

Abstract
With the emergence of multidrug-resistant bacterial strains, there is a dire need for new drug targets for antibacterial drug discovery and development. Filamentous temperature sensitive protein Z (FtsZ), is a GTP-dependent prokaryotic cell division protein, sharing less than 10% sequence identity with the eukaryotic cell division protein, tubulin. FtsZ forms a dynamic Z-ring in the middle of the cell, leading to septation and subsequent cell division. Inhibition of the Z-ring blocks cell division, thus making FtsZ a highly attractive target. Various groups have been working on natural products and synthetic small molecules as inhibitors of FtsZ. This review summarizes the recent advances in the development of FtsZ inhibtors, focusing on those in the last 5 years, but also includes significant findings in previous years.
Keywords: Antibacterial agent, Multidrug resistance, FtsZ, Z-ringProtofilament, Septation, Cell division, Inhibitor
Graphical abstract
1. Introduction
Emergence and growing prevalence of multidrug resistance (MDR) in bacterial strains such as Methicillin-resistant Staphylococcus aureus (MRSA), drug-resistant tuberculosis, vancomycin-resistant enterococci (VRE) and other bacteria has caused a serious threat to human health worldwide.1 MRSA is a variant of Staphylococcus aureus (S. aureus) that is resistant to methicillin as well as other β-lactam antibiotics, such as nafcillin and oxacillin.2 It is responsible for numerous illnesses, ranging from skin infections to pneumonia. In 2011, the CDC estimated 11,285 MRSA related deaths in the U.S.2 Tuberculosis (TB) is an infection, typically in the lungs, caused by Mycobacterium tuberculosis (M. tuberculosis). This disease has claimed 1.5 million lives worldwide in 2014 alone, and most fatalities are prevalent in developing countries.3 Multi-drug resistant (MDR-TB) and extensively-drug resistant (XDR-TB) strains exist and are becoming more frequent. The CDC classifies antibiotic-resistant threat levels as “concerning,” “serious,” and “urgent,” with “concerning” as the lowest level and “urgent” being the highest level.2 MRSA and MDR/XDR-TB cause devastating infections and are ranked as threat level “serious” by the CDC.2 Thus, urgent action needs to be taken to ensure these threats do not elevate to the “urgent” level.
The top three on the CDC’s list of “urgent” drug-resistant threats are Clostridium difficile (C. difficile), carbapenem-resistant enterobacteriaceae (CRE), and drug-resistant Neisseria gonorrhoeae (N. gonorrhoeae).2 C. difficile infections result in life-threatening diarrhea. There are 250,000 infections a year resulting in 14,000 deaths annually, and at least $1 billion are spent annually on medical costs for treating C. difficile infections. CRE are a class of bacteria that are resistant to many antibiotics, including carbapenems, which are considered last-resort drugs. Drug-resistant N. gonorrhoeae causes the sexually transmitted disease, gonorrhea. The CDC estimated 246,000 cases of antibiotic-resistance in 2011.2 These bacteria cause diseases that are virtually untreatable, and immediate action is needed to discover new ways to combat these pathogens. There is a dire need for next-generation antibiotics with unexploited mechanisms of action, directed at novel targets. One such target is filamenting temperature-sensitive protein Z (FtsZ), an essential bacterial cell division protein.
FtsZ is a prokaryotic cell division protein with GTPase activity, which plays a vital role in cell division. The protein is highly conserved throughout eubacteria. It is present in bacteria with cell walls, such as E. coli, B. subtilis, and M. tuberculosis, as well as in bacteria lacking cell walls, such as Mycoplasma pulmonis (M. pulmonis) and Acholeplasma laidlawii (A. laidlawaii).4–7 Even Halobacterium salinarium (H. salinarium) and Pyrococcus woesei (P. woesei), from the Archaea domain, contain genes for FtsZ.8,9 With FtsZ present across a wide range of bacterial species, it is an attractive target for the drug discovery of new-generation antibacterial agents.
1.1. Structure and function of FtsZ
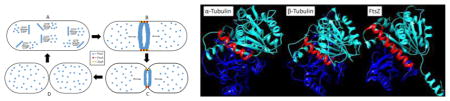
FtsZ is a structural homologue of tubulin, a eukaryotic cell division protein. Both proteins polymerize in a GTP dependent manner, forming cytoskeletal filaments that are utilized in cell division, but each protein has a distinct function.10,11 Tubulin, polymerized into microtubules, is responsible for migration of the genetic material to the poles of a dividing cell, while FtsZ, polymerized into the Z-ring, is responsible for division of a bacterial cell into daughter cells.10–12 Tubulin and FtsZ share only 7% sequence identity, but share high structural similarity.11 Both proteins contain two domains, N-terminal and C-terminal, connected by an α-helix (H7).11,13 The two domains on the FtsZ are a series of β-sheets surrounded by α-helices. The N-terminal domain, consisting of helices H1-H6 and strands S1-S6, contains the nucleotide binding site, and the C-terminal domain, consisting of helices H8-H10 and strands S7-S10, houses the vital T7 loop. The GTPase active site is located at the junction of two FtsZ monomers, i.e., the nucleotide-binding site of a FtsZ monomer combined with the T7 loop of another FtsZ monomer forms the active site (Figure 1).14,15
Figure 1.

Comparisons of α-tubulin, β-tubulin, and FtsZ monomer. Cyan colored-region is the N-terminal domain. The red-colored region is the H7 helix. The yellow-colored region is the T7 loop. The blue-colored region is the C-terminal domain. The magenta-colored molecule is GDP/GTP, which sits in the nucleotide binding site. (PDB entries 1TUB for tubulins and 2VAP for FtsZ).
There are approximately 15,000 copies of FtsZ in a single E. coli cell. Upon binding of GTP, FtsZ polymerizes to form protofilaments, which align at the center of a dividing cell.16,17 FtsZ polymerization directionality is not fully understood. Observations from immunofluorescence microscopy experiments on MCZ26 E. coli cells showed centrally located and symmetrical Z-spirals.18 From these observations, Addinal and Lutkenhaus proposed an initial nucleation site at the center of cells that expanded outwards from both ends, forming protofilaments in a bi-directional manner.18 However, Jindal and Panda proposed a uni-directional mechanism based on the polymerization assays using B. subtilis FtsZ (Bs-FtsZ) and demonstrated that truncated N-terminal constructs inhibited polymerization, while C-terminal constructs had no effect on polymerization.19 These results might suggest that the directionality of polymerization differs among bacterial species. However, it appears too early to conclude because of the fact that two different techniques were employed in two different bacterial species. Thus, further study is required to clarify this intriguing issue.
1.2. Formation and localization of Z-ring
Regardless of polymerization directionality, protofilaments localize at the middle of the cell to form the Z-ring, which is a key structure that recruits other cell division proteins to form the divisome. About 30% of the available FtsZ is sequestered to the Z-ring, while the remaining FtsZ monomers float around the cytoplasm and are readily exchanged with polymerized FtsZ molecules in the protofilaments of the Z-ring.20 Fluorescence recovery after photobleaching (FRAP) experiments highlighted the dynamic nature of the Z-ring, i.e., photobleaching recovery halftime was about 30 seconds, demonstrating FtsZ monomers constantly exchange with polymerized FtsZ molecules.20 In E. coli, the Z-ring is a single layer of FtsZ protofilaments that runs parallel to the inner cell membrane and is five to ten protofilaments thick.21 FtsZ protofilament interaction with ZipA and FtsA anchors the Z-ring to the cell membrane.22–24 ZipA is a protein, containing a transmembrane domain and a FtsZ binding domain.22 FtsA is a peripheral membrane protein, containing an amphipathic helix that binds to the cell membrane and a FtsZ binding domain.23 The binding domains of both proteins attach to the 17-amino acid sequence on the C-terminal of FtsZ. The Z-ring is tethered to the cell membrane through ZipA and FtsA (Figure 2).
Figure 2.

A) FtsZ monomers polymerize in the presence of GTP, forming protofilaments. B) Protofilaments line up at the center of the cell to form the highly dynamic Z-ring, which is anchored to the bacterial cell membrane by FtsA and ZipA. FtsZ monomers readily exchange with FtsZ molecules incorporated into the Z-ring. C) Contraction of the Z-ring leads to invagination of the cell membrane. D) Septation is completed and the Z-ring dissipates.
In E. coli and B. subtilis, localization of the Z-ring is regulated by the Min system and nucleoid occlusion proteins. The Min system, in E. coli, is a series of proteins, MinC, MinD and MinE, that ensure correct formation of the Z-ring at the midcell. Deletion of the Min system overall in bacterial cells leads to formation of minicells, which do not contain any genetic material.25 Deletion of MinE, while leaving MinC and MinD intact, leads to filamentation of bacterial cells.25 MinC, a protein that prevents FtsZ polymerization, is MinD-dependent, and together, they form a MinCD complex. This complex binds to the cell membrane, particularly at the poles, and hampers FtsZ polymerization and Z-ring construction.26 MinE, a protein that suppresses MinCD activity, forms a dynamic ring structure that oscillates from pole to pole.27,28 Oscillation of the MinE ring prevents MinCD buildup at the midcell. FtsZ polymerization and subsequent Z-ring formation can occur at the midcell, but not poles of the cell because MinCD remains active in those regions.27
In B. subtilis cells, there is no equivalent to the E. coli MinE.29 DivIVA, a protein with affinity to the poles of the bacterial cell, is thought to localize the MinCD complex at the poles of the cell through a bridging protein, MinJ.29,30 Sequestration of MinCD to the poles inhibits FtsZ polymerization in those regions and allows for the formation of the Z-ring at the midcell. The Min system ensures the proper placement of the Z-ring during cell division, and this system is crucial for the proper formation of the divisome. However, in Min system mutants, bacterial cells can properly localize the Z-ring and divide, albeit in a decreased capacity compared to wildtype cells.31 This indicates that there are multiple regulatory factors involved in the correct placement of the Z-ring.
Another regulating factor for the localization of the Z-ring is nucleoid occlusion. Bacterial cells have a mechanism that prevents the destruction of the genetic material caused by cell division through the nucleosome. In E. coli, SlmA is the protein responsible for protecting the bacterial DNA during cell division.32 This protein has DNA-binding and FtsZ-inhibiting properties, i.e., it inhibits FtsZ polymerization within its proximity through binding to the nucleosome.32 In B. subtilis, Noc is a nonspecific DNA-binding protein, which is found throughout the nucleosome, with FtsZ-inhibiting properties. Like SmlA, this protein prevents FtsZ polymerization near the nucleosome.31 Both proteins are part of the nucleoid occlusion regulation of Z-ring formation.
Since GTP-bound protofilaments have slight curvatures and GDP bound protofilaments are highly curved, it is speculated that the hydrolysis of GTP to GDP contracts the Z-ring and generates the force required for septation. Molecular dynamics simulations of FtsZ dimers predicted the force generated by GTP hydrolysis to be 30 pN per FtsZ monomer.33 The calculated force required to invaginate the cell wall is 8–80 pN for E. coli.34,35 Another molecular dynamics simulation, performed on the GTP-bound and GDP-bound FtsZ filaments, found that GTP hydrolysis caused conformational changes in the filaments.36 These two experiments support the hypothesis that Z-ring contraction is initiated by GTP hydrolysis to GDP. However, this does not explain how bacterial cells expressing FtsZ mutants, with hampered or no GTPase activity, can still divide. Since GTP-bound protofilaments are slightly curved, they generate an inward force, estimated to be 10 pN per FtsZ monomer.33 These estimates suggest that invagination of the cell wall can be achieved without hydrolysis of GTP to GDP.33
2. FtsZ inhibitors
With the rise of antibiotic-resistant strains of bacteria, there is much enthusiasm for producing drugs with novel mechanisms of action. FtsZ is a well-studied, yet unexploited, protein. It is essential for cell division in bacteria, making it an attractive target for antibiotic research. Inhibition of the protein prevents proper formation of the divisome, which leads to filamentation and eventual cell death (Figure 3).37,38 The highly conserved nature of the protein among numerous prokaryotic and archea species provides an opportunity for the development of broad spectrum-antibiotics.39,40 Currently, there are no drugs on the market that target this protein, but many research labs have made great headway in studying inhibitors of FtsZ and have demonstrated that FtsZ inhibition leads to bacterial cell death. These inhibitors are summarized below.
Figure 3.

