

Abstract
Altered inhibition from parvalbumin-containing GABA neurons is thought to contribute to impaired gamma frequency oscillations and cognitive deficits in schizophrenia. Crabtree and colleagues report that proline dehydrogenase deficits produce excessive cytosolic levels of the GABA-mimetic L-proline which impairs GABA synthesis and gamma oscillations in a manner that mimics schizophrenia.
Keywords: 22q11, proline dehydrogenase, parvalbumin, GAD, cognition, postmortem, prefrontal cortex
In schizophrenia, fast-spiking inhibitory neurons that express the calcium-binding protein parvalbumin exhibit disturbances, including lower mRNA and protein levels for the 67 kD isoform of the GABA synthesizing enzyme glutamic acid decarboxylase (GAD67), in the prefrontal cortex (PFC) [1] (Figure 1). Parvalbumin-containing basket neurons, which provide perisomatic inhibitory input to pyramidal neurons, generate cortical oscillatory activity at gamma (30–80 Hz) frequencies that are thought to be critical for PFC-dependent cognitive processes, such as working memory [2]. Accordingly, working memory impairments in schizophrenia have been associated with deficits in PFC gamma power [3]. Thus, understanding the pathogenic processes that may affect GABA synthesis, such as altering GAD levels or activity, in parvalbumin neurons, may inform the mechanisms leading to deficient gamma oscillations and cognitive dysfunction in schizophrenia.
Figure 1.
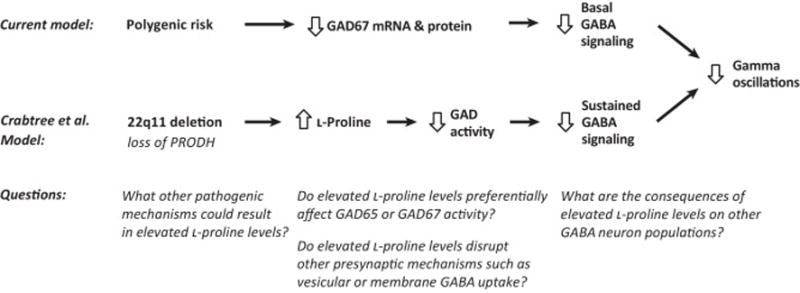
Schizophrenia, and other psychiatric disorders, are highly prevalent in individuals with chromosome 22q11 deletion syndrome. This deletion site contains the gene encoding proline dehydrogenase (PRODH), which degrades the amino acid L-proline. Recently, Crabtree et al, noting the structural similarities between L-proline and GABA, hypothesized that PRODH deficiency would increase L-proline levels and thus adversely affect GABA signaling in a manner relevant for schizophrenia [4]. They found that L-proline may indeed act as a GABA-mimetic and activate GABA receptors, although only at very high concentrations. Using electrophysiology in cortical slices from PRODH-deficient mice, the authors showed that GABA synaptic transmission onto pyramidal cells showed increased synaptic depression during sustained stimulation which suggests a presynaptic mechanism. Consistent with the idea that PRODH deficiency could alter GABA neurotransmission from parvalbumin-containing basket neurons, the authors found reduced oscillation power in the gamma band, but not in other frequencies, in local field potentials from the PFC of PRODH-deficient mice. Building on previous findings suggesting that in PRODH-deficient mice L-proline is primarily elevated intracellularly, the authors hypothesized that L-proline could affect GABA synthesis. Using a combination of studies in PRODH-deficient mice and cell transfection experiments, they found that elevated L-proline levels selectively and competitively inhibited GABA synthesis by GAD67. They also found that excessive L-proline levels primarily impair the response of GABA synapses during sustained stimulation, when demand for rapid GABA synthesis is high. Indeed, the effect on synaptic transmission could be reversed by an inhibitor of GABA transaminase which metabolizes GABA. Interestingly, high L-proline concentrations did not affect baseline GABA release, GABA receptors, or glutamatergic signaling, suggesting a specific effect on inhibiting GAD activity.
