

Abstract
The discovery of driver genes is a major pursuit of cancer genomics, usually based on observing the same mutation in different patients. But the heterogeneity of cancer pathways plus the high background mutational frequency of tumor cells often cloud the distinction between less frequent drivers and innocent passenger mutations. Here, to overcome these disadvantages, we grouped together mutations from close kinase paralogs under the hypothesis that cognate mutations may functionally favor cancer cells in similar ways. Indeed, we find that kinase paralogs often bear mutations to the same substituted amino acid at the same aligned positions and with a large predicted Evolutionary Action. Functionally, these high Evolutionary Action, non-random mutations affect known kinase motifs, but strikingly, they do so differently among different kinase types and cancers, consistent with differences in selective pressures. Taken together, these results suggest that cancer pathways may flexibly distribute a dependence on a given functional mutation among multiple close kinase paralogs. The recognition of this “mutational delocalization” of cancer drivers among groups of paralogs is a new phenomena that may help better identify relevant mechanisms and therefore eventually guide personalized therapy.
1. Introduction
A major focus of recent cancer sequencing projects, such as the TCGA, is to identify causal driver mutations responsible for tumorigenesis (1). To this end, many computational tools have been produced to predict the impact of mutations on protein function in order to screen out null function or low impact mutations (2). The efforts of these approaches have identified many proteins and mutations driving cancer progression. Unfortunately, the inherent mutational heterogeneity displayed within cancer often limits the statistical power of these methods so as to capture only the most frequent driver mutations in a large cohort of patients (3). By contrast, low frequency drivers or smaller patient cohorts suffer from a lack of statistical significance and are therefore easily missed.
While infrequent mutations in a single gene may, at first glance, appear to indicate insignificance in cancer progression, this may be an oversimplification. Driver mutations in cancer may not only target a single gene but rather groups of genes or functional pathways, distributing the mutational burden across many functionally related genes (4, 5); while a single gene may lack significance, combining mutations across a regulatory pathway can increase the power of the analysis and identify gene groups driving cancer progression (3, 6). Prior studies have taken these groups from databases such as KEGG (7), Reactome (8), and analyses of gene association networks like STRING (9). However, these approaches are not limited to functional or hierarchical pathways but rather could be applied to any group of proteins that share functionality such as, Gene Ontology terms or even groups of protein homologs sharing significant functional overlap.
Further confounding the prediction of cancer drivers, single gene analyses group mutations regardless of their structural location and, therefore, do not account for the functional heterogeneity of these mutations. To account for these difference, an analysis in Colon and Breast Cancers grouped mutations from various genes occurring in homologous protein domains, finding specific domains enriched for high frequency mutations across many individual proteins (10). Furthermore, an analysis of disease-related mutations across all human kinases showed that these mutations preferentially localized in specific structural domains, affected certain residues types, and had conserved amino acid substitutions (11). These studies show disease-related mutations can preferentially occur at specific structural domains in homologous proteins, such as kinases, and that kinase mutations share conserved patterns of substitution. Here, we expand upon this work and ask whether there are mutational biases in individual positions in the context of cancer.
For the purpose of this study, we focus on human kinases in order to better understand this essential protein family and how it contributes to cancer. There are over 500 human kinases sharing substantial homology in both the kinase structure and the catalytic mechanism (12). The kinase family has been further subdivided into 7 classes based on substrate specificity and evolutionary lineage. Kinases are ubiquitous proteins involved in a diverse array of cellular functions; as a result, numerous perturbations in kinase coding regions, translation, and expression lead to disease and cancer progression (13). Moreover, after G protein-coupled receptors, kinases are the second most drugged protein family (11). While some kinases such as BRAF, EGFR, and PI3-kinase demonstrate a remarkably high mutation rate within cancer (14, 15), many kinases are mutated at a much lower frequency making it difficult to access their influence on cancer progression
Here, we hypothesize that some closely related kinases may act as a single functional group from the perspective of a cancer type. That is, mutations at the same (cognate) position across a group of kinases may have a similar functional effect and fulfill the same selective pressure, leading to positional enrichment of impactful mutations within the cancer. To test this possibility, we used kinase alignments and exomic mutations from the TCGA to group all mutations occurring at the same sequence position and then quantified the predicted functional impact using Evolutionary Action (EA). We identified highly conserved, functionally related positions with a significantly increased mutation rate in a pan-cancer and pan-kinase analysis. Additionally, mutational differences are clear between the various kinase subclasses and additional differences across cancer types. This work shows a novel method that moves beyond a single gene approach and which suggests that functionally related homologous proteins may bear driver mutations that substitute for each other to support cancer progression.