(A) Normal cells divide and remain a short length. (B) and (C) Inhibition of FtsZ causes filamentation. Cells continue to grow and elongate, but cannot divide. Reprinted with permission from Huang, Q.; Kirikae, F.; Kirikae, T.; Pepe, A.; Amin, A.; Respicio, L.; Slayden, R. A.; Tonge, P. J.; Ojima, I., Targeting FtsZ for Antituberculosis Drug Discovery: Noncytotoxic Taxanes as Novel Antituberculosis Agents. J. Med. Chem. 2006, 49 (2), 463–466. Copyright 2012 American Chemical Society.
2.1 Natural Products (Figure 4)
Figure 4.

Naturally occurring FtsZ inhibitors
2.1.1. Curcumin
Curcumin is a naturally-occurring compound extracted from Curcuma longa. It has been used as a spice and a coloring agent in Indian and Southeast Asian cooking for ages, and it is known to have antioxidant, anticancer and anti-bacterial properties.41 Curcumin has been shown to bind to tubulin and exhibit antiproliferative activity by destabilizing microtubules.42 Rai et al. showed that curcumin inhibits FtsZ in a dose-dependent manner using light scattering assay. Curcumin displayed an IC50 of 17 μM against B. subtilis 168 and 58 μM against E. coli K12 MG1655.41 GTPase assay suggested that curcumin inhibited FtsZ by increasing the GTPase activity, hence destabilizing FtsZ polymers.41 Utilizing a computational docking program, Molecular Electrostatic Potential (MEP), and cavity depth analysis, Kaur et al. identified possible curcumin binding sites in E. coli-FtsZ (Ec-FtsZ) and Bs-FtsZ, which were confirmed by mutagenesis studies.43 The curcumin binding site was found to overlap with the GTP binding site, as several amino acid residues were common to both binding sites.42
2.1.2. Coumarins
Coumarins are phytochemicals, consisting of a benzopyrone core and are mostly known for their anticoagulant properties.44 Duggirala et al. reported that coumarins inhibited Bs-FtsZ polymerization in a dose-dependent manner.44 For example, scopoletin inhibited FtsZ polymerization with an IC50 of 41 μM and daphnetin with an IC50 of 73 μM. Scopoletin and daphnetin inhibited the GTPase activity with IC50 values of 23 μM and 57 μM, respectively. Coumarin, 6-methylcoumarin, and hydroxylcoumarin showed negligible activity against GTPase and FtsZ polymerization.44 Docking studies of Ec-FtsZ revealed that coumarins bind to an allosteric site, located in the T7 loop.44 Chiang et al. reported that coumarins such as umbelliferone, scopoletin and phellodenol-A inhibited the growth of M. tuberculosis H37Rv with MIC values of 58, 42, and 60 μg/mL, respectively.45 The results of Duggirala et al. suggest that coumarins exhibit antibacterial activity against M. tuberculosis H37Rv by inhibiting polymerization of FtsZ.
2.1.3. Plumbagin
Plumbagin is a naphthoquinone derivative found as a secondary metabolite in the root of Plumbago zeylanica.46 Acharya et al. reported that plumbagin inhibited the polymerization of microtubules by binding to tubulin at the colchicine binding site.47 Since FtsZ is a homolog of tubulin, Bhattacharya et al. examined the effect of plumbagin on FtsZ. Their results show that plumbagin inhibits the FtsZ polymerization of B. subtilis 168 in a dose-dependent manner. Thus, 2, 5, and 10 μM of plumbagin reduced the assembly of the Bs-FtsZ by 26, 33, and 45 %, respectively. Plumbagin descreased the hydrolysis of GTP by reducing the FtsZ’s GTPase activity. Thus, 24 μM plumbagin inhibited 58 % of the GTPase activity. Computational analysis suggested that plumbagin did not bind to the GTP binding site and the plumbagin-binding site was located near the C terminal of Bs-FtsZ. Although plumbagin clearly inhibited the polymerization of Bs-FtsZ, it did not show any effect on the polymerization or GTPase activity of Ec-FtsZ. This indicates that there is a substantial difference in the structures of FtsZ proteins from different bacteria.48
2.1.4. Resveratrol
Resveratrol is a phytoalexin that is produced mainly in plants, such as grapevines, legumes, and pines as a defense mechanism49 in response to attack by pathogens such as bacteria or fungi.49,50 It has been shown to have antioxidant, antimicrobial, and anti-proliferative activities.51 Hwang et al. investigated the mechanism of action of resveratrol for its antibacterial activity.51 Then, it was found that the Z-ring formation was affected, to a great extent, in E. coli treated with resveratrol. In order to examine if resveratrol inhibited FtsZ, PNA-FtsZ, an RNA silencer that selectively targeted the mRNA of FtsZ, was used. The treatment consisting of PNA-FtsZ and resveratrol showed a synergistic antibacterial effect while PNA-fabI, an antisense inhibitor of the fabI gene, did not show any synergistic effect. Confocal microscopy analysis showed that the E. coli cells treated with PNA-FtsZ and resveratrol induced cell elongation, whereas PNA-fabI treatment did not show any cell elongation. This result led to the conclusion that the antibacterial activity of resveratrol is ascribed to the inhibition of the Ec-FtsZ gene expression.51
2.1.5. Chrysophaentins
Chrysophaentins are polyhalogenated, polyoxygenated, bisdiarylbutene ether macrocycles isolated from algae Chrysophaeum taylori. Plaza et al. reported that chrysophaentins were effective against several drug-sensitive and drug-resistant strains of gram-positive bacteria.52 Using saturation transfer difference NMR (STD-NMR), chrysophaentin A was found to bind to Ec-FtsZ, and NMR competition experiments using GTPγS suggested, chrysophaentin A is a competitive inhibitor of Ec-FtsZ.52 Keffer et al. synthesized hemi-chrysophaentin and found that its mechanism of action was similar to that of chrysophaentin A.53 Both chrysophaentin A and hemi-chrysophaetin did not exhibit antimicrobial activity against gram-negative bacteria, but inhibited Ec-FtsZ in vitro, which could be ascribed to these compounds’ inability to pass through the outer membrane of gram-negative bacteria. Thus, in order to test this hypothesis, the activities of chrysophaentin A and hemi-chrysophaetin were determined against E. coli envA1, a strain with increased compound permeability, and found the IC50 values to be 27 μM and 84 μM, respectively, which proved the hypothesis.53
2.1.6. Berberine and its derivatives
Berberine is a plant alkaloid isolated from various species of Breberis. Damodia et al. reported that berberine targeted Ec-FtsZ by inhibiting the FtsZ assembly (IC50 10 μM) and GTPase activity (IC50 16 μM) in a dose-dependent manner. Computational docking studies revealed that the berberine binding site overlapped with the GTP binding pocket in Ec-FtsZ.54
Based on the crystal structure of S. aureus-FtsZ (Sa-FtsZ), Sun et al. designed and synthesized a series of 9-phenoxyalkylberberine derivatives, which were predicted to bind to the interdomain cleft of Sa-FtsZ. Compounds from this series exhibited MIC values of 2–8 μg/mL against MRSA and methicillin-sensitive Staphylococcus aureus (MSSA), and 4–16 μg/mL against vancomycin-sensitive Enterococcus faecalis (VRE) and vancomycin-sensitive Enterococcus faecalis (VSE). These berberine analogs also inhibited the growth of gram-negative bacteria such as E. coli and Klebsiella pneumoniae (K. pneumoniae) with MIC values of 32–128 μg/mL. Compound 1 was the most potent compound in this series, which inhibited Sa-FtsZ GTPase (IC50 38 μg/mL) and inhibited FtsZ polymerization in a dose-dependent manner. Transmission electron microscopy (TEM) images of Sa-FtsZ with compound 1, revealed that it significantly reduced the thickness and size of FtsZ polymers, as well as the bundling of FtsZ protofilaments.55
Parhi et al. synthesized a series of substituted dibenzo[a,g]quinolizin-7-ium salts and tested them against MRSA, MSSA, VSE and VRE. Compounds consisting of an aryl substituent at the 2- or 12-position of the tetramethoxydibenzoquinolizinium moiety, exhibited better anti-staphylococcal and anti-enterococcal activity compared to berberine. Compound 2 was the most potent in this series (MIC values: 0.5 μg/mL against MSSA and MRSA and 2 μg/mL against VRE and VSE). Light scattering assay of compound 2 using Sa-FtsZ revealed that substituted dibenzo[a,g]quinolizin-7-ium compounds act by promoting FtsZ polymerization. Since benzamides like PC190723 act by promoting FtsZ polymerization, it was hypothesized that these compounds may have similar mechanism of action.56
2.1.7. Phenylpropanoids
Phenylpropanoids are secondary metabolites produced by plants to protect plants against predators and pathogens.57 It has been shown that these compounds possess antibiotic activities and inhibit FtsZ.57,58 Eight phenylpropanoids were assayed against Ec-FtsZ and were found to inhibit FtsZ polymerization (IC50 69 μM~>250 μM). Chlorogenic acid, being the most active, had an IC50 of 69.55±3.6 μM. Caffeic acid, 2,4,5-trimethoxycinnamic acid, p-coumaric acid, and cinnamic acid had IC50 of 105.96±6.3 μM, 148.59±4.3 μM, 189.53±3.7 μM, and 238.91±7.1 μM, respectively. 3,4-Dimethoxycinnamic acid, 2,4,5-trimethoxycinnamic acid, and eugenol were the least active with IC50 >250 μM. Most of the eight phenylpropanoids inhibited GTPase activity. Light scattering analysis showed a dose-dependent reduction in the polymerization of FtsZ. B. subtilis 168 cells were elongated, compared to the control when exposed to phenylpropanoids.59
2.1.8. Cinnamaldehyde and its derivatives
Cinnamaldehyde, a phenylpropanoid chalcone is the major constituent of the bark extract of Cinnamomum verum. It has been shown that cinnamaldehyde is effective against a variety of bacteria, viruses and fungi.60 Cinnamaldehyde exhibited the MIC values of 0.1–0.5 μg/mL against E. coli (0.1 μg/mL), B. subtilis (0.5 μg/mL) and MRSA (0.25 μg/mL). Cinnamaldehyde inhibited the GTPase activity thus inhibiting the assembly of FtsZ protofilaments.61 Confocal imaging of E. coli cells containing GFP-tagged Ec-FtsZ treated with cinnamaldehyde showed reduction in the Z ring formation.61
Li et al. synthesized a library of cinnamaldehyde derivatives and tested in vitro for their antibacterial activity against a variety of gram-positive and gram-negative bacteria.62 Several compounds exhibited MIC values of 0.25–4 μg/mL against S. aureus ATCC25923. Cinnamaldehyde derivatives containing a 2-methylbenzimidazolyl substitution at 1-position and 3-chlorophenyl, 2,4-dichlorophenyl, 4-clorophenyl, 4-fluorophenyl, or 4-nitrophenyl at the 3-position exhibited the best activity (Compounds 3 and 4). They also carried out polymerization and GTPase assays to confirm Sa-FtsZ as the target.62
2.2. Synthetic small molecules
2.2.1 Benzamides
Ohashi et al. assayed the antibacterial activity of 3-methoxybenzamide (3-MBA) on B. subtilis UOT1285 as well as FtsZ mutant strains of B. subtilis UOT1285 such as RIK7 (brgA1 mutation), RIK8 (spb-1 mutation), RIK9 (brgA1 and spb-1 mutations). The growth of B. subtilis UOT1285 was inhibited in the presence of ≥5 mM 3-MBA leading to filamentation followed by cell death, whereas the growth of B. subtilis with mutant FtsZ strains was not inhibited by 3-MBA even at 35 mM. This led to the hypothesis that 3-MBA inhibited the growth of B. subtilis UOT1285 by inhibiting FtsZ.63
Since 3-MBA displays poor anti-bacterial activity (MIC value of 4000 μg/mL against B. subtilis cells),64 Haydon et al. performed the SAR study of 3-MBA based on over 500 3-MBA congeners, which identified PC190723, a compound with better potency than 3-MBA.65 PC190723 exhibited antibacterial activity against B. subtilis and several strains of staphylococci, including MRSA and multidrug resistant Staphylococcus aureus (MDRSA), with MIC values of 0.5–1 μg/mL. PC190723 inhibited the GTPase activity of Sa-FtsZ in a dose-dependent manner (IC50 55 ng/mL). Treatment of B. subtilis bearing green fluorescent protein-labeled FtsZ (GFP-FtsZ) with PC190723 showed mislocalization of FtsZ, wherein FtsZ was distributed as discrete foci throughout the elongated cell (Figure 5). The in vivo efficacy of PC190723 was tested in a murine septicaemia model of staphylococcal infection. There was 100 % survival of mice that were inoculated with a lethal dose of S. aureus following iv or PO administration of 30 mg/kg of PC190723.65 A PC190723-resistant mutant strain of S. aureus was isolated when an 8 times higher concentration than the MIC value was employed.65
Figure 5.