The results of Crabtree et al. suggest specific mechanisms by which altered GABA function could contribute to cognitive deficits in schizophrenia. First, their findings provide proof-of-concept evidence that deficient PRODH function, such as low protein levels due to 22q11 deletion, results in excessive cortical L-proline levels which then inhibit GAD activity (Figure 1). Interestingly, approximately half of schizophrenia subjects have severe deficits in PFC GAD67 mRNA levels in the PFC, while the remaining half have relatively normal GAD67 mRNA levels [5]. Consequently, these results suggest a new pathogenic mechanism by which GAD inhibition could lead to lower GABA levels in schizophrenia subjects with otherwise normal GAD levels (Figure 1). Second, they demonstrate that L-proline-mediated inhibition of GAD reduces GABA synthesis in a manner that selectively disrupts GABAergic signaling during high demand states, such as gamma oscillations. Consequently, in the illness, pathogenic processes that disrupt GAD function, such as deficits in GAD67 mRNA levels or high L-proline levels, could have their greatest effect during gamma oscillations when the need for GABA synthesis is high (Figure 1). Consistent with this hypothesis, in schizophrenia, deficits in gamma oscillation power worsen with greater cognitive load [6] and lower cortical GABA levels are associated with poorer cognitive performance [7]. Finally, Crabtree and colleagues provide evidence that the deficit in GABAergic function during high demand states is reversible with agents that reduce GABA degradation, such as GABA transaminase inhibitors, a finding with potential therapeutic implications for the treatment of schizophrenia.
The experiments of Crabtree et al suggest additional studies regarding the potential role of excessive L-proline levels in the disease process of schizophrenia (Figure 1). First, many of the results of this study come from homozygous PRODH-deficient mice. However, 22q11 deletion syndrome in humans results in only a heterozygous loss of PRODH function, and relatively few cases of schizophrenia are attributable to 22q11 deletion syndrome. Consequently, additional studies are needed to investigate other pathogenic mechanisms that may disrupt PRODH function, perhaps through deficient gene expression or inhibition of its enzymatic activity, in schizophrenia. Second, the authors demonstrate that high L-proline levels inhibit GAD67 activity in cell transfection assays. However, the authors also show that in PRODH-deficient mice, baseline GABA neurotransmission, which is regulated by the GAD67 isoform in parvalbumin neurons [8], is not altered, while sustained, high frequency GABA activity, which is regulated by the GAD65 isoform [9], is deficient. In schizophrenia, GAD67, but not GAD65, mRNA levels have been reported to be reduced in the PFC, whereas in schizoaffective disorder mRNA and protein levels of both GAD67 and GAD65 are reduced in the PFC [1, 10]. Consequently, it is important to determine if L-proline generally inhibits all GAD activity, preferentially affects GAD65 or GAD67, or perhaps also interferes with other presynaptic GABA mechanisms such as vesicular or plasma membrane GABA transporters. Third, the authors found that PRODH is also expressed by multiple neuronal populations. Thus, it is critical to determine whether excessive L-proline affects GAD function in other inhibitory neuron populations in addition to parvalbumin-containing basket neurons. Taken together, the results of Crabtree and colleagues raise the possibility that a combination of screening for dysfunction of metabolic enzymes that may impact GABA function (e.g., PRODH) and imaging studies of cortical GABA levels may help identify schizophrenia subjects who might benefit from novel personalized therapeutic approaches to restore metabolic and GABAergic function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Curley AA, et al. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: Clinical, protein, and cell type-specific features. Am J Psychiatry. 2011;168:921–929. doi: 10.1176/appi.ajp.2011.11010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sohal VS, et al. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Burgos G, et al. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry. 2015;77:1031–1040. doi: 10.1016/j.biopsych.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crabtree GW, et al. Cytosolic Accumulation of L-Proline Disrupts GABA-Ergic Transmission through GAD Blockade. Cell Rep. 2016;17:570–582. doi: 10.1016/j.celrep.2016.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volk DW, et al. Deficits in transcriptional regulators of cortical parvalbumin neurons in schizophrenia. Am J Psychiatry. 2012;169:1082–1091. doi: 10.1176/appi.ajp.2012.12030305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho RY, et al. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad of Sci USA. 2006;103:19878–19883. doi: 10.1073/pnas.0609440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowland LM, et al. Frontal Glutamate and gamma-Aminobutyric Acid Levels and Their Associations With Mismatch Negativity and Digit Sequencing Task Performance in Schizophrenia. JAMA Psychiatry. 2016;73:166–174. doi: 10.1001/jamapsychiatry.2015.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazarus MS, et al. GAD67 deficiency in parvalbumin interneurons produces deficits in inhibitory transmission and network disinhibition in mouse prefrontal cortex. Cereb Cortex. 2015;25:1290–1296. doi: 10.1093/cercor/bht322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian N, et al. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci USA. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glausier JR, et al. Lower glutamic acid decarboxylase 65-kDa isoform messenger RNA and protein levels in the prefrontal cortex in schizoaffective disorder but not schizophrenia. Biol Psychiatry. 2015;77:167–176. doi: 10.1016/j.biopsych.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]