2. Methods
2.1. Evolutionary Trace and Action Analysis
To identify evolutionarily important residues, we performed Evolutionary Trace (ET) analysis on each of the kinase sub-families as previously described (16). ET utilizes changes in genotype and corresponding phenotypic divergences in the phylogenetic tree to score the evolutionary importance of each residue in a protein sequence. In previous work, ET has identified functional sites and their determinants so as to guide mutational engineering in case studies (17, 18).
Evolutionary Action (19) builds upon ET to predict the impact a mutation has on protein function by multiplying the importance of the position (ET) by the magnitude of the substitution (evolutionary substitution odds). Prediction scores are then normalized for each individual kinase so the range falls between a predicted effect that is null, 0, to one that is most impactful, 100. EA has been repeatedly validated. It was shown to correctly predict mutation impact in multiple systems (e.g. P53, RecA, bacteriophage T4 lysozyme, etc.), it also outcompeted state of the art methods in the past 3 CAGI challenges (Critical Assessment of Genome Interpretation) (19), and in a clinical context, it can stratify patients with head and neck cancer based on their p53 mutational status (28). Using this technique we score each mutations predicted impact.
2.2. Kinase Alignment, Mutation Acquisition and Mapping
In order to compare mutations across all human kinases, we aligned separately each of the 7 major subclasses from The Human Kinome project (20). These alignments were used as a translation tool, in order to map mutations across human kinases onto canonical protein sequences. Representative crystallized structures were selected for each sub-family to visualize analysis. Representative proteins can be found in the supplement and were manually chosen based on: 1) the availability of a high resolution crystal structure 2) their similarity to other proteins within that class and finally 3) with a focus on longer proteins so as to limit the number of blank alignment positions when mapping other proteins onto the structure.
Mutation data was acquired from the TCGA for 21 major cancer types using the computationally annotated calls. Chromosome positions were converted to protein position using ANNOVAR (21) and then were each mapped onto the representative sequence within the alignments. In this way we were able to measure how mutations within kinases distribute throughout the conserved kinase domain.
Unless otherwise stated, all mutation numbering is in relation to the representative structure from TKL kinases (ACTR2B-2QLU). For visualization purposes on the structure, sphere size of each position was scaled based on frequency of high impact mutations (EA>40) according to the equation:
| (1) |
Initially this analysis was performed on each of the seven kinase subclasses (358 individual kinases total) using separate alignments and representative structures for each subclass. CK1 kinases were dropped from the analysis due to insufficient mutations. The remaining six individual representative structures were then aligned and merged into a complete pan-cancer analysis.
2.3. Random Controls
See Supplement for additional Methods at http://mammoth.bcm.tmc.edu/GallionEtAlPSB/
3. Results
3.1. Evolutionary Trace Identifies Functionally Important and Divergent Kinase Positions
In order to gauge the impact of kinase mutations we first sought to identify key functional residues and sites in kinases. This was done using Evolutionary Trace (ET). Figure 1 shows the ET ranks from most to least important (red to blue) mapped onto the structure of ACTR2B, 2QLU (PDB-ID). As expected, functionally essential motifs, such as the magnesium binding DFG motif and the catalytic HRD motif emerge as ET hotspots. ET also suggests functionally relevant residues throughout the substrate pocket and allosteric sites consistent with known protein functionality. Positions predicted to be the least important tend to cluster near the edges of helices, the loop regions, and near solvent exposed positions. Repeating ET analysis on each individual class, we are able to identify positions important to each group. These results confirm that in kinases, ET is able to identify both universally important positions as well as the positions that are evolutionarily divergent among subfamilies correlating to divergent functionalities.
Fig 1.

Evolutionary Trace Analysis of ACTR2B (2QLU) identifies evolutionarily important residues corresponding to known motifs.
