal health and density.
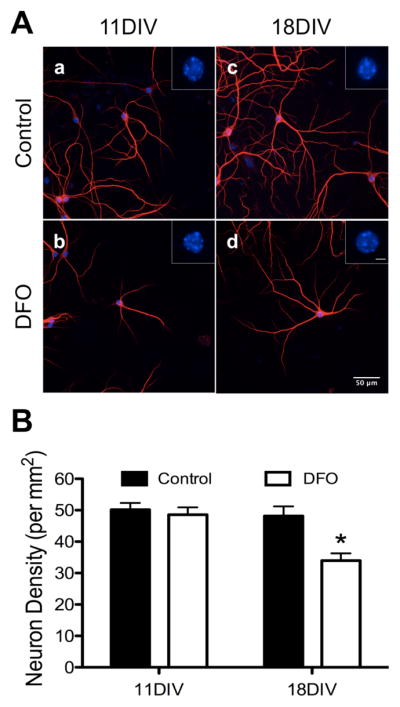
Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3DIV. At 11DIV and 18DIV cultures were fixed, the nuclei were stained with DAPI and the dendrites stained with MAP2, and images were taken at 20x magnification. (A) Representative images of 11DIV (a,b) and 18DIV (c,d) neurons showing healthy nuclei (DAPI, blue) and dendrites (MAP2, red) in 0 μM (a,c) and 10 μM (b,d) treated cultures. Scale bar in (A, d) = 50 μm. Insets show digital zoom of DAPI-stained nucleus for each representative neuron. Scale bar in (A, d inset) = 5 μm. (B) The number of neuronal nuclei was counted in merged MAP2/DAPI images and neuronal density was calculated. The data from 2–3 independent cultures were pooled and are presented as mean ± SEM. Asterisks indicate statistical difference between groups at a given neuronal age by Student’s t-test (p < 0.05). 11DIV: n=102–105 images per group, 18DIV: n=80 images per group.
Gene expression indexing iron status, neuron development and mitochondrial function
Expression of genes/proteins involved in cellular iron uptake is regulated at level of mRNA stability through iron-regulatory proteins (IRPs) binding to iron-response element (IRE) regions, providing sensitive regulation of cellular iron status [44]. To determine the effect of DFO treatment on functional, neuronal iron status, qPCR was performed for Tfr1 and Slc11a2 (i.e., Dmt1), IRE/IRP-regulated genes involved in neuronal iron uptake. Titrating DFO dosage from 1 to 10 μM demonstrated that 10 μM DFO induces a similar degree Tfr1 and Slc11a2 upregulation at 18DIV (Supplemental Figure 1) as seen in rodent ID models and human iron-deficient infants [24,31,45]. At 11DIV, DFO treatment increased Tfr1 and Slc11a2 mRNA levels by 42% and 23%, respectively (Figure 2A). By 18DIV, Tfr1 and Slc11a2 mRNA levels were 118% and 43% higher in DFO-treated compared to untreated hippocampal cultures (Figure 2B). DFO has its highest affinity for iron but can also chelate other divalent metals (e.g., zinc) [46]. However, DFO treatment did not alter Znt-1 mRNA levels in 11DIV neurons (Supplemental Figure 2), indicating neuronal zinc status was preserved [47].
Figure 2. The effect of DFO treatment on gene expression.

Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3DIV. At 11DIV and 18DIV, cells were collected, total RNA was extracted, and cDNA was synthesized. Quantitative real-time PCR (qPCR) was performed for genes indexing (A, B) neuronal iron status (i.e., Tfr1 and Slc11a2), (C, D) synaptic development, function, and plasticity (i.e., BdnfVI, Camk2a, Vamp1, Psd95, and Egr1), (E, F) cytoskeletal structural development (i.e., Cfl1, Pfn1, Pfn2, and Gda), and (G, H) mitochondrial health (i.e., Ucp2, Pink1, and Cox6a1). Relative mRNA levels are calculated relative to an internal control cDNA sample and a reference gene (i.e. Tbp). The data from 2–3 independent cultures were pooled and are presented as mean ± SEM. Asterisks indicate statistical difference between groups at a given neuronal age by Student’s t-test or Mann-Whitney U test (p < 0.05). 11DIV: n=8–13, 18DIV: n=9.
DFO treatment reduced mRNA levels for genes indexing synaptic plasticity/function and cytoskeletal structural development (Figure 2C–F). BdnfVI, Camk2a, Vamp1, and Psd95 mRNA levels were 20%, 53%, 52%, and 46% lower, respectively, in 11DIV DFO-treated cultures (Figure 2C) and 40%, 38%, 32%, and 33% lower, respectively, in 18DIV DFO-treated cultures (Figure 2D). Egr1 mRNA levels were not significantly altered at either time point. DFO treatment reduced Cfl1, Pfn2, and Gda mRNA levels by 30%, 44%, and 67% at 11DIV (Figure 2E) and by 20%, 27%, and 60% at 18DIV (Figure 2F). Pfn1 mRNA levels were 12% lower at 11 DIV, but not significantly altered at 18DIV.