PC190723 inhibits localization of FtsZ. GFP-FtsZ of B. subtilis 2020 cells, in the absence (a) and presence of 8 μg/mL of PC190723 (b). Adapted with permission from reference 64.
Comparison of the protein sequences of FtsZ from six different bacteria, i.e., B. subtilis, S. aureus, Enterococcus faecalis (E. faecalis), E. coli, Pseudomonas aeruginosa (P. aeruginosa), and Streptococcus. pneumoniae (S. pneumoniae), and mammalian β-tubulin showed that the PC190723 mutations overlapped with the residues of the taxane-binding site in β-tubulin, suggesting a similar mechanism of action.65 Andreu et al. reported that Bs-FtsZ treated with PC190723 showed a reduced GTPase activity, which resulted in the formation of straight bundles and ribbons.66 Then, it was suggested that PC190723 inhibited FtsZ by stabilizing the FtsZ polymer.66 Elsen et al. carried out several structural and biochemical analyses of Sa-FtsZ and PC190723, which revealed that PC190723 shifted the equilibrium towards the high-affinity FtsZ conformation (Figure 6) and reduced the transition slope for polymerization.67 The co-crystal structure of Sa-FtsZ with PC190723 indicated that the C-terminal domain moved in such a manner that FtsZ protofilaments were stabilized over FtsZ monomers.67
Figure 6.
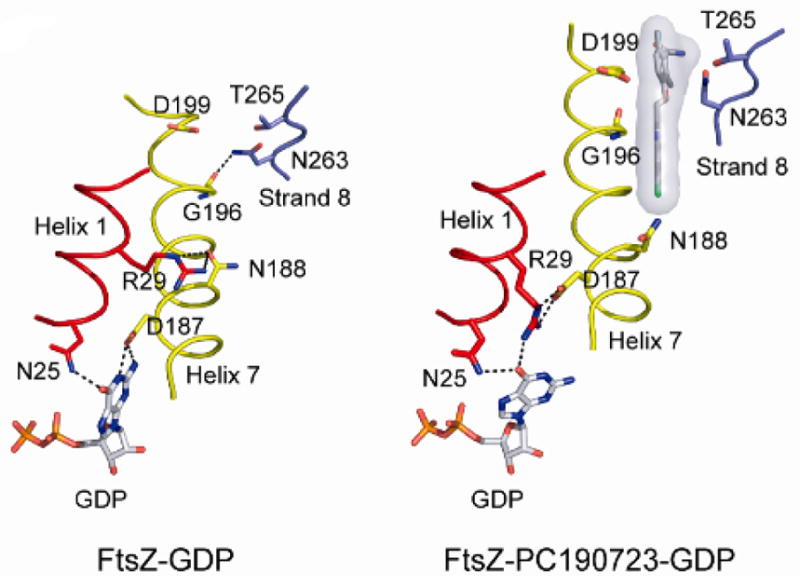
Movement of helix 7 favors the high affinity state over the low affinity state. Reprinted with permission from Elsen, N. L.; Lu, J.; Parthasarathy, G.; Reid, J. C.; Sharma, S.; Soisson, S. M.; Lumb, K. J., Mechanism of action of the cell-division inhibitor PC190723: modulation of FtsZ assembly cooperativity. J. Am. Chem. Soc. 2012, 134 (30), 12342–5. Copyright 2012 American Chemical Society.
Stokes et al. developed PC190723 analogs, bearing a phenyloxazole group instead of a thiazolopyridine group and a substituted methylene group connecting the oxazole and benzamide moieties.68 A series of (4,5-disubstituted oxazolyl)-CH(R)-O-benzamides were found to improve the potency significantly against wild type S. aureus (Compound 5, MIC as low as 0.03 μg/mL) The C5 position of the oxazole moiety tolerated a variety of alkyl, aryl and halogen substituents to keep a good potency. Among them, 5-Bromo- and 5-chlorooxazolyl analogs exhibited high activity against a mutant strain of S. aureus, G196A. In order to improve the pharmacokinetic parameters, polar groups, e.g., alcohol, amine, carboxylic acid, heterocycles, etc., were introduced to the pseudo benzylic position, i.e., -CH(R)-O- moiety, which improved the solubility and metabolic stability. Introduction of a gem-dimethyl group at the pseudobenzylic position resulted in loss of activity, suggesting the importance of chirality for antibacterial activity. Thus, two enantiomers of compound 6, bearing a -CH(CH2OH)-O- group, were isolated by chiral chromatography and their antibacterial activity examined, which revealed that (R)-(+)-enantiomer is 128 times more potent than the corresponding (S)-(−)-enantiomer. In order to enhance the solubility of compound 6, they synthesized a prodrug 7, which was twice as soluble as the parent compound. 68
In order to improve the pharmacological properties of PC190723, Kaul et al. designed and synthesized a 1-methylpiperidine-4-carboxamide TXY541, a prodrug of PC190723, which was found to be effective against both MRSA and MSSA.69 TXY541 was 143 times more soluble in an aqueous acidic vehicle (10 mM citrate, pH 2.6) than PC190723 and exhibited efficacy in vivo via intravenous (i.v.) and oral (p.o) administration utilizing mouse peritonitis model of systemic infection with S. aureus using female Swiss-Webster mice. TXA541 showed the MBC/MIC ratio of 1~2 against MSSA (strains 8325-4 and 19636) as well as MRSA (strains ATCC 33951 and 443300), indicating bactericidal activity.69 In order to overcome the CYP-mediated dechlorination/oxygenation of TXY541, TXA707, bearing a trifluoromethyl group in place of the chloro group in TXY541, as well as its prodrug TXA709 were developed.70 The introduction of a trifluoromethyl group substantially improved metabolic stability. The modal MIC of TXA707 was found to be 1 μg/mL against MRSA, VISA, VRSA, DNSSA, LNSSA, and MSSA.
Transmission electron microscopy (TEM) images of S. aureus cells displayed absence of septa within 1 hour of treatment with TXA707 (Figure 7). The t1/2 of TXA707 following i.v. administration of TXA709 was 3.65 hours, while the t1/2 of PC190723 following the i.v. administration of TXA541 was 0.56 hours.70 Thus, the prodrugs of PC190723 exhibited better pharmacokinetic properties by maintaining the strong antibacterial potency. The in vivo efficacy study of TXA709 was carried out using mouse tissue (thigh) model of infection with MRSA ATCC 33591. 120 or 160 mg/kg of TXA709 administered orally, reduced the bacterial load of MRSA by nearly 2 log10 CFU in the thigh, while 200 mg/kg of orally administered PC190723 reduced only 0.5 log10 CFU.70 (Figure 8)
Figure 7.

Transmission electron micrographs of MSSA 8325-4 bacteria treated with DMSO vehicle or 4 μg/ml TXA707 for 1, 4, and 9 hours. Adapted with permission from reference 69.
Figure 8.