3.2. Kinase Mutations Demonstrate Non-Random Structural Pattern In TCGA
To explore structural biases of kinase mutations in cancer, we next conducted a pan-cancer analysis of TCGA data. This analysis grouped mutations occurring at the same sequence position across kinase evolutionary history. This broad pan-cancer analysis identifies 77 residues with a statistically significant mutation rate (p-value<0.01) compared to control (See Supplementary). Then, in order to focus on the subset of impactful mutations and screen out low impact polymorphisms, we repeated the above analysis only using mutations with EA scores greater than 40, and mapped them onto the ET analysis of ACTR2B (Figure 2A). All positions are numbered based on the 2QLU structure unless otherwise specified. For example, the well-known driver mutations from BRAF-V600 (equivalent position V344 in figure) and CHEK2-K373 (R345 in figure) are the most frequently mutated, high impact mutations. Other frequently mutated positions with high impact substitutions occur at known functional residues, such as the glycine-rich region G199, the DFG motif D339, the HRD domain R320 and D321, and a conserved ion-pairing residue R468. Since these mutations involve positions with large ET scores, they are likely to impair protein function. By contrast, and as seen in Figure 2B, there are 54 residues mutated at a lower rate than expected (p-value<0.01). These seldom mutated positions, shown by the small sphere size in Figure 2A fall preferentially in the solvent exposed loop regions of the kinase that are evolutionarily less important according to ET and thus unlikely to have much functional consequence. These data show that kinase mutations in cancer are not evenly distributed throughout the structure. Rather many mutations preferentially fall non-randomly so as to recurrently involve functionally important cognate positions within conserved motifs, where they are likely to be disruptive; conversely, in the loop regions, which are less important, mutations are more rare and involve positions of lesser importance.
Fig 2.

Kinase Mutation Pattern Pancancer (A) Pan-cancer mutations, with an EA>40, mapped onto ACTR2B structure where sphere size=frequency, color=ET importance. (B) Actual mutation frequency significantly varies from Poisson Distribution.
3.3. Frequently Mutated Positions are Enriched for Mutations Predicted to Have a Significant Impact on Protein Function
To further explore the functional consequences of these mutations, we used EA to predict the functional impact of each mutation on protein function. EA combines the evolutionary importance of the position (ET) with the likelihood of that substitution, based on all evolutionary history, in order to predict the impact of a mutation on protein function. We compared the EA score distribution of frequent positions and infrequent positions (p-value<0.01) to the distribution of all kinase domain mutations from the TCGA using a two-sided t-test (Figure 3A). In agreement with the structural and ET biases, the frequently mutated positions are predicted to have a higher impact on protein function (p-value=10−28) while the infrequently mutated positions are biased towards lower impact mutations (p-value= 10−5). These data show the frequently mutated positions from the TCGA are further enriched for high impact mutations, while those positions infrequently mutated are predicted to have little functional effect.
Fig 3.

(A) High frequency mutations are significantly biased towards high impact mutations (Pvalue=5*10−28) while low frequency mutations are biased towards low predicted impact (Pvalue 2.5*10−05). Mean=diamond Median=red line Whisk=2STD (B) Observed kinase mutation rate compared to computer simulation of random mutations. BRAF and CHEK2 (550 and 160 mutations, respectively) not shown on plot.
3.4. Frequently Mutated Positions Occur in Many Different Kinases at a Low Individual Frequency
While these cancer somatic mutations demonstrate site specificity, we next investigated which individual kinases carried these mutations and whether specific proteins drove this pattern. The mutation frequency of each individual kinase is displayed in Figure 3B and is compared against a random simulation in which the same number of mutations were randomly distributed to an equal number of proteins. The random distribution had a mean value of 21.4 mutations per kinase while the experimental distribution, after dropping out the outliers BRAF and CHEK2 (550 and 160 mutations, respectively), had a mean of 19.5. We note that the mutation rate in individual kinases is more variable than expected. Overall the distribution is leftward shifted compared to control with a select number of proteins hypermutated: 29% of kinases were mutated at a decreased frequency (p-value<0.05) while only 14% of kinases were significantly hypermutated (p-value<0.05). Of the hypermutated kinases, nine were mutated at an exceptionally high rate (>50 mutations/protein); many of these however, represent known, high frequency driver mutations occurring at the same location in the same kinase (e.g. BRAF, CHEK2, and EGFR). These data show that within cancer cells, certain kinases experience a remarkably increased mutation rate while the majority of the remaining kinases are hypomutated, typically with fewer that 20 SNVs across a pan-cancer analysis.