As a preliminary measure of the effect of neuronal ID on mitochondrial function, mRNA levels were quantified for genes encoding mitochondrial proteins important for regulating oxidative stress (i.e., Ucp2 [48]), mitophagy (i.e., Pink1 [49]), and electron transport (i.e., Cox6a1 [50]). Ucp2 and Pink1 mRNA levels were 31% and 41% lower, respectively, in 11DIV DFO-treated cultures but no longer altered in 18DIV DFO-treated cultures (Figure 2G and H). Cox6a1 mRNA levels were ~18% lower in both 11DIV and 18DIV DFO-treated cultures (Figure 2G and H).
Neuronal energy metabolism
To further assess the effect of neuronal ID on mitochondrial function, real-time OCR and ECAR were measured at baseline and following oligomycin, FCCP, and antimycin A/rotenone treatments (Figure 3A). DFO treatment impaired overall mitochondrial respiration (Figure 3A), reducing basal respiration, maximal respiration, ATP production and spare respiratory capacity by ~70% (Figure 3B), without affecting ATP coupling efficiency (data not shown). Likewise, neuronal ID significantly reduced glycolytic rate (40% lower) and capacity (84% lower) with a trend towards less glycolytic reserve (Figure 3C).
Figure 3. ID reduces mitochondrial respiration and glycolysis in hippocampal neurons at 18DIV.
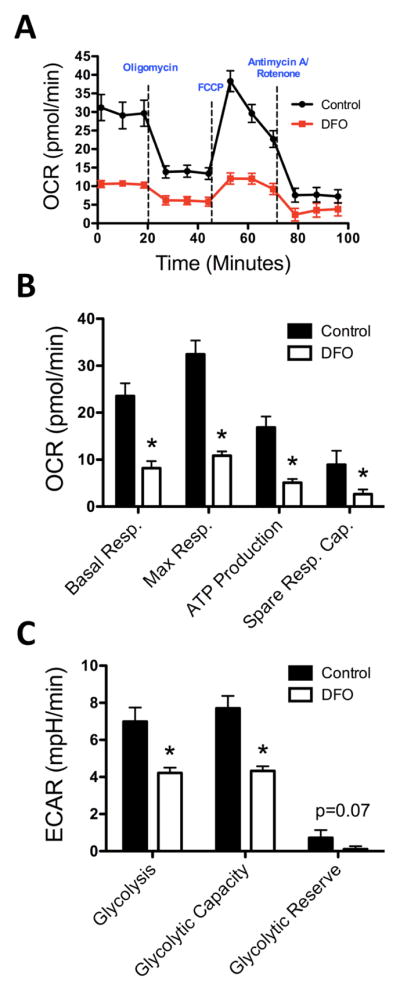
Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3DIV. (A) At 18DIV, real-time OCR and ECAR (not shown) were measured for iron-sufficient (black) and iron-deficient (red) hippocampal cultures following treatment with oligomycin, FCCP, and antimycin A/rotenone. (B) Basal respiration, maximal respiration, ATP production, and spare respiratory capacity were calculated from OCR measurements. (C) Glycolytic rate, capacity, and reserve were calculated from ECAR measurements. The data from 3 independent cultures were pooled and are presented as mean ± SEM. Asterisks indicate statistical difference between groups by Student’s t-test (p < 0.05). n=11–15.
Hippocampal pyramidal neuron dendrite morphology
Representative images (and dendrite tracings) of hrGFP-labeled hippocampal neurons from untreated and DFO-treated cultures are shown in Figure 4A and 4B. Sholl analysis revealed a significant decrease in overall distal dendritic arbor complexity in DFO-treated neurons (Figure 4C and D). The number of crossings was significantly lower between 70 μm and 170 μm at 11DIV and between 90 μm and 210 μm at 18DIV.
Figure 4. Iron deficiency impairs dendritic arbor complexity of cultured hippocampal neurons.

Hippocampal neurons cultured from E16 mice were transfected with an hrGFP-expressing plasmid and then treated with DFO and 5-FU beginning at 3DIV. At 11DIV and 18DIV cultures were fixed and processed for hrGFP and MAP2 immunocytochemistry. Images were taken at 20x magnification and merged to allow visualization of dendrites on individual neurons. Representative (A) 11DIV and (B) 18DIV images of hrGFP-expressing neurons and tracings [primary dendrite (red), secondary branch (blue), tertiary branch (green)] of untreated (a,c) and DFO-treated (b,d) neurons. (C) 11DIV (n=74–84 neurons per group) and (D) 18DIV (n=83–90 neurons per group) Sholl analysis curves. The data from 2–3 independent cultures were pooled and are presented as mean ± SEM. For a given distance from the soma, asterisks indicate statistical difference by two-way ANOVA and Sidak’s post-hoc test (p < 0.05). Scale bar in (B, d) = 50 μm.
To distinguish between effects on dendrite number and/or lengths, detailed analysis of dendrite tracings (Figure 4A and B, panels c and d) was performed. The average number of primary dendrites per neuron (Figure 5A) was not altered at 11DIV but was 11% lower in DFO-treated neurons at 18DIV (4.98 ± 0.13 in untreated vs. 4.41 ± 0.12 in DFO-treated). DFO treatment reduced the length of primary dendrites by 20% and 17% at 11DIV and 18DIV, respectively (Figure 5B). The length of the longest dendrite (an in vitro surrogate for pyramidal neuron apical dendrite length) was 19% and 14% shorter in 11DIV and 18DIV DFO-treated neurons (Figure 5C).