PC190723, its analogs and their prodrugs
2.2.2 2-Nitro-vanillin-aniline Schiff bases
Vanillin, the principal component of vanilla and a widely used flavoring agent,71 is also known to possess antibacterial properties and has been used as a food preservative.72 Sun et al. synthesized a series of 2-nitro-vanillin-aniline Schiff base derivatives and examined their antibacterial activities against E. coli, P. aeruginosa, B. subtilis, and S. aureus.71 Then, a dozen compounds exhibited strong antibacterial activities, and it was found that compounds with two substituents on the aniline moiety showed better activities than their monosubstituted counterparts (Figure 11). The presence of electron-donating groups on the aniline moiety increased the antibacterial potency, while electron-withdrawing groups reduced potency. For example, compound 8 was found to be the most potent against E. coli in the series (MIC 0.28 μg/mL), which was more potent than the kanamycin (MIC 0.40 μg/mL) used as a positive control. This compound was reported to have inhibited FtsZ polymerization (ID50 2.1 μM), although it was not clearly stated which bacterial FtsZ was used.71
2.2.3 Arene-diol digallates
In order to find small molecule inhibitors that replace GTP in FtsZ, Andreu and collaborators screened over 4 million compounds by docking into Bs-FtsZ GTP binding site. Hits from this screen were further tested via mant-GTP anisotropy method for Bs-FtsZ. Hit compounds included UCM05, UCM44, and UCM53, which inhibited bacterial growth with MIC values of 100 μM, 25 μM, and 13 μM, respectively. However, these compounds were not effective against gram-negative bacteria such as E. coli.
The binding modes of UCM05 and UCM44 suggest that the phenolic groups and the naphthalene core occupy the phosphate and nucleic base binding sites of GTP, hence leading to their inhibitory action. The bacterial cell growth was inhibited via filamentation by all three compounds, which is a phenotypic response of FtsZ inhibition.73 Building upon hit compounds, UCM05 and UCM44, a series of small molecule inhibitors was synthesized and examined for their potencies. This series of compounds interacted with the GTP binding site of Bs-FtsZ with Kd values of 0.4–0.8 μM. The lead compounds blocked the bacterial cell division by inhibiting proper assembly of FtsZ, leading to filamentation (Figure 9). The most potent compound from this series, compound 9 displayed strong binding (Kd 0.5 μM) and antibacterial activity against MRSA (MIC 7 μM). Most of the compounds did not inhibit tubulin assembly at 100 μM, showing high specificity to FtsZ.74 (Figure 11)
Figure 9.
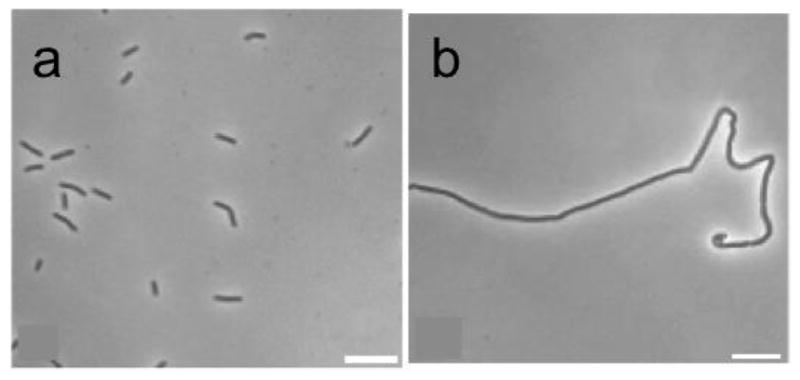
Phase contrast microscopy of B. subtilis 168 cells in the absence (a) and presence (b) of 5 μM of compound 21. Reprinted with permission from Artola, M.; Ruiz-Avila, L. B.; Vergonos, A.; Huecas, S.; Araujo-Bazan, L.; Martin-Fontecha, M.; Vazquez-Villa, H.; Turrado, C.; Ramirez-Aportela, E.; Hoegl, A.; Nodwell, M.; Barasoain, I.; Chacon, P.; Sieber, S. A.; Andreu, J. M.; Lopez-Rodriguez, M. L., Effective GTP-replacing FtsZ inhibitors and antibacterial mechanism of action. ACS Chem. Biol. 2015, 10 (3), 834–43. Copyright 2015 American Chemical Society.
2.2.4 Rhodanine derivatives
OTBA, a rhodanine derivative, was found to promote the assembly and stabilization of Ec-FtsZ and Bs-FtsZ protofilaments, thus inhibiting the Z-ring dynamics.75 Singh et al. screened a library of 151 rhodanine compounds for their antibacterial activity on B. subtilis cells.76 It was found that two compounds inhibited cell proliferation at 2 μM and three compounds increased the bacterial cell length, indicating FtsZ as the target. One of these three compounds, CCR11, bound to FtsZ (Kd 1.5 μM) and inhibited FtsZ assembly as well as GTPase activity in vitro. Docking study of CCR-11 to Bs-FtsZ suggested that CCR-11 would bind to FtsZ in a cavity adjacent to the T7 loop. CCR-11 inhibited the proliferation of B. subtilis cells (MIC 3 μM), and perturbed the Z-ring formation, but did not affect the nucleoid segregation or membrane integrity of B. subtilis cells (Figure 10) (Figure 11).76
Figure 10.
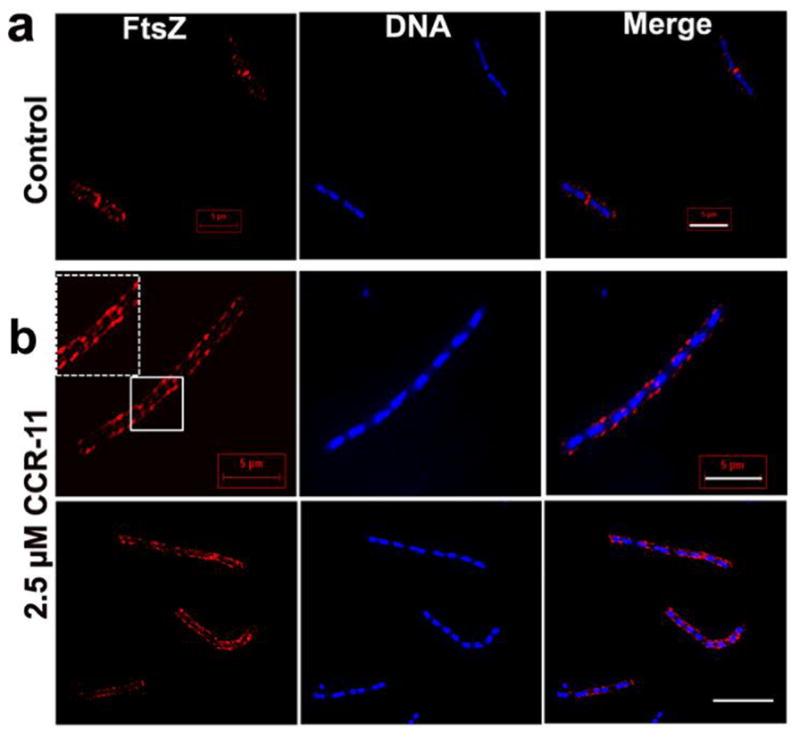
Effect of CCR-11 on Z-ring and nucleoid of B. subtilis cells in the absence (a) and presence of 2.5 μM CCR-11 (b). Reprinted with permission from Singh, P.; Jindal, B.; Surolia, A.; Panda, D., A rhodanine derivative CCR-11 inhibits bacterial proliferation by inhibiting the assembly and GTPase activity of FtsZ. Biochemistry. 2012, 51 (27), 5434–42. Copyright 2012 American Chemical Society.
Figure 11.

2-Nitro-vanillin-aniline Schiff bases, arene-dial digallates and a rhodanine derivative
2.2.5 Benzo[g]quinazolines, Quinazolines, quinoxalines and 1,5-naphthyridines
Based on zantrin Z3(1), a known GTPase inhibitor of Bs-FtsZ (IC50 24 μM),77 Nepomuceno et al. synthesized several quinazoline analogs as inhibitors of FtsZ and their potency examined.77 Compound ZZ3(2), the N,N-dimethyl analog of ZZ3(1), was found to be twice as potent as ZZ3(1) (IC50 12 μM). Further SAR study revealed that benzo[g]quinazoline of ZZ3(1) and ZZ3(2) could be replaced by a smaller quinazoline, bearing an ammonium salt side chain, to form the most active compound 10 in this series (IC50 9 μM). It was found that 4-chlorostyryl moiety was essential for good inhibitory activity.77
Parhi et al. synthesized a series of quinoxalines, quinazolines and 1,5-naphthyridines and evaluated their potencies against MRSA, MSSA, VSE, and VRE. Introduction of hydrophobic substitutions such as phenyl, 3-biphenyl, 4-fluorophenyl, 4-t-butylphenyl at the 3-position of 1-mehtylquinoxalinium iodide (Compounds 11, 11a) increased the antibacterial activity. In a similar manner, the introduction of a carbomethoxy or 5-carboxamido group to the quinoxaline skeleton increased potency (compound 12). Guanidinomethyl derivatives of quinazoline and 1,5-naphthiridine showed better activity than their amidinomethyl counterparts. Compounds 13, 13A, 14 and 15 displayed good MIC values (4–8 μg/mL) against MRSA and MSSA, and those compounds were also found to be bactericidal (Figure 12).78
Figure 12.

Quinoline, quinoxalines, quinazolines and 1,5-naphthyridine derivatives
2.2.6 Pyrimidine-quinuclidine derivatives
For the identification of hit compounds that target the GTP binding site of FtsZ, Chan et al. screened in silico a library of over 20,000 compounds, consisting of natural products and their semisynthetic analogs, using the crystal structure of Methanococcus jannaschii-FtsZ (Mj-FtsZ) for docking.79 Top-ranked 10 compounds from this in silico screening were purchased, and tested in vitro for their GTPase inhibitory activity, as well as MIC against E. coli and S. aureus.79 Then, compound 14 (Figure 13), containing a pyrimidine-quinuclidine moiety as the core structure, was found to display moderate GTPase inhibitory activity (IC50 317 μM) and antibacterial activity against E. coli and S. aureus (MIC 449 μM and 897 μM, respectively). Thus, 16 was selected as the hit compound for further in silico optimization, library synthesis and SAR study. The most potent compound from this series was compound 17 (Figure 13), which showed more than 10 times higher GTPase inhibitory activity (IC50 37.5 μM) and antibacterial activity against S. aureus (MIC 24.6 μM) and E. coli (MIC 49.6 μM). Compound 15 selectively inhibited FtsZ over tubulin, hence not cytotoxic.79
Figure 13.