However, while this analysis recapitulates known drivers such as L858R within EGFR, it further identifies mutations at a single residue that individually occur at a low frequency but, taken as a whole, occur at a high frequency. For instance, Table 1 displays a random selection of mutations occurring at the Asp residue of the HRD domain (p-value=2×10−4). While each individual mutation has a conserved amino acid transition, individual proteins are mutated infrequently with a median value of 1 and a maximal value of 5 mutations (occurring within MAP2K7). Of the original 54 positions with a p-value<0.01 only 6 are at least partially driven by a single protein (1 protein with >20% of the mutations), while all remaining positions were significant only through this combination. These data show that while individual mutations may occur at low frequency, they frequently occur at homologous structural positions with the same native residue and amino acid substitution. Furthermore this pattern is distributed across many individual kinases without a single driver protein.
Table 1.
Random sample of mutations occurring at catalytic Asp residue from the HRD domain.
| Protein | Substitution | EA Score | Kinase Class | Cancer Type |
|---|---|---|---|---|
| PRKCI | D378N | 64.36 | AGC | PAAD |
| PRKG2 | D576Y | 98.63 | AGC | READ |
| CHEK1 | D130Y | 98.73 | CAMK | LUSC |
| STK17B | D158N | 74.06 | CAMK | SKCM |
| CLK4 | D286N | 63.19 | CMGC | COAD |
| MAPK4 | D149G | 90.95 | CMGC | STAD |
| MAP2K3 | D190N | 71.77 | STE | SKCM |
| MAP2K7 | D243N | 61.93 | STE | COAD |
| MAP2K7 | D243N | 61.93 | STE | PAAD |
| PAK7 | D568N | 75.15 | STE | SKCM |
| EPHA3 | D746N | 43.15 | TK | SKCM |
| FES | D683E | 67.14 | TK | BRCA |
| ROR2 | D615N | 74.82 | TK | SKCM |
| MAP3K7 | D156Y | 96.77 | TKL | LIHC |
3.5. Individual Kinase Classes Show Unique Mutational Patterns
Individual subclasses of kinases display marked functional and structural differences corresponding to their target specialization (12). To test if our conclusions held true despite these differences, we repeated the above analysis for each kinase class. As an example, three of these classes are displayed in Figure 4A. While the general location of these residues tend to stay near the catalytic site, the frequently mutated positions from each class vary. Nine residues in CMGC kinases form a statistically significant cluster (z-score=4.58) roughly localized around and occurring within the HRD domain. Seven residues in TK kinases are more broadly distributed throughout the structure with the three most frequent near the HRD domain. Finally, STE kinases seem to show two distinct areas of mutation, the HRD region and the ATP-binding hinge region. In all three cases, similar to the pan-cancer analysis, the most frequent positions tend to occur at evolutionarily important residues in functional motifs with high impact mutations. In addition to these differences, significant positions from the pan-kinase analysis are still significant in multiple classes (e.g. R320 (HRD motif) and R468 (Ion pair)). These data indicate that within cancer, certain positions are preferentially enriched in select kinase subclasses while other positions demonstrate broad enrichment across many or all kinase types.
Fig 4.

Individual kinase subclasses are frequently mutated at distinct positions (Left to Right: CMGC, TK, STE kinases). Sphere Size=Frequency, Color= ET importance from high to low (red to blue) for each representative kinase: ERK1, EphA5, PAK1 (respectively). All labels are based on ACTR2B numbering. (B) The protein mutation rate for each kinase class was compared against a simulated random distribution specific to the total number of mutations and proteins in each class. BRAF (TKL) and CHEK2 (CAMK) are not shown on their respective figures
We further note differences in the mutation frequency of proteins from each of the kinase classes (Figure 4B). In each class, some proteins are mutated at a significantly higher rate than expected. Proteins from the AGC kinase class are normally distributed with an exaggerated variance compared to random simulation, indicating that mutations within this class are fairly distributed to many proteins. Likewise, the mutation rate in CMGC and TK kinases is even more varied but still follow a roughly normal distribution centered around the expected mean. The distribution from CAMK, STE, and TKL kinases match the pan-kinase analysis with a leftward shifted distribution displaying many hypomutated proteins and several hypermutated proteins. As 43% of all mutations within TKL kinases occur in BRAF, we have removed these mutations from this analysis. However, creating a random distribution for this class without first removing this outlier shifts the random distribution right (mean=30). This data shows that, in addition to structural differences, the individual kinase classes are mutated at different rates, with some classes having broadly distributed mutations to many individual proteins while other classes are primarily mutated in a select few proteins.