Figure 5. Iron deficiency impairs primary dendrite development in cultured hippocampal neurons.
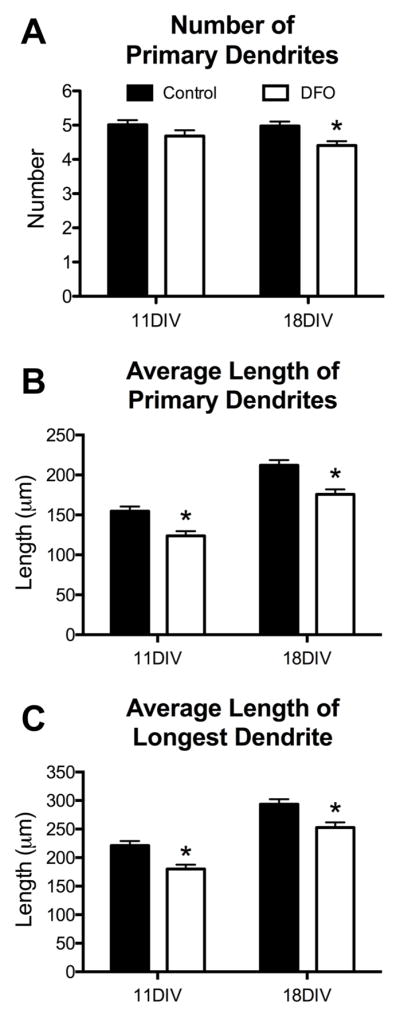
Hippocampal neurons cultured from E16 mice were transfected with an hrGFP-expressing plasmid and then treated with DFO and 5-FU beginning at 3DIV. At 11DIV and 18DIV cultures were fixed and processed for hrGFP and MAP2 immunocytochemistry. Images were taken at 20x magnification and merged to allow manual tracing of dendrites on individual neurons. The (A) number and (B) average length of primary dendrites were quantified. (C) As an in vitro surrogate for apical dendrite length, the average length of the longest dendrite was quantified. The data from 2–3 independent cultures were pooled and are presented as mean ± SEM. Asterisks indicate statistical difference between groups at a given neuronal age by Student’s t-test (p < 0.05). 11DIV: n=72–84 neurons per group, 18DIV: n=83–90 neurons per group.
The total number of dendrite branches was 28% lower in DFO-treated neurons at 11DIV but was no longer significantly different at 18DIV (Figure 6A). However, DFO treatment reduced branch length by 20% and 13% at 11DIV and 18DIV, respectively (Figure 6D). Secondary and tertiary branches were similarly affected by DFO treatment at 11DIV but not 18DIV. At 11DIV, DFO treatment resulted in 25% fewer and 21% shorter secondary branches and 38% fewer and 36% shorter tertiary branches (Figure 6B, C, E, F). At 18DIV, there was a 14% reduction in secondary branch length (Figure 5E) and a trend (p = 0.06) toward reduced secondary branch number (Figure 6B) in DFO-treated neurons. There was no significant difference in tertiary branch number or length at 18DIV (Figure 6C and F). The sum length of all branches was significantly less in both 11DIV (41%) and 18DIV (22%) DFO-treated neurons (Figure 7A). To give an indication of the total dendrite output of DFO-treated and untreated neurons, the sum length of all dendrites (primary, secondary, and tertiary) was calculated. DFO treatment reduced the sum dendrite length by 33% and 25% at 11DIV and 18DIV, respectively (Figure 7B).
Figure 6. Iron deficiency impairs dendrite branching in cultured hippocampal neurons.

Hippocampal neurons cultured from E16 mice were transfected with pVC1.2-hrGFP and then treated with DFO and 5-FU beginning at 3DIV. At 11DIV and 18DIV cultures were fixed and processed for hrGFP and MAP2 immunocytochemistry. Images were taken at 20x magnification and merged to allow manual tracing of dendrites on individual neurons. (A) Quantification of the total number of branches per neuron. The total number of (B) secondary and (C) tertiary branches per neuron was quantified. (D) Quantification of the average length of all branches. The average length of (E) secondary and (F) tertiary branches was quantified. The data from 2–3 independent cultures were pooled and are presented as mean ± SEM. Asterisks indicate statistical difference between groups at a given neuronal age by Student’s t-test or Mann-Whitney U test (p < 0.05). δ indicates a statistical trend (p = 0.06). 11DIV: n=72–84 neurons per group, 18DIV: n=83–90 neurons per group.
Figure 7. Effects of iron deficiency on secondary and tertiary branches in cultured hippocampal neurons.