Pyrimidine-quinuclidine and pyridopyrazine analogs
2.2.7 Pyridopyrazine and pyrimidothiazine analogs
Based on compounds 18 and 19 (Figure 13) that were reported to inhibit Mtb-FtsZ,80 Mathew et al. synthesized a series of pyridopyrazine and pyrimidothiazine analogs and their biological activities evaluated.80 It was found that (a) pyridopyrazine analogs, bearing heteroaromatic substituents at the C6 and C7 positions, were more potent than those bearing aliphatic substituents, (b) the C4 position tolerated various dialkylamino groups, and (c) a carbamate group at the C2 position was essential for activity. Compounds 18 and 20 exhibited an excellent antibacterial activity against M. tuberculosis H37Rv (IC90 <0.19 μM) and a moderate inhibitory activity for M. tuberculosis-FtsZ (Mtb-FtsZ) polymerization (IC50 ~34 μM). Both compounds 18 and 20 were evaluated in vivo for their efficacy in an acute TB model in immune-compromised GKO mice. Compound 18 reduced the bacterial load in lungs by 0.86 log10 CFU, but did not have any effect on the spleen. Compound 20 (Figure 13) did not reduce bacterial load in both lungs and spleen.80 Pyrimidothiazine analogs of compound 19 were synthesized and evaluated, but were found to be 10-fold less effective than 19.80
2.2.8 Taxanes
Paclitaxel, a microtubule-stabilizing agent known as a very important drug for cancer chemotherapy displayed moderate inhibitory activity (MIC 40 μM) against both drug-sensitive and drug-resistant strains of M. tuberculosis, although it was highly cytotoxic against human cancer cell lines (IC50 0.019 – 0.028 μM).81 Based on the functional similarity between tubulin and FtsZ, it was reasonable to hypothesize that taxanes could serve as the starting point to identify FtsZ inhibitors.81 Thus, real time PCR assay was used to screen a library of taxanes, including cytotoxic taxanes and taxane multidrug-resistance reversal agents (TRAs).81
SB-RA-2001 (TRA) (Figures 14 and 15) was found to exhibit good antitubercular activity against drug-sensitive (H37Rv: MIC 5 μM) and multidrug-resistant (IMCJ946.K2: MIC 2.5 μM) strains of M. tuberculosis, but it was still cytotoxic (A589: IC50 15.7 μM).81 Antiangiogenic C-seco-taxane IDN5390, bearing C-seco-baccatin moiety, was reported to be less cytotoxic than paclitaxel.82–83 Thus, C-seco-baccatin analogs of SB-RA-2001 were synthesized and evaluated.81,84 Then, it was found that C-seco-TRAs, SB-RA-5001 and analogs (Figure 15), exhibited strong antitubercular activity (MIC99 1.25~5 μM) with substantially reduced cytotoxicity (IC50 >80 μM). Scanning electron microscopy (SEM) analysis of M. tuberculosis cells treated with SB-RA-2018 and SB-RA-5001 showed filamentation, a phenotypic response of FtsZ inhibition.81 Transition electron microscopy (TEM) analysis of Mtb-FtsZ treated with SB-RA-5001 indicated that SB-RA-5001 stabilized Mtb-FtsZ polymers.85
Figure 14.
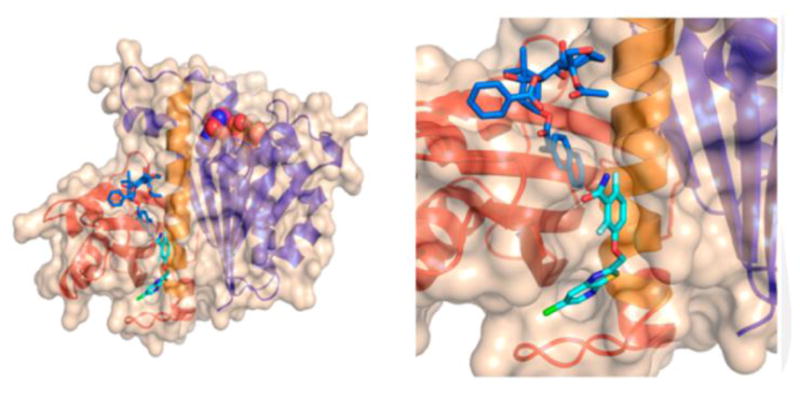
Analysis of the presumed binding of SB-RA-2001 and PC190723 on Bs-FtsZ. SB-RA-2001 is indicated in blue while PC190723 is indicated in cyan. Reprinted with permission from Singh, D.; Bhattacharya, A.; Rai, A.; Dhaked, H. P.; Awasthi, D.; Ojima, I.; Panda, D., SB-RA-2001 inhibits bacterial proliferation by targeting FtsZ assembly. Biochemistry. 2014, 53 (18), 2979–92. Copyright 2014 American Chemical Society.
Figure 15.

A lead Taxane and C-seco-taxanes
Singh et al. reported that SB-RA-2001 inhibited the growth of B. subtilis and Mycobacterium smegmatis (M. smegmatis) cells (MIC 30 μM and 60 μM, respectively).86 SB-RA-2001 substantially increased the bacterial cell length, indicating FtsZ as the target. SB-RA-2001 disturbed the formation of Z-ring in B. subtilis and affected the midcell localization of another cell division protein DivIVA. SB-RA-2001 also inhibited the GTPase activity of Bs-FtsZ, and promoted the assembly and bundling of Bs-FtsZ protofilaments. Docking analysis of the binding site of SB-RA-2001 in Bs-FtsZ suggested its proximity to that of PC190723 (Figure 14).86
2.2.9. Benzimidazoles
2.2.9.1. Trisubstituted benzimidazoles
Researchers at Southern Research Institute (SRI) identified several pyridopyrazine and pteridine based FtsZ inhibitors with antitubercular activity.87–88 Albendazole and thiabendazole are known tubulin inhibitors, and Sarcina et al. found that thiabendazole caused elongation of E. coli cells, suggesting FtsZ as the target.89 Slayden et al. demonstrated albendazole and thiabendazole inhibited septation of M. tuberculosis cells leading to filamentation and cell death.37
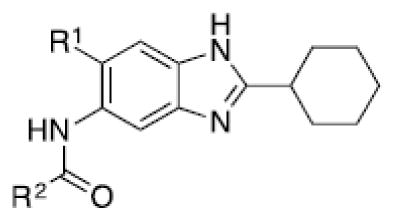
Through structural analysis of these heterocycles and substitution patterns, Ojima and Lee designed a library of trisubstituted benzimidazoles as FtsZ inhibitors.90 Libraries of 2,5,6- and 2,5,7-trisubstiutued benzimidazoles (~1,100 compounds) were synthesized and evaluated for their activity against M. tuberculosis H37Rv using microplate alamar blue assay in a 96-well format, which identified 204 hit compounds that showed antibacterial activity at ≤5 μg/mL.85,91,92 Then, 56 compounds out of the 204 hits were selected, resynthesized and their potencies determined (MIC 0.06–6.1 μg/mL).85, 91–92 SAR studies indicated that a cyclohexyl group at the 2-position is critical for high activity although other groups were tolerated. The 6-position tolerated various dialkylamino and aryloxy groups, but a dimethylamino group was found to provide a series of the most potent compounds.91–93 Compounds with a carbamate group at the 5-position were more potent than the 5-amido counterparts.91–92 The most potent compound of this series in the cell-based assay was SB-P17G-C2 (MIC 0.06 μg/mL).92 Selected lead compounds inhibited the polymerization of Mtb-FtsZ in a dose-dependent manner and increased the GTPase activity of Mtb-FtsZ, thus decreasing the stability of FtsZ polymers and protofilaments. TEM analysis of Mtb-FtsZ protein treated with SB-P17G-C2 and others indicated that these compounds not only inhibited the polymerization of FtsZ monomers into protofilaments but also destabilized and depolymerized FtsZ polymers preformed in the presence of GTP (Figure 16) (Table 1).92
Figure 16.
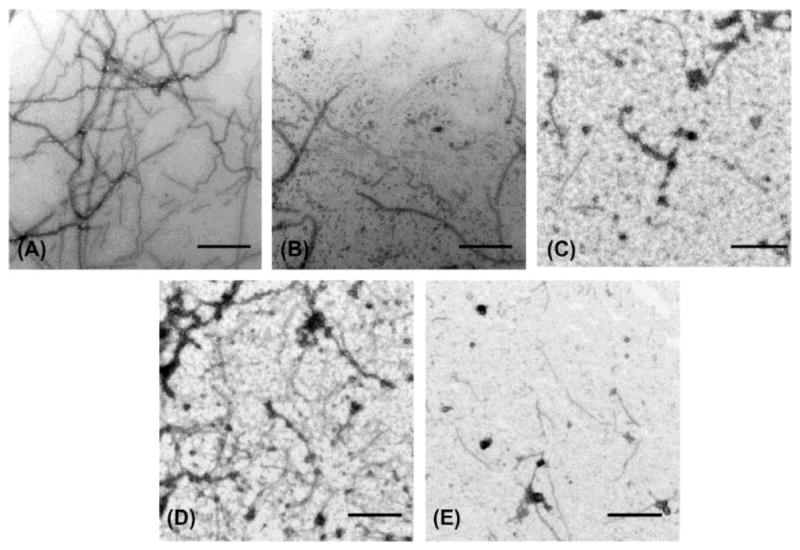
Transmission electron microscopy (TEM) Images of Mtb-FtsZ. Mtb-FtsZ (5 μM) was polymerized by GTP (25 lM) in the absence (A and D) and presence of 80 μM of SB-P3G2 (B) and SB-P17G-C2 (C). Image (E) displays the effect of SB-P17G-C2 (80 μM) on preformed polymers, wherein FtsZ polymers formed after addition of GTP were treated with compound. Images are at 49,000 magnification (scale bar 500 nm). Reprinted with permission from Awasthi, D.; Kumar, K.; Knudson, S. E.; Slayden, R. A.; Ojima, I., SAR Studies on Trisubstituted Benzimidazoles as Inhibitors of Mtb FtsZ for the Development of Novel Antitubercular Agents. J. Med. Chem. 2013, 56 (23), 9756–9770. Copyright 2013 American Chemical Society.
Table 1.
Antitubercular activity of lead benzimidazoles
| |||
|---|---|---|---|
| Compound | R1 | R2 | MIC99 (μg/mL) H37Rv |
| SB-P3G2 |
|
|
0.63 |
| SB-P8B2 |
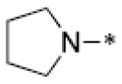
|
|
0.39 |
| SB-P17G-C2 |
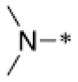
|
|
0.06 |
| SB-P17G-C4 |
|
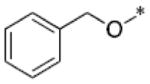
|
0.16 |
| SB-P20G3 |
|
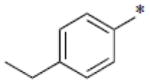
|
0.16 |
| SB-P17G-A20 |
|
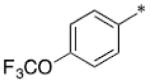
|
0.16 |
| SB-P21G4 |
|
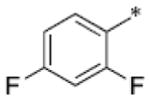
|
0.31 |
| SB-P21G7 |
|
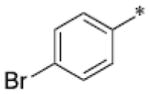
|
0.31 |
| SB-P17G-C8 |
|
|
0.31 |
| SB-P17G-A28 |
|
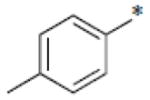
|
0.31 |
| SB-P17G-C12 |
|
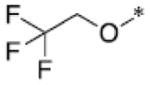
|
0.63 |
| SB-P17G-A33 |
|
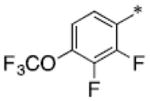
|
0.39 |
| SB-P17G-A38 |
|
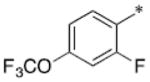
|
0.31 |
| SB-P17G-A42 |
|
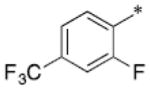
|
0.18 |
| SB-P26D2 |
|
|
0.63 |
Selected lead compounds were examined for their potencies against drug-sensitive (W210) and drug-resistant clinical strains (NHN382 and TN587) of M. tuberculosis and were found to be equally effective against both strains, as anticipated (Table 2).94 SB-P3G2 and SB-P8B2 were found to have modest activity against non-replicating M. tuberculosis cells grown under low oxygen conditions, which suggested that this series of compounds might have efficacy against latent TB infections.95 In the in vivo efficacy study of SB-P3G2 and SB-P17G-A20 using an acute TB infection model in GKO mice showed SB-P3G2 reduced the bacterial load of M. tuberculosis H37Rv by 0.7 ± 0.17 log10 CFU in the lungs and 0.41 ± 0.36 log10 CFU in spleen,95 while SB-P17G-A20 reduced the bacterial load by 1.73 ± 0.24 log10 CFU and 2.68 ± 0.48 log10 CFU in lungs and spleen, respectively.94
Table 2.
Antitubercular activity of lead benzimidazoles against drug sensitive and drug resistant strains
| Compound | (MIC99 μg/mL) M. tuberculois strains
|
||||
|---|---|---|---|---|---|
| H37Rv | NHN382 | TN587 | W210 | NHN20 | |
|
| |||||
| SB-P3G2 | 0.63 | 1.25 | 1.25 | 1.25 | - |
| SB-P8B2 | 0.39 | 0.31 | 0.16 | 0.63 | - |
| SB-P17G-C2 | 0.06 | 0.06 | 0.13 | 0.06 | - |
| SB-P20G3 | 0.16 | 0.16 | 0.16 | 0.16 | - |
| SB-P17G-A20 | 0.16 | 0.16 | 0.16 | 0.16 | - |
| SB-P17G-A33 | 0.39 | 0.37 | 0.31 | 0.47 | 0.39 |
| SB-P17G-A38 | 0.31 | 0.31 | 0.31 | 0.47 | 0.24 |
| SB-P17G-A42 | 0.18 | 0.16 | 0.24 | 0.31 | 0.16 |
In order to improve the plasma metabolic stability, SB-P17G-A33, SB-P17G-A38 and SB-P17G-A42 that contained additional fluorine atoms on the benzamido moiety were synthesized.96 These compounds inhibited drug sensitive and drug resistant strains of M. tuberculosis with MIC values ranging between 0.18 – 0.39 μg/mL (Table 2). SB-P17G-A33 and SB-P17G-A38 were stable in human and murine plasma after 4 hours of incubation.96 SB-P17G-A33, SB-P17G-A38 and SB-P17G-A42 displayed limited lability in the presence of liver microsomes. In the in vivo efficacy study of SB-P17G-A33, SB-P17G-A38 and SB-P17G-A42 using an acute TB infection model in GKO mice showed SB-P17G-A38 and SB-P17G-A42 reduced the bacterial load of M. tuberculosis H37Rv by y 5.7–6.3 log10 CFU in the lungs and 3.9–5.0 log10 CFU in the spleen, while SB-P17G-A33 reduced the bacterial load by 1.7–2.1 log10 CFU in the lungs and 2.5–3.4 log10 CFU in the spleen.96 The efficacy exhibited by SB-P17G-A38 and SB-P17G-A42 is equivalent to that of isoniazid (INH), and those lead compounds are active against INH-resistant patient-derived M. tuberculosis strains. Thus, these two lead benzimidazoles are highly promising to become drug candidates.
Kumar et al. screened libraries of 2,5,6- and 2,5,7-trisubstituted benzimidazoles against Francisella tularensis (F. tularensis) LVS strain, which resulted in 23 hits with >90 % growth inhibition at 10 μg/mL.97 Out of the 23 hits, 7 compounds displayed 40~50 % growth inhibition at 0.2 μg/mL, and 21 compounds exhibited MIC90 values of 0.35 48.6 μg/mL. Compound 21 (Figure 17) was the most potent compound in this series (MIC90 0.35 μg/mL). Ex-vivo efficacy assay of compounds 22e, 22f, 22g and 22h (Figure 17) was carried out using RAW macrophages infected by F. tularensis LVS, which disclosed that these benzimidazoles reduced CFU by 2–3 log units at 10–50 μg/mL except for 22f.97
Figure 17.