3.6. Kinases Further Demonstrate Cancer Type Specific Mutational Patterns
Variances between kinase subtypes led us to next speculate that certain protein positions could have varying functional importance to specific cancer types as well. The above analysis was repeated, now grouping all kinases together and instead performing a cancer-specific analysis for 7 cancer types within the TCGA [Breast invasive carcinoma (BRCA), Bladder Urothelial Carcinoma (BLCA), Colon adenocarcinoma (COAD), Head and Neck squamous cell carcinoma (HNSC), Lung adenocarcinoma (LUAD), Skin Cutaneous Melanoma (SKCM), and Stomach adenocarcinoma (STAD)]. The most frequently mutated position for all but BRCA and STAD was 345 and 346, driven by the high frequency driver mutations BRAF-V600 and CHEK2-K373 (respectively); these mutations were then removed from this analysis in order to search for novel other positions. Figure 5 shows a selection of positions that were significantly mutated within specific cancer types. Interestingly, the analyses from LUAD and STAD resulted in clusters of mutations within the kinase domain. Some positions were significant in two cancer types, such as L325 in LUAD and BRCA. In agreement with the pan-cancer analysis, R468 was frequently mutated in many cancer types including STAD and COAD. These data indicate that individual cancer types are enriched for varying structural positions across many individual kinases.
Fig 5.
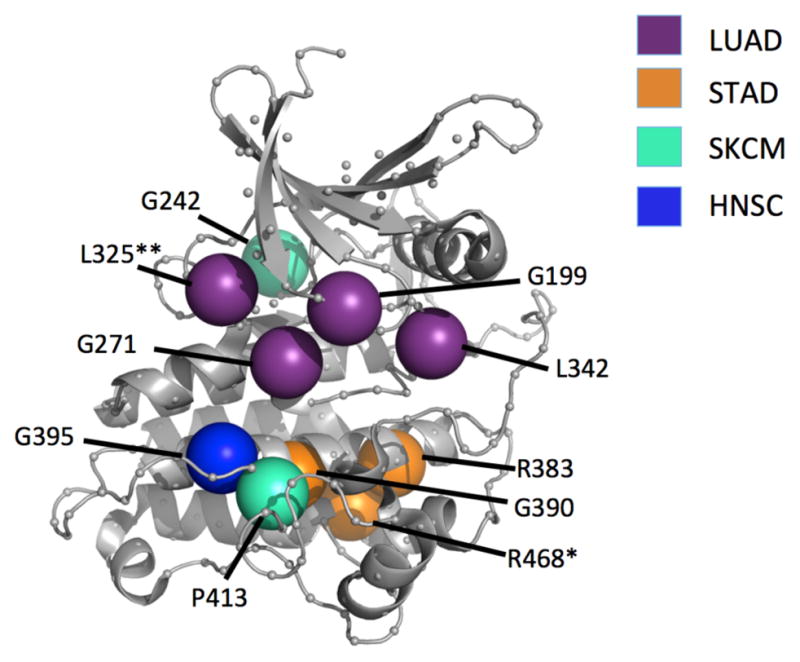
Cancer types demonstrate some specificity towards certain mutation positions. *Occurs in STAD and COAD **Occurs in both LUAD and BRCA
4. Discussion
In order to better predict driver mutations within cancer, computational methods have been extended from gene-by-gene analyses to consider instead groupings of mutations in functional pathways or subnetworks (3, 6, 22). In this manner, driver proteins mutated at a low frequency due to the heterogeneity within cancer that are missed by a single gene analysis can still be identified despite their low individual frequency. Being able to predict these diverse infrequent drivers of cancer helps move medicine closer to personalized diagnoses and care. Here, as an alternate way to group genes, we explored protein homology rather than curated hierarchical pathways and gene interactions. Strikingly, we find that among kinases, mutations are structurally biased to functional motifs and evolutionarily important residues.