Hippocampal neurons cultured from E16 mice were transfected with pVC1.2-hrGFP and then treated with DFO and 5-FU beginning at 3DIV. At 11DIV and 18DIV cultures were fixed and processed for hrGFP and MAP2 immunocytochemistry. Images were taken at 20x magnification and merged to allow manual tracing of dendrites on individual neurons. (A) Quantification of the sum length of all branches. (B) Quantification of the sum length of all dendrites. The data from 2–3 independent cultures were pooled and are presented as mean ± SEM. Asterisks indicate statistical difference between groups at a given neuronal age by Student’s t-test (p < 0.05). 11DIV: n=72–84 neurons per group, 18DIV: n=83–90 neurons per group.
Discussion
In order to determine the specific effects of neuronal ID on hippocampal neuron dendrite structure during the period of rapid dendrite maturation, we developed a novel primary hippocampal neuron culture model of chronic ID. Chronic iron chelation with DFO allows easy manipulation of the amount of iron available to neurons, without modifying neuronal growth media that is already optimized for long-term survival of hippocampal neuron cultures. It is also preferable over culturing neurons from transgenic mice with hippocampal neuron-specific ID where cells from individual embryos would have to be cultured separately (a prohibitively time-consuming process) to ensure genotype homogeneity. A limitation of this approach is that DFO, despite having its highest affinity for iron, can also chelate other divalent cations (e.g., zinc) at high DFO doses [46]. To account for this, we show that zinc transporter-1 mRNA levels, a measure of cellular zinc status [47], are not altered in 10 μM DFO-treated 11DIV neurons. This culture model of neuronal ID shows a 2-fold increase in Tfr1 mRNA levels at 18DIV (Figure 2B), a time point approximately equivalent to P15, in vivo. This magnitude of Tfr1 upregulation is similar to the effect seen in P15 rats following dietary-induced fetal/neonatal IDA [24] and in non-anemic P15 mice with a late fetal, neuronal specific knockout of DMT-1 [31], indicating a similar degree of ID as in vivo models that demonstrate dendrite structure abnormalities in the hippocampus. The P15 IDA rat has a 60–70% loss of hippocampal iron content and a 40% loss of whole brain iron content [24,28,51], which is commensurate with the degree of brain ID documented in human newborns [45]. The altered mRNA expression for genes involved in synaptic development and function (i.e., BdnfVI, Camk2a, Vamp1, and Psd95) and actin and microtubule dynamics (i.e., Cfl1, Pfn1, Pfn2, and Gda) is consistent with alterations in these pathways in the neonatal rodent hippocampus following early-life ID with or without anemia [8,24,26,31,52]. Thus, our culture model of chronic hippocampal neuron ID during development provides a physiologically- and translationally-relevant model for studying the molecular, cellular, and structural developmental processes affected by neuronal iron loss.
Declarative learning and memory processes depend on proper functioning of hippocampus-dependent circuits [7], which in turn are dependent on the degree of dendritic arborization available for synaptic connectivity [53]. Prior to this study, the specific effect of neuronal iron loss on hippocampal neuron dendrite growth and branching was unknown since previous analyses were qualitative and only assessed in adults. Adult transgenic mice with hippocampal neuron-specific ID (knockout of Slc11a2 (i.e., DMT-1) or conditional overexpression of a dominant negative form of TfR1 beginning at E18.5) have aberrant CA1 dendritic arbor organization [8,31]. In the dominant negative TfR1 model, these structural impairments remain present with late iron repletion (i.e., P42) but are rescued with early iron repletion (i.e., P21), indicating a critical postnatal period for intervention [8]. Consistent with the neonatal effects of nutritional IDA on hippocampal neuron dendrite arborization [26,27,51], our 11DIV and 18DIV morphology data demonstrate that chronic neuronal ID during the proposed critical period [6,8] blunts dendritic growth and branching, resulting in an overall reduced dendritic field in developing iron-deficient neurons. Thus, the long-term defects in hippocampal neuron dendritic arborization observed in both IDA and neuronal-specific ID models [8,26,31,51] likely stem from chronic neonatal ID impairing dendrite growth and branch during a period of rapid maturation early in life. These findings also indicate that the early-life effects of ID on dendrite branching are driven by loss of neuronal iron, without the confounding effects of anemia and whole body/brain ID. This demonstrates that the neuron is the site of action and a potential, specific target for intervention, which is important translationally, as non-anemic ID is 2–3 times more common than IDA in children [2] and is associated with impaired auditory cortex electrophysiology and recognition memory in human infants [54]. In addition, low maternal iron intake during pregnancy is associated with impaired gray matter organization in infant offspring, perhaps driven through early-life iron-dependent deficits in dendritic growth and branching [55].
Between 11DIV and 18DIV chronically iron-deficient neurons appeared to adapt their structural development to the lack of iron. At 18DIV, compared to 11DIV, there were slightly less severe effects of DFO treatment on neurodevelopmental gene transcript levels and some measures of dendrite branching despite a greater degree of ID. In particular, the number of dendrite branches recovered to near control levels between 11DIV and 18DIV. However at 18DIV, branches remained shorter and there was an accompanying significant decrease in the number of primary dendrites. This suggests either a process of primary dendrite retraction or that iron-deficient neurons with fewer primary dendrites are more likely to survive, as neuronal density was significantly lower in iron-deficient neurons at 18DIV but not 11DIV. These data are consistent with a prioritization of the limited iron to branch and synapse maturation (at the expense of neuronal and primary dendrite number) between 11DIV and 18DIV, when these important neurodevelopmental processes are ramping up.