Trisubstituted benzimidazoles active against F. tularensis.
2.2.9.2. 2, 4-Disubstituted benzimidazoles
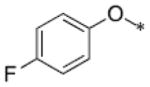
Ray et al. screened a library of 100 benzimidazole derivatives against B. subtilis cells and identified 2 hits, BT-benzo-29 (Figure 18) and BT-benzo-81.98 BT-benzo-29 caused cell elongation, inhibited Bs-FtsZ GTPase activity, and inhibited the Z-ring formation. BT-benzo-29 inhibited the proliferation of B. subtilis and M. smegmatis cells (IC50 1 and 1.6 μM, respectively). It bound to tubulin with very weak affinity. Docking study indicated that the binding site of BT-benzo-29 would be at the C-terminal domain near the T7 loop in Bs-FtsZ, which was confirmed by site-directed mutagenesis.98
Figure 18.
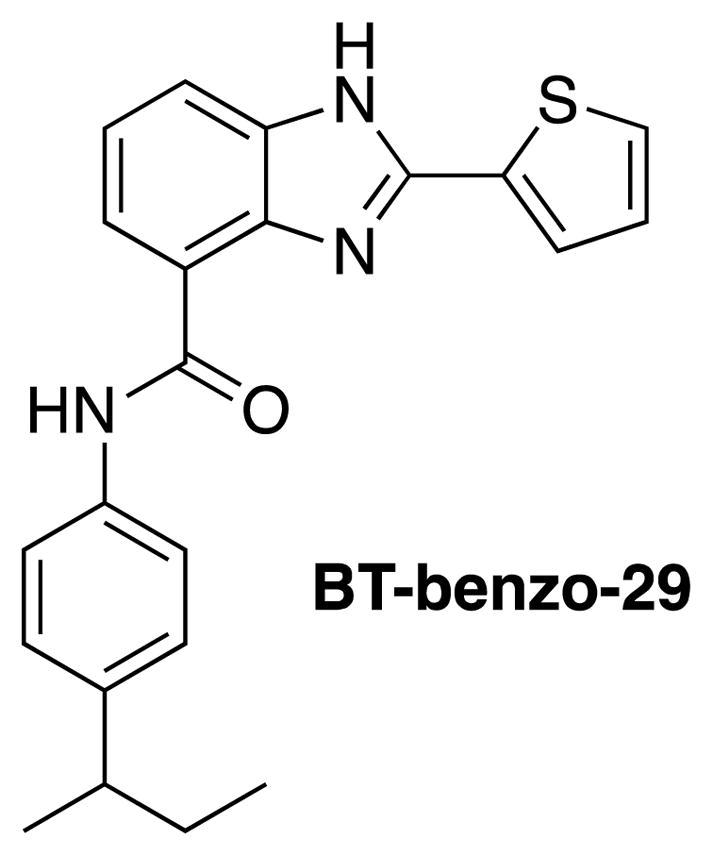
BT-benzo-29.
2.3. Polypeptides
2.3.1. Cathelin-related antimicrobial peptide (CRAMP)
Cathelin-related antimicrobial peptide is a murine defense peptide with 37 amino acid residues that bolsters the immune system by binding to bacterial cell surfaces and increasing the permeability of the outer membrane.99,100 This protein was active against various species and strains of bacteria, such as E. coli, Salmonella typhimurium (S. typhimurium), MRSA, and Bacillus megaterium (B. megaterium) with MIC values ranging from 0.5 μM to 64 μM.100 Recent studies using a truncated version of the peptide, CRAMP (16–33), exhibited FtsZ activity.101 Light scattering analysis of FtsZ and tubulin treated with CRAMP showed a dose-dependent inhibition of FtsZ polymerization, while tubulin polymerization was unaffected.101 This peptide also decreased the GTPase activity of FtsZ, but it did not share the same binding site as GTP, and B. subtilis cells exposed to this protein were elongated and lacked Z-ring formation. A docking study using Bs-FtsZ indicated a crevice near the T7 loop where CRAMP (16–33) might bind (Figure 19).101 The proposed mechanism is that CRAMP (16–33) binds to FtsZ near the T7 loop and disrupts monomer-monomer interaction of FtsZ, hence preventing polymerization.101
Figure 19.

Interaction of CRAMP (16–33) with FtsZ. The N-terminus of FtsZ is blue. The H7 helix is green. The C-terminus is red, and CRAMP (16–33) is magenta. A) Proposed CRAMP (16–33) binding site located near the T7 loop. B) Magnification of CRAMP (16–33) binding site. C) Residue interactions forming salt bridges (green) and hydrogen bonds (orange). Reprinted with permission from SocietyRay, S., Dhaked, H. P., Panda, D., Antimicrobial peptide CRAMP (16–33) stalls bacterial cytokinesis by inhibiting FtsZ assembly. Biochemistry. 2014, 53 (41), 6426–9. Copyright 2014 American Chemical Society.
2.3.2. Edeine
Edeine is a polypeptide antibiotic obtained from Bacillus brevis (B. brevis) strain Vm4.102 The antibiotic consists of a pentapeptide linked to a base moiety. It exists in two forms, edeine A and edeine B, which differ in the base moiety. Edeine A and edeine B are each further differentiated into two reversible isomers, edeine A1, A2 and edeine B1, B2, respectively. Edeine A1 and edeine B1 are active, while the other isomers are inactive.103 The pentapeptide moiety, which is identical for both forms of edeine, is Gly-isoSer-(2,6-diamino-7-hydroxyazelayl)-β-Tyr-(α,β-diaminopropanoyl).102 The base component of edeine A and edeine B are spermidine and N-guanyl-N′-(3-aminopropyl)-1,4-diaminobutane(guanylspermidine), respectively.102 Edeine A1 inhibits DNA and protein synthesis within bacteria, resulting in a filamentous phenotype.104–106 Cells cannot divide when DNA synthesis is halted. Recently, the target of edeine B1 (Figure 20) was found to be FtsZ in B. subtilis cells.103 Z-ring formation was inhibited in cells expressing FtsZ-GFP, but DNA replication was unhindered.103
Figure 20.