Mutations providing a benefit to cancer cells become clonally enriched, as that cell proliferates more efficiently than others in the tumor population (5). From the pan-kinase analysis, we identified positions frequently mutated across many individual kinases. While the known high frequency driver genes were captured in this analysis, an additional 39 positions were mutated at a low frequency in any given kinase but were significantly mutated across the kinase family. These high frequency positions were preferentially biased for high impact mutations, strongly suggesting a significant effect on protein function. In contrast, the infrequently mutated positions all occurred at evolutionarily unimportant loop regions with a bias towards low impact mutations. These data indicate that enrichment is correlated to functional impact. Presumably, the high-impact mutations across many kinases provide a functional benefit within the cancer cell and are therefore enriched, whereas low-impact mutations, providing little benefit to the cancer cell, are lost from the population resulting in a low mutation rate at those positions.
Previous work in kinases has demonstrated that identical mutations in two different kinases can result in the same phenotype (23, 24). For instance, mutations conferring resistance to kinase inhibitors in EGFR occur at the same position as drug resistance mutations in BCR-ABL, PDGFRA and KIT (25, 26). A systematic study of mutation locations built upon these observations and demonstrated the existence of ‘domain hotspots’: frequently mutated regions in many proteins leading to the same functional consequence (22). In the context of this analysis of exomic mutations from TCGA, these frequently mutated positions, across many different kinases, with the exact same substitution strongly suggests a conserved functional mechanism driving enrichment: the same mutation in two different kinases likely producing a similar benefit in cancer.
The kinase catalytic mechanism itself is highly conserved across all kinases and is orchestrated by groups of functional motifs; these same positions in all kinases are responsible for the same functions (12). These motifs are themselves frequently mutated at a high frequency within cancer; in fact, the majority of the frequently mutated positions occur at or nearby conserved motifs. These positions are often studied in the context of kinases enabling us to speculate on their functional consequences within cancer. For instance, the catalytic Asp and Arg residues of the HRD domain are both mutated in many diverse kinases and are furthermore mutated to the same types of residues (D→N and R→Q/W/H respectively) in each case. Previous characterization of the D→N mutations within the Drosophila Src64 kinase indicate this mutation is equivalent to a gene knockout (27). Cancer cells carrying this SNV would therefore experience a loss of kinase activity within this protein, possibly suggesting a tumor suppressing mechanism. What remains to be determined is how far these characterization studies can be extrapolated to other kinases. Further experimental studies are needed in which the same mutation is characterized in multiple proteins to assess how universal these conclusions are. However, given 1) the initial conserved function of these positions, 2) the significant enrichment of the same substitution across many proteins, and 3) the sizeable predicted consequence of these mutations: it becomes tantalizing to suggest that the same mutation in different kinases may produce the same functional benefit in cancer, regardless of the kinase where it occurs.
This hypothesis is further supported by the kinase subclass and cancer type specific analyses. The individual kinase sub-families are evolved to phosphorylate different types of proteins (12). As a result, they have diverged. While the overall structure is conserved, some positions are specific for given target proteins and therefore differ among kinase classes. Likewise, while some positions are broadly mutated, the class specific analyses demonstrate appreciable differences; some positions are enriched in one class but do not occur in another. These variations between kinase classes likely stem from their functional divergence. Mutations occurring at important positions in one class may be beneficial, while the same position in a different class may not, resulting in differential enrichment. Furthermore, cancer types themselves display heterogeneity among their causal driver mutations (5), a heterogeneity reflected within kinase mutations as well. Different cancer types are enriched for different kinase positions, again suggesting that some positions may be preferentially beneficial for one cancer type more so than another, and therefore clonally enriched. When the selection pressure varies, either by differing cancer types or by the different kinase classes, the positions of the enriched mutations also vary. This further suggests that a conserved functional mechanism drives this mutational enrichment across many individual kinases.