The mechanisms underlying this prioritization phenomenon may be cell intrinsic or extrinsic. Cell intrinsically, mitochondrial respiration and energy metabolism is a potential candidate, as mitochondrial function is directly dependent on iron and is also critical for dendrite growth [9]. Abnormalities in hippocampal energy metabolism have been documented in the IDA rat while iron-deficient [13,24,56] and in adulthood long after iron repletion [57]. Our neuronal bioenergetics findings demonstrate for the first time that neuronal ID impairs both mitochondrial respiration and glycolysis, indicating a complete blunting of neuronal energetic capacity. These data also suggest that neuronal iron loss, and not anemia or whole body/brain ID, is responsible for these ID-induced hippocampal energy metabolism deficits. Our 11DIV gene expression data demonstrate impairments in mRNA levels for genes implicated in oxidative stress (i.e., Ucp2) and mitophagy (i.e., Pink1), two other important mitochondrially-regulated pathways. Ucp2 and Pink1 mRNA levels were recovered to control levels by 18DIV, leading us to speculate that homeostatic mechanisms regulating mitochondrial health (including mitophagy and regulation of mitochondrial reactive oxygen species) may be responsible for partial recovery of dendrite branching observed in 18DIV iron-deficient neurons. At the same time, impaired mitochondria energy production likely contributes to the continued deficits in dendrite growth in iron-deficient neurons.
Hippocampal neuron dendritic branching is also highly dependent on afferent innervation, especially after the first week in culture [58]. In addition, both axonal arborization and functional synapse formation increase during development, peaking between 14DIV and 21DIV [34,59]. The fact that Pfn1, which regulates dendritic spine formation but not dendritic arborization [16], was the lone cytoskeletal gene altered by DFO at 11DIV to completely recover at 18DIV implies that extrinsic signaling mechanisms may also contribute to the observed compensation. It is possible that intercellular connection-dependent dendrite maturation is not altered in iron-deficient neuronal cultures, accounting for the partial recovery observed between 11DIV and 18DIV. Despite these potential homeostatic mechanisms, iron-deficient neurons remain fundamentally abnormal during this developmental time period with blunted dendritic fields and impaired neurodevelopmental gene expression.
The most troubling aspect of early-life ID is that the cognitive deficits (including learning and memory impairments) persist into adulthood despite iron treatment in infancy, implying that iron therapy alone is not sufficient for full recovery [6]. Understanding the iron-dependent molecular/cellular mechanisms that directly cause early-life neurological dysfunction will allow future design of more effective therapies to prevent the long-lasting effects of early-life ID. Our findings with a novel culture model of chronic hippocampal neuron ID will allow further interrogation of the specific iron-dependent mechanisms (e.g., mitochondrial respiration) underlying the effects of neuronal iron loss on dendrite maturation.
Supplementary Material
Supplemental Table 1: Seahorse Assay Protocol
Supplemental Figure 1: The effect of various DFO doses on Tfr1 and Slc11a2 gene expression at 18DIV. Hippocampal neurons cultured from E16 mice were treated with various DFO doses (i.e., 0, 1, 5, 8, or 10 μM) and 5-FU beginning at 3DIV. At 18DIV, cells were collected, total RNA was extracted, and cDNA was synthesized. Quantitative real-time PCR (qPCR) was performed for Tfr1 and Slc11a2, two IRE/IRP-regulated genes involved in neuronal iron uptake that index neuronal iron status. Relative mRNA levels are calculated relative to an internal control cDNA sample and a reference gene (i.e., Tbp). The data from 1–3 independent cultures were pooled and are presented as mean ± SEM. Groups not sharing a common letter (a > b > c) are significantly different by one-way ANOVA and Tukey’s post-hoc multiple comparison test (p < 0.05). n=4–10.
Supplemental Figure 2: The effect of DFO treatment on neuronal zinc status. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3DIV. At 11DIV, cells were collected, total RNA was extracted, and cDNA was synthesized. Quantitative real-time PCR (qPCR) was performed for Znt-1, a gene coding for a zinc transporter, to index neuronal zinc status. Relative mRNA levels are calculated relative to an internal control cDNA sample and a reference gene (i.e., Tbp). The data are presented as mean ± SEM. There was no statistical difference between groups by Student’s t-test (p < 0.05). n=5.
Acknowledgments
Grants and fellowships supporting the writing of this paper:
Grant Sponsor: NIH; Grant number: R01 HD029421
We thank the members of the Georgieff and Lanier labs for their invaluable assistance with culture preparation and tracing analysis. In particular we would like to thank Justin Campagna, Lanka Dasanayaka, and George Michalopoulos. Grants supporting this research included NIH R01 HD029421.
Footnotes
Disclosure statement: The authors have nothing to declare.