Comparison of peptide, PNA, LNA, and DNA. PNA and LNA can readily associate through nucleotide base pairing with DNA.
2.4. Nucleic Acids
2.4.1. Peptide nucleic acids (PNA)
Peptide nucleic acids (PNAs) are oligonucleotide analogues with a polypeptide-like backbone instead of a ribose-phosphate backbone (Figure 20).107,108 PNAs are neutral and highly resistant to degradation by nucleases.107 PNAs can silence genes by binding to complimentary strands of RNA to prevent them from expression. Recently, ten regions free of secondary structures were found in ftsZ-mRNA using a prediction software, Mfold web server and RNAstructure 5.5. Then, complimentary oligonucleotides for each viable region were synthesized and then examined through dot-blot hybridization.
A PNA oligomer was synthesized based on the best result from dot-blot hybridization, and conjugated to the cell penetrating peptide, (RXR)4XB, for improved internalization into cells. The resulting conjugate was PPNA1, which was complimentary to nucleotides 309–323 of ftsZ-mRNA.109 A second conjugate, PPNA2, was synthesized from the translation initiation and ribosome binding sites of ftsZ-mRNA.109 Both PPNA1 and PPNA2 inhibited the growth of MRSA CY-11 strain in a dose-dependent manner, and complete growth inhibition was observed at 30 μM for PPNA1 and 40 μM for PPNA2.109 Cell viability assays, at 40 μM of both compounds, revealed that PPNA1 was bactericidal, while PPNA2 was not.109 RT-PCR experiments, on bacterial cells exposed to PPNA1 and PPNA2, showed a dose-dependent decrease in ftsZ-mRNA, indicating an antisense gene knock-down mechanism.109
2.4.2. Locked nucleic acids (PLNA)
Locked nucleic acids (LNA) are nucleic acid analogues with the ribose ring locked by a 2′-O, 4′-C-methylene bridge (Figure 20).110,111 Oligomeric chains of LNA have high affinity to complimentary strands of RNA or DNA, and act by silencing target genes.111 Gene silencing with LNA has been effective in killing E. coli cells by targeting RNase P expression.110 Recently, a peptide-LNA hybrid was synthesized and exhibited activity against MRSA-FtsZ. A peptide-LNA compound, PLNA787, consisted of a cell-penetrating peptide, (KFF)3K, attached to a cysteine-succinimidyl trans-4-(maleimidylmethyl)cyclohexane-1-carboxylate-C6 (Cys-SMCC-C6) linker and an LNA oligomer.111 The LNA portion of LNA787 was complimentary to the nucleotides 787–808 fragment of MRSA ftsZ-mRNA.
PLNA787 and its various components were examined against 10 strains of S. aureus for their antibacterial activity, wherein one strain was MSSA ATCC29213 strain, and nine others, Mu50, WHO-2, and MRSA01 to MRSA07, were MRSA strains. It was found that LNA787 itself exhibited no antibiotic activity against those strains in this assay, but (KFF) 3K-PLNA787 conjugate was active, which indicates that the conjugation to a cell penetrating peptide was required for efficient delivery of PLNA787 into the cell.111 (KFF)3K-PLNA787 conjugate was active against all 10 strains of S. aureus with MIC values of 1.56~12.5 μM.111 When human gastric mucosa epithelial cells were infected with Mu50, PLNA787 conjugate protected the eukaryotic cells against infection.
PLNA787 conjugate also rescued Mu50-infected mice, while untreated and ampicillin-treated mice died within two days. A mechanistic study showed a decline in ftsZ-mRNA expression as well as diminished FtsZ protein expression.111 TEM of Mu50 cells treated with PLNA787 conjugate showed increased size compared to untreated cells, and some cells exhibited diploid and tetraploid phenotypes. PLNA787 was demonstrated to inhibit cell division in various MRSA strains by silencing the ftsZ gene and preventing protein expression.111 The conjugate was also shown to be noncytotoxic and effective in bacterial growth assays, epithelial cell infection assays, and mouse infection models.111
2.5. Summary of FtsZ inhibitors, their (putative) binding sites and mechanism of action
Table 3 summarizes the FtsZ inhibitor’s compounds types, bacterial species studied, (putative) binding sites in FtsZ and their mechanisms of action. Since the protein X-ray crystallography of FtsZ-inhibitor has been successful only for PC190723- Sa-FtsZ co-crystals,67 the structural biology of FtsZ inhibitors needs to be advanced to promote rational structure-based design. Nevertheless, putative binding sites have been indicated and reasonable mechanisms of action have been proposed based on various biochemical and molecular biology studies.
Table 3.
Summary of inhibitors and their mechanisms of action.
| Compound | Bacteria studied | Binding site | Mechanism of action |
|---|---|---|---|
|
| |||
| Curcumin | E.coli, S.aureus | close to GTP binding site42 | increases GTPase activity and destabilizes FtsZ polymers41 |
| Coumarin | M. tuberculosis, B.subtilis, E.coli | allosteric site near T7 loop44 | inhibition of GTPase activity44 |
| Plumbagins | B.subtilis, E.coli | near the C-terminal48 | inhibition of GTPase activity48 |
| Resveratrol | E.coli | unknown | inhibition of FtsZ gene expression51 |
| Chrysophaentin | E.coli | GTP binding site52 | inhibition of GTPase activity53 |
| Berberine | E.coli | close to GTP binding site54 | inhibition of GTPase activity54 |
| Berberine derivative (2) | S.aureus | interdomain cleft56 | promotes FtsZ polymerization and stabilizes FtsZ polymers56 |
| Phenylpropanoids | B.subtilis, E.coli | T7 loop59 | inhibition of FtsZ polymerization59 |
| Cinnamaldehyde and cinnamides | B.subtilis, E.coli, MRSA | C terminal region around the T7 loop61 | inhibition of GTPase activity61 |
| PC190723 | MRSA, MDRSA | between C terminal domain and helix 7 (co-crystal X-ray)67 | stabilize FtsZ polymers65 |
| Arene-diol digallates | B.subtilis, MRSA | GTP binding site73 | inhibition of GTPase activity73 |
| Rhodanine derivatives | B.subtilis, E.coli | cavity adjacent to T7 loop76 | promotes assembly and stabilizes FtsZ polymers76 |
| Pyrimidine quinuclidine derivatives | S.aureus, E.coli | GTP binding site79 | inhibition of GTPase activity79 |
| Taxanes (SB-RA-5001, SB-RA-2001) | M. tuberculosis, B.subtilis | close to the PC-190723 binding site86 | promotes FtsZ polymerization and stabilizes FtsZ polymers85 |
| Trisubstituted benzimidazoles | M. tuberculosis | unknown | increases GTPase activity, destabilizing FtsZ polymers91,92 |
| BT-Benzo-29 | B.subtilis | C terminal domain near T7 loop98 | inhibition of GTPase activity98 |
| CRAMP (16–33) | B.subtilis | near T7 loop101 | disrupts monomer- monomer interaction101 |
| PNA | MRSA | FtsZ-mRNA109 | antisense gene knockdown109 |
| (KFF)3K-PLNA787 | S.aureus, MRSA | FtsZ-mRNA111 | antisense gene knockdown111 |
3. Conclusion
Emergence of drug resistance to currently used antibiotics and antimicrobials drugs, especially multidrug resistance, is an imminent threat to human health. The vast majority of clinically used antibacterial drugs target cell wall biosynthesis, nucleic acid synthesis and protein synthesis. Thus, identification of a novel drug target that is critical for the bacterial survival to overcome such drug resistance, is urgently needed. In the last 15 years, FtsZ, an essential bacterial cell division protein and a homologue of tubulin, emerged as a highly promising new molecular target for new-generation antimicrobial drug discovery. FtsZ is a highly conserved and ubiquitous bacterial protein, playing a vital role in bacterial cytokinesis. Thus, the inhibition of proper FtsZ assembly causes the disruption of septum formation and bacterial cell division, leading to cell death. Therefore, FtsZ inhibitors have been actively investigated for broad-spectrum or pathogen-specific antibacterial agents. In addition, extensive studies have been performed on the FtsZ structures and functions based on structural biology as well as cell and microbiology, which can be translated into structure-based or fragment-based drug discovery by exploiting computational biology tools.
As described above, a variety of natural products and their derivatives, as well as synthetic small molecules have been investigated and several classes of compounds, targeting FtsZ, have been found effective against various pathogens, including M. tuberculosis, MRSA and VRE, as well as B. subtilis, S.aureus, MSSA and VSE. Among various natural products and their derivatives, berberine derivatives, cinnamaldehyde and cinnamamides exhibited MIC values as low as 0.1 μg/mL against MRSA, S. aureus, E. coli and S. subtilis, but so far no in vivo efficacy data have been reported. However, it should be noted that curcumin, rhodamine, and their derivatives, for example, are categolized as “Pan Assay INterference compoundS (PAINS)”112, which display promiscuous behavior that result in interference of a variety of assays like metal chelation, redox cycling, and protein reactivity. Such behavior may either result in false positives while carrying out high throughput screening.112 Curcumin could perturb membranes and alter the function of membrane proteins such as membrane-anchored metalloproteases, mechanosensitive ion channels, and voltage-dependent potassium and sodium channels.113 Also, the promiscuous interaction of rhodanine with other proteins could cause toxicity while the color of rhodanine itself could interfere in colorimetric assays.114 Accordingly, compounds like curcumin and rhodanine may not provide a good starting point for the development of FtsZ inhibitors.
Among the small molecule FtsZ inhibitors investigated, a benzamide derivative, PC190723, exhibited very strong potency against S. aureus (MIC 0.03 μg/mL). Although PC190723 is highly potent, the compound has rather poor pharmacological properties. Accordingly, TX707, which has an improved solubility, was developed, and also prodrugs of PC190723 and TX707 were explored. Thus, TX541 (prodrug of PC190723) and TX709 (prodrug of TX707) were developed, which exhibited efficacy in the MRSA infected mice model (up to 2 log10 CFU reduction in thigh). At present, TX709 appears to be the most advanced lead compound in this series.65,69,70 These compounds, however, showed poor activity against other Gram-positive and Gram-negative pathogens. Accordingly, the discovery of broad-spectrum antibacterial agents may require another round of lead discovery efforts.