Cellular homeostasis and function is often maintained by a complex network of proteins with significant functional overlap and crosstalk between functional homologues. For this reason, a single gene approach to predicting driver mutations in cancer may be overly simplistic, therefore requiring a methodology to combine mutations based on functional similarity. Here, we propose that in addition to curated pathways, mutations can also be grouped across homologous protein families. Within kinases, we have demonstrated that individual proteins are enriched for mutations occurring at cognate positions utilizing the same substitutions. These results suggest that the selection pressure within certain cancers may be specific to the mutation’s location and not differentiate between which kinase carries the mutation. Taken together, these data show individual kinases may behave in a functionally redundant manner in cancer and that a combined analysis of their mutations could identify individually infrequent driver mutations, previously missed, that occur frequently across the entire class. The conserved nature of these mutations allows speculation as to their predicted functional effect by extrapolating previous characterization studies, in a single protein, to the other kinases. Finally, while these results are specific to kinases, similar analyses could be broadly applicable across many protein families, thereby shifting focus from a ‘protein specific’ to a ‘paralog-wide, cognate position specific’ analysis of cancer driver mutations.
Supplementary Material
Contributor Information
JONATHAN GALLION, Structural Computational Biology and Molecular Biophysics, Baylor College of Medicine, One Baylor Plaza Houston, TX, 77030, USA.
ANGELA D. WILKINS, Immunology, Lichtarge Laboratory BCM, One Baylor Plaza Houston, TX, 77030, USA
OLIVIER LICHTARGE, Structural Computational Biology and Molecular Biophysics, Baylor College of Medicine, One Baylor Plaza Houston, TX, 77030, USA.
References
- 1.Kaminker JS, et al. Cancer Res. 2007;67:465–473. doi: 10.1158/0008-5472.CAN-06-1736. [DOI] [PubMed] [Google Scholar]
- 2.Katsonis P, et al. Protein Sci. 2014;23:1650–1666. doi: 10.1002/pro.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vandin F, Clay P, Upfal E, Raphael BJ. Pac Symp Biocomput. 2012:55–66. [PubMed] [Google Scholar]
- 4.Vogelstein B, Kinzler KW. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Jia P, Zhao Z. PLoS Comput Biol. 2014;10:e1003460. doi: 10.1371/journal.pcbi.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanehisa M, Goto S. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croft D, et al. Nucleic Acids Res. 2014;42:D472–477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szklarczyk D, et al. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nehrt NL, Peterson TA, Park D, Kann MG. BMC Genomics. 2012;13(Suppl 4):S9. doi: 10.1186/1471-2164-13-S4-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torkamani A, Schork NJ. Genomics. 2007;90:49–58. doi: 10.1016/j.ygeno.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Endicott JA, Noble ME, Johnson LN. Annu Rev Biochem. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- 13.Greenman C, et al. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies H, et al. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 15.Lengyel E, Sawada K, Salgia R. Curr Mol Med. 2007;7:77–84. doi: 10.2174/156652407779940486. [DOI] [PubMed] [Google Scholar]
- 16.Wilkins A, Erdin S, Lua R, Lichtarge O. Methods Mol Biol. 2012;819:29–42. doi: 10.1007/978-1-61779-465-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang HJ, Wilkins AD, Lichtarge O, Wensel TG. J Biol Chem. 2015;290:2870–2878. doi: 10.1074/jbc.M114.622233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson SM, et al. Proc Natl Acad Sci U S A. 2015;112:7097–7102. doi: 10.1073/pnas.1502742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katsonis P, Lichtarge O. Genome Res. 2014;24(12):2050–2058. doi: 10.1101/gr.176214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yue P, et al. Hum Mutat. 2010;31:264–271. doi: 10.1002/humu.21194. [DOI] [PubMed] [Google Scholar]
- 23.Marks JL, et al. PLoS One. 2007;2:e426. doi: 10.1371/journal.pone.0000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies H, et al. Cancer Res. 2005;65:7591–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- 25.Pao W, et al. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi S, et al. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 27.Strong TC, Kaur G, Thomas JH. PLoS One. 2011;6:e28100. doi: 10.1371/journal.pone.0028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neskey, et al. Cancer Research. 2015;75(7):1527–36. doi: 10.1158/0008-5472.CAN-14-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.