References
- 1.McLean E, Cogswell M, Egli I, Wojdyla D, de Benoist B. Worldwide prevalence of anaemia, who vitamin and mineral nutrition information system, 1993–2005. Public Health Nutr. 2009;12:444–454. doi: 10.1017/S1368980008002401. [DOI] [PubMed] [Google Scholar]
- 2.Yip R. Iron deficiency: Contemporary scientific issues and international programmatic approaches. J Nutr. 1994;124:1479S–1490S. doi: 10.1093/jn/124.suppl_8.1479S. [DOI] [PubMed] [Google Scholar]
- 3.Cogswell ME, Looker AC, Pfeiffer CM, Cook JD, Lacher DA, Beard JL, Lynch SR, Grummer-Strawn LM. Assessment of iron deficiency in us preschool children and nonpregnant females of childbearing age: National health and nutrition examination survey 2003–2006. Am J Clin Nutr. 2009;89:1334–1342. doi: 10.3945/ajcn.2008.27151. [DOI] [PubMed] [Google Scholar]
- 4.Mei Z, Cogswell ME, Looker AC, Pfeiffer CM, Cusick SE, Lacher DA, Grummer-Strawn LM. Assessment of iron status in us pregnant women from the national health and nutrition examination survey (nhanes), 1999–2006. Am J Clin Nutr. 2011;93:1312–1320. doi: 10.3945/ajcn.110.007195. [DOI] [PubMed] [Google Scholar]
- 5.Taylor EM, Morgan EH. Developmental changes in transferrin and iron uptake by the brain in the rat. Brain Res Dev Brain Res. 1990;55:35–42. doi: 10.1016/0165-3806(90)90103-6. [DOI] [PubMed] [Google Scholar]
- 6.Fretham SJB, Carlson ES, Georgieff MK. The role of iron in learning and memory. Advances in Nutrition: An International Review Journal. 2011;2:112–121. doi: 10.3945/an.110.000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Squire LR. Memory systems of the brain: A brief history and current perspective. Neurobiol Learn Mem. 2004;82:171–177. doi: 10.1016/j.nlm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Fretham SJ, Carlson ES, Wobken J, Tran PV, Petryk A, Georgieff MK. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus. 2012;22:1691–1702. doi: 10.1002/hipo.22004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukumitsu K, Fujishima K, Yoshimura A, Wu YK, Heuser J, Kengaku M. Synergistic action of dendritic mitochondria and creatine kinase maintains atp homeostasis and actin dynamics in growing neuronal dendrites. J Neurosci. 2015;35:5707–5723. doi: 10.1523/JNEUROSCI.4115-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oruganty-Das A, Ng T, Udagawa T, Goh EL, Richter JD. Translational control of mitochondrial energy production mediates neuron morphogenesis. Cell Metab. 2012;16:789–800. doi: 10.1016/j.cmet.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheng ZH. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol. 2014;204:1087–1098. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dallman PR. Biochemical basis for the manifestations of iron deficiency. Annu Rev Nutr. 1986;6:13–40. doi: 10.1146/annurev.nu.06.070186.000305. [DOI] [PubMed] [Google Scholar]
- 13.de Ungria M, Rao R, Wobken JD, Luciana M, Nelson CA, Georgieff MK. Perinatal iron deficiency decreases cytochrome c oxidase (cytox) activity in selected regions of neonatal rat brain. Pediatr Res. 2000;48:169–176. doi: 10.1203/00006450-200008000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Koleske AJ. Molecular mechanisms of dendrite stability. Nat Rev Neurosci. 2013;14:536–550. doi: 10.1038/nrn3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akum BF, Chen M, Gunderson SI, Riefler GM, Scerri-Hansen MM, Firestein BL. Cypin regulates dendrite patterning in hippocampal neurons by promoting microtubule assembly. Nat Neurosci. 2004;7:145–152. doi: 10.1038/nn1179. [DOI] [PubMed] [Google Scholar]
- 16.Michaelsen K, Murk K, Zagrebelsky M, Dreznjak A, Jockusch BM, Rothkegel M, Korte M. Fine-tuning of neuronal architecture requires two profilin isoforms. Proc Natl Acad Sci U S A. 2010;107:15780–15785. doi: 10.1073/pnas.1004406107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosario M, Schuster S, Juttner R, Parthasarathy S, Tarabykin V, Birchmeier W. Neocortical dendritic complexity is controlled during development by noma-gap-dependent inhibition of cdc42 and activation of cofilin. Genes Dev. 2012;26:1743–1757. doi: 10.1101/gad.191593.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parrish JZ, Emoto K, Kim MD, Jan YN. Mechanisms that regulate establishment, maintenance, and remodeling of dendritic fields. Annu Rev Neurosci. 2007;30:399–423. doi: 10.1146/annurev.neuro.29.051605.112907. [DOI] [PubMed] [Google Scholar]
- 19.Qin X, Jiang Y, Tse Y, Wang Y, Wong T, Paudel H. Early growth response 1 (egr-1) regulates nmda receptor-dependent transcription of psd-95 and ampa receptor trafficking in hippocampal primary neurons. J Biol Chem. 2015 doi: 10.1074/jbc.M115.668889. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zimmermann J, Trimbuch T, Rosenmund C. Synaptobrevin 1 mediates vesicle priming and evoked release in a subpopulation of hippocampal neurons. J Neurophysiol. 2014;112:1559–1565. doi: 10.1152/jn.00340.2014. [DOI] [PubMed] [Google Scholar]
- 21.Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by camkii. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- 22.El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. Psd-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- 23.Siddappa AJ, Rao RB, Wobken JD, Leibold EA, Connor JR, Georgieff MK. Developmental changes in the expression of iron regulatory proteins and iron transport proteins in the perinatal rat brain. J Neurosci Res. 2002;68:761–775. doi: 10.1002/jnr.10246. [DOI] [PubMed] [Google Scholar]
- 24.Carlson ES, Stead JD, Neal CR, Petryk A, Georgieff MK. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus. 2007;17:679–691. doi: 10.1002/hipo.20307. [DOI] [PubMed] [Google Scholar]
- 25.Pokorny J, Yamamoto T. Postnatal ontogenesis of hippocampal ca1 area in rats. I. Development of dendritic arborisation in pyramidal neurons. Brain Res Bull. 1981;7:113–120. doi: 10.1016/0361-9230(81)90075-7. [DOI] [PubMed] [Google Scholar]
- 26.Brunette KE, Tran PV, Wobken JD, Carlson ES, Georgieff MK. Gestational and neonatal iron deficiency alters apical dendrite structure of ca1 pyramidal neurons in adult rat hippocampus. Dev Neurosci. 2010;32:238–248. doi: 10.1159/000314341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greminger AR, Lee DL, Shrager P, Mayer-Proschel M. Gestational iron deficiency differentially alters the structure and function of white and gray matter brain regions of developing rats. J Nutr. 2014;144:1058–1066. doi: 10.3945/jn.113.187732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jorgenson LA, Sun M, O’Connor M, Georgieff MK. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area ca1 of the developing rat hippocampus. Hippocampus. 2005;15:1094–1102. doi: 10.1002/hipo.20128. [DOI] [PubMed] [Google Scholar]
- 29.Felt BT, Beard JL, Schallert T, Shao J, Aldridge JW, Connor JR, Georgieff MK, Lozoff B. Persistent neurochemical and behavioral abnormalities in adulthood despite early iron supplementation for perinatal iron deficiency anemia in rats. Behav Brain Res. 2006;171:261–270. doi: 10.1016/j.bbr.2006.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kennedy BC, Dimova JG, Siddappa AJ, Tran PV, Gewirtz JC, Georgieff MK. Prenatal choline supplementation ameliorates the long-term neurobehavioral effects of fetal-neonatal iron deficiency in rats. J Nutr. 2014;144:1858–1865. doi: 10.3945/jn.114.198739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson ES, Tkac I, Magid R, O’Connor MB, Andrews NC, Schallert T, Gunshin H, Georgieff MK, Petryk A. Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr. 2009;139:672–679. doi: 10.3945/jn.108.096354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pisansky MT, Wickham RJ, Su J, Fretham S, Yuan LL, Sun M, Gewirtz JC, Georgieff MK. Iron deficiency with or without anemia impairs prepulse inhibition of the startle reflex. Hippocampus. 2013;23:952–962. doi: 10.1002/hipo.22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strasser GA, Rahim NA, VanderWaal KE, Gertler FB, Lanier LM. Arp2/3 is a negative regulator of growth cone translocation. Neuron. 2004;43:81–94. doi: 10.1016/j.neuron.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- 35.Penrod RD, Campagna J, Panneck T, Preese L, Lanier LM. The presence of cortical neurons in striatal-cortical co-cultures alters the effects of dopamine and bdnf on medium spiny neuron dendritic development. Frontiers in Cellular Neuroscience. 2015:9. doi: 10.3389/fncel.2015.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes alzheimer’s pathology in female mouse model of alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zolotukhin S, Potter M, Hauswirth WW, Guy J, Muzyczka N. A “Humanized” Green fluorescent protein cdna adapted for high-level expression in mammalian cells. J Virol. 1996;70:4646–4654. doi: 10.1128/jvi.70.7.4646-4654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider CA, Rasband WS, Eliceiri KW. Nih image to imagej: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popko J, Fernandes A, Brites D, Lanier LM. Automated analysis of neuronj tracing data. Cytometry A. 2009;75:371–376. doi: 10.1002/cyto.a.20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- 42.Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. 2. Cambridge, MA: The MIT Press; 1998. pp. 339–370. [Google Scholar]
- 43.Nguyen SM, Lieven CJ, Levin LA. Simultaneous labeling of projecting neurons and apoptotic state. J Neurosci Methods. 2007;161:281–284. doi: 10.1016/j.jneumeth.2006.10.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: Regulation of mammalian iron metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 45.Petry CD, Eaton MA, Wobken JD, Mills MM, Johnson DE, Georgieff MK. Iron deficiency of liver, heart, and brain in newborn infants of diabetic mothers. J Pediatr. 1992;121:109–114. doi: 10.1016/s0022-3476(05)82554-5. [DOI] [PubMed] [Google Scholar]
- 46.Liu ZD, Hider RC. Design of clinically useful iron(iii)-selective chelators. Med Res Rev. 2002;22:26–64. doi: 10.1002/med.1027. [DOI] [PubMed] [Google Scholar]
- 47.Chowanadisai W, Kelleher SL, Lonnerdal B. Zinc deficiency is associated with increased brain zinc import and liv-1 expression and decreased znt-1 expression in neonatal rats. J Nutr. 2005;135:1002–1007. doi: 10.1093/jn/135.5.1002. [DOI] [PubMed] [Google Scholar]
- 48.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins ucp2 and ucp3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 49.Matsuda S, Kitagishi Y, Kobayashi M. Function and characteristics of pink1 in mitochondria. Oxid Med Cell Longev. 2013;2013:601587. doi: 10.1155/2013/601587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fornuskova D, Stiburek L, Wenchich L, Vinsova K, Hansikova H, Zeman J. Novel insights into the assembly and function of human nuclear-encoded cytochrome c oxidase subunits 4, 5a, 6a, 7a and 7b. Biochem J. 2010;428:363–374. doi: 10.1042/BJ20091714. [DOI] [PubMed] [Google Scholar]
- 51.Jorgenson LA, Wobken JD, Georgieff MK. Perinatal iron deficiency alters apical dendritic growth in hippocampal ca1 pyramidal neurons. Dev Neurosci. 2003;25:412–420. doi: 10.1159/000075667. [DOI] [PubMed] [Google Scholar]
- 52.Tran PV, Carlson ES, Fretham SJ, Georgieff MK. Early-life iron deficiency anemia alters neurotrophic factor expression and hippocampal neuron differentiation in male rats. J Nutr. 2008;138:2495–2501. doi: 10.3945/jn.108.091553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McAllister AK. Cellular and molecular mechanisms of dendrite growth. Cereb Cortex. 2000;10:963–973. doi: 10.1093/cercor/10.10.963. [DOI] [PubMed] [Google Scholar]
- 54.Siddappa AM, Georgieff MK, Wewerka S, Worwa C, Nelson CA, Deregnier RA. Iron deficiency alters auditory recognition memory in newborn infants of diabetic mothers. Pediatr Res. 2004;55:1034–1041. doi: 10.1203/01.pdr.0000127021.38207.62. [DOI] [PubMed] [Google Scholar]
- 55.Monk C, Georgieff MK, Xu D, Hao X, Bansal R, Gustafsson H, Spicer J, Peterson BS. Maternal prenatal iron status and tissue organization in the neonatal brain. Pediatr Res. 2015 doi: 10.1038/pr.2015.248. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rao R, Tkac I, Townsend EL, Gruetter R, Georgieff MK. Perinatal iron deficiency alters the neurochemical profile of the developing rat hippocampus. J Nutr. 2003;133:3215–3221. doi: 10.1093/jn/133.10.3215. [DOI] [PubMed] [Google Scholar]
- 57.Rao R, Tkac I, Schmidt AT, Georgieff MK. Fetal and neonatal iron deficiency causes volume loss and alters the neurochemical profile of the adult rat hippocampus. Nutr Neurosci. 2011;14:59–65. doi: 10.1179/1476830511Y.0000000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kossel AH, Williams CV, Schweizer M, Kater SB. Afferent innervation influences the development of dendritic branches and spines via both activity-dependent and non-activity-dependent mechanisms. J Neurosci. 1997;17:6314–6324. doi: 10.1523/JNEUROSCI.17-16-06314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basarsky TA, Parpura V, Haydon PG. Hippocampal synaptogenesis in cell culture: Developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buratowski S. The basics of basal transcription by rna polymerase ii. Cell. 1994;77:1–3. doi: 10.1016/0092-8674(94)90226-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Seahorse Assay Protocol
Supplemental Figure 1: The effect of various DFO doses on Tfr1 and Slc11a2 gene expression at 18DIV. Hippocampal neurons cultured from E16 mice were treated with various DFO doses (i.e., 0, 1, 5, 8, or 10 μM) and 5-FU beginning at 3DIV. At 18DIV, cells were collected, total RNA was extracted, and cDNA was synthesized. Quantitative real-time PCR (qPCR) was performed for Tfr1 and Slc11a2, two IRE/IRP-regulated genes involved in neuronal iron uptake that index neuronal iron status. Relative mRNA levels are calculated relative to an internal control cDNA sample and a reference gene (i.e., Tbp). The data from 1–3 independent cultures were pooled and are presented as mean ± SEM. Groups not sharing a common letter (a > b > c) are significantly different by one-way ANOVA and Tukey’s post-hoc multiple comparison test (p < 0.05). n=4–10.
Supplemental Figure 2: The effect of DFO treatment on neuronal zinc status. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3DIV. At 11DIV, cells were collected, total RNA was extracted, and cDNA was synthesized. Quantitative real-time PCR (qPCR) was performed for Znt-1, a gene coding for a zinc transporter, to index neuronal zinc status. Relative mRNA levels are calculated relative to an internal control cDNA sample and a reference gene (i.e., Tbp). The data are presented as mean ± SEM. There was no statistical difference between groups by Student’s t-test (p < 0.05). n=5.