Another series of promising FtsZ inhibitors is trisubstituted benzimidazoles, especially against drug-resistant M. tuberculosis. 2,5,6-Trisubstituted benzimidazoles inhibit the nucleation/aggregation/polymerization of Mtb-FtsZ quite effectively in a dose-dependent manner, by enhancing the GTPase activity of Mtb-FtsZ. Several lead compounds have been identified, which show highly promising efficacy in the acute animal models as well as ADME profiles in the preclinical drug development. Especially, SB-P17G-A38 and SB-P17G-A42 reduced the bacterial load of M. tuberculosis H37Rv by y 5.7~6.3 log10 CFU in the lungs and 3.9~5.0 log10 CFU in the spleen in the in vivo efficacy study using an acute TB infection model in GKO mice.85,91,92,94–96 It is noteworthy that the efficacy exhibited by these two benzimidazoles is equivalent to that of isoniazid (INH), and those lead compounds are also active against INH-resistant patient-derived M. tuberculosis strains. Consequently, these two leads are likely to become anti-TB drug candidates.
Quite recently, peptide-based FtsZ inhibitors, such as CRAMP and edeine B1, have exhibited good activities against various bacteria, including MRSA.101,103 This is certainly a new trend and other peptide-based FtsZ-inhibitors will certainly be explored in coming years.
Collectively, it has been shown that (i) the disruption of Z-ring formation, either by stabilizing or destabilizing FtsZ polymers, leads to bacterial cell death and (ii) the FtsZ-interacting agents do not have any appreciable resistance to the drug-resistant strains for clinically used antibacterial drugs at present since FtsZ is an entirely new target for antibacterial agents.
In addition to the inhibition of FtsZ protein function, a different approach is showing promise, which is the suppression of the ftsZ gene expression by nucleic acid-based agents. PNA and LNA can silence ftsZ gene by using complementary oligonucleotides to ftsZ-mRNA, but delivery to cells is an issue. Thus, those were conjugated to Arg-rich cell-penetrating peptides for enhancing internalization. PPNA1 is active against MRSA CY-11 strain and bactericidal.109 LNA787 is complimentary to the nucleotides 787–808 fragment of MRSA ftsZ-mRNA. Although LNA can silence gene expressions effectively, LNA787 does not show any antibacterial activity, due to the delivery problem. However, PLNA787 conjugate, consisting of a cell-penetrating peptide, (KFF)3K, and LNA787, is active against 10 strains of S. aureus with very good MIC values and has been able to rescue Mu50-infected mice, while untreated and ampicillin-treated mice died within two days.111 Thus, this new approach appears highly promising.
As discussed above, FtsZ has proven to be an excellent molecular target for antibacterial drug discovery and there has been extensive studies on the discovery and development of efficacious FtsZ inhibitors as next-generation antibacterial agents. Accordingly, it appears likely that a good number of clinical candidates will be developed in the near future.
Acknowledgments
The authors acknowledge a grant support from the National Institutes of Health (AI 078251 to I.O.) and the support from Sanofi as a collaboration partner for a part of the work in this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Who. Antimicrobial Resistance Global Report on Surveillance. WHO Press; 2014. [Google Scholar]
- 2.Cdc. Antibiotic resistance threats. 2013. [Google Scholar]
- 3.Who. Global Tuberculosis Report 2015. WHO Press; 2015. [Google Scholar]
- 4.Beall B, Lutkenhaus J. Genes Dev. 1991;5:447. doi: 10.1101/gad.5.3.447. [DOI] [PubMed] [Google Scholar]
- 5.Dai K, Lutkenhaus J. J Bacteriol. 1991;173:3500. doi: 10.1128/jb.173.11.3500-3506.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leung AKW, Lucile White E, Ross LJ, Reynolds RC, DeVito JA, Borhani DW. J Med Chem. 2004;342:953. doi: 10.1016/j.jmb.2004.07.061. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Lutkenhaus J. J Bacteriol. 1996;178:2314. doi: 10.1128/jb.178.8.2314-2319.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margolin W, Wang R, Kumar M. J Bacteriol. 1996;178:1320. doi: 10.1128/jb.178.5.1320-1327.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumann P, Jackson SP. Proc Natl Acad Sci USA. 1996;93:6726. doi: 10.1073/pnas.93.13.6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukherjee a, Lutkenhaus J. J Bacteriol. 1994;176:2754. doi: 10.1128/jb.176.9.2754-2758.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nogales E, Wolf SG, Downing KH. Nature. 1998;391:199. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 12.Amos LA, van den Ent F, Löwe J. Curr Opin Cell Biol. 2004;16:24. doi: 10.1016/j.ceb.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Ma S. Eur J Med Chem. 2015;95:1. doi: 10.1016/j.ejmech.2015.03.026. [DOI] [PubMed] [Google Scholar]
- 14.Oliva MA, Trambaiolo D, Löwe J. J Med Chem. 2007;373:1229. doi: 10.1016/j.jmb.2007.08.056. [DOI] [PubMed] [Google Scholar]
- 15.Kumar K, Awasthi D, Berger WT, Tonge PT, Slayden RA, Ojima I. Future Med Chem. 2010;2:1305. doi: 10.4155/fmc.10.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goehring NW, Beckwith J. Curr Biol. 2005;15:514. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 17.Lu C, Stricker J, Erickson HP. Cell Motil Cytoskeleton. 1998;40:71. doi: 10.1002/(SICI)1097-0169(1998)40:1<71::AID-CM7>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 18.Addinall SG, Lutkenhaus J. Mol Microbiol. 1996;22:231. doi: 10.1046/j.1365-2958.1996.00100.x. [DOI] [PubMed] [Google Scholar]
- 19.Jindal B, Panda D. Biochemistry. 2013;52:7071. doi: 10.1021/bi400129j. [DOI] [PubMed] [Google Scholar]
- 20.Stricker J, Maddox P, Salmon ED, Erickson HP. Proc Natl Acad Sci USA. 2002;99:3171. doi: 10.1073/pnas.052595099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szwedziak P, Wang Q, Bharat TAM, Tsim M, Löwe J. eLife. 2014;3:1. doi: 10.7554/eLife.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hale Ca, De Boer Pa. J Cell. 1997;88:175. doi: 10.1016/s0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- 23.Pichoff S, Lutkenhaus J. Mol Microbiol. 2007;64:1129. doi: 10.1111/j.1365-2958.2007.05735.x. [DOI] [PubMed] [Google Scholar]
- 24.Pichoff S, Lutkenhaus J. EMBO J. 2002;21:685. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Boer Pa, Crossley RE, Rothfield LI. Cell. 1989;56:641. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- 26.Raskin DM, de Boer Pa. Cell. 1997;91:685. doi: 10.1016/s0092-8674(00)80455-9. [DOI] [PubMed] [Google Scholar]
- 27.Raskin DM, de Boer Pa. Proc Natl Acad Sci USA. 1999;96:4971. doi: 10.1073/pnas.96.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu X, Shih YL, Zhang Y, Rothfield LI. Proc Natl Acad Sci USA. 2001;98:980. doi: 10.1073/pnas.031549298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cha JH, Stewart GC. J Bacteriol. 1997;179:1671. doi: 10.1128/jb.179.5.1671-1683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Baarle S, Bramkamp M. PLoS One. 2010;5:e9850. doi: 10.1371/journal.pone.0009850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu LJ, Errington J. Cell. 2004;117:915. doi: 10.1016/j.cell.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 32.Bernhardt TG, de Boer PA. J Mol Cell. 2005;18:555. doi: 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsin J, Gopinathan A, Huang KC. Proc Natl Acad Sci USA. 2012;109:9432. doi: 10.1073/pnas.1120761109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allard JF, Cytrynbaum EN. Proc Natl Acad Sci USA. 2009;106:145. doi: 10.1073/pnas.0808657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan G, Wolgemuth CW, Sun SX. Proc Natl Acad Sci USA. 2007;104:16110. doi: 10.1073/pnas.0702925104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez-Aportela E, Lopez-Blanco JR, Andreu JM, Chacon P. Biophys J. 2014;107:2164. doi: 10.1016/j.bpj.2014.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slayden RA, Knudson DL, Belisle JT. Microbiology. 2006;152:1789. doi: 10.1099/mic.0.28762-0. [DOI] [PubMed] [Google Scholar]
- 38.Addinall SG, Bi E, Lutkenhaus J. J Bacteriol. 1996;178:3877. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margolin W. FEMS Microbiol Rev. 2000;24:531. doi: 10.1111/j.1574-6976.2000.tb00554.x. [DOI] [PubMed] [Google Scholar]
- 40.Rajagopalan M, Atkinson MA, Lofton H, Chauhan A, Madiraju MV. Biochem Biophys Res Commun. 2005;331:1171. doi: 10.1016/j.bbrc.2005.03.239. [DOI] [PubMed] [Google Scholar]
- 41.Rai D, Singh Jay K, Roy N, Panda D. Biochem J. 2008;410:147. doi: 10.1042/BJ20070891. [DOI] [PubMed] [Google Scholar]
- 42.Gupta KK, Bharne SS, Rathinasamy K, Naik NR, Panda D. FEBS J. 2006;273:5320. doi: 10.1111/j.1742-4658.2006.05525.x. [DOI] [PubMed] [Google Scholar]
- 43.Kaur S, Modi NH, Panda D, Roy N. Eur J Med Chem. 2010;45:4209. doi: 10.1016/j.ejmech.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 44.Duggirala S, Nankar RP, Rajendran S, Doble M. Appl Biochem Biotechnol. 2014;174:283. doi: 10.1007/s12010-014-1056-2. [DOI] [PubMed] [Google Scholar]
- 45.Chiang C-C, Cheng M-J, Peng C-F, Huang H-Y, Chen I-S. Chem Biodivers. 2010;7:1728. doi: 10.1002/cbdv.200900326. [DOI] [PubMed] [Google Scholar]
- 46.Jamal MS, Parveen S, Beg MA, Suhail M, Chaudhary AG, Damanhouri GA, Abuzenadah AM, Rehan M. PLoS One. 2014;9:e87309. doi: 10.1371/journal.pone.0087309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Acharaya BR, Battacharyya B, Chakrabarti G. Biochemistry. 2008;47:7838. doi: 10.1021/bi800730q. [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharya A, Jindal B, Singh P, Datta A, Panda D. FEBS J. 2013;280:4585. doi: 10.1111/febs.12429. [DOI] [PubMed] [Google Scholar]
- 49.Burns J, Yokota T, Ashihara H, Lean MEJ, Crozier A. J Agric Food Chem. 2002;50:3337. doi: 10.1021/jf0112973. [DOI] [PubMed] [Google Scholar]
- 50.Soleas JG, Diamandis PE, Goldberg MD. Clin Biochem. 1997;30:91. doi: 10.1016/s0009-9120(96)00155-5. [DOI] [PubMed] [Google Scholar]
- 51.Hwang D, Lim YH. Sci Rep. 2015;5:10029. doi: 10.1038/srep10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plaza A, Keffer JL, Bifulco G, Lloyd JR, Bewley CA. J Am Chem Soc. 2010;132:9069. doi: 10.1021/ja102100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keffer JL, Huecas S, Hammill JT, Wipf P, Andreu JM, Bewley CA. Bioorg Med Chem. 2013;21:5673. doi: 10.1016/j.bmc.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Domadia PN, Bhunia A, Sivaraman J, Swarup S, Dasgupta D. Biochemistry. 2008;47:3225. doi: 10.1021/bi7018546. [DOI] [PubMed] [Google Scholar]
- 55.Sun N, Chan FY, Lu YJ, Neves MA, Lui HK, Wang Y, Chow KY, Chan KF, Yan SC, Leung YC, Abagyan R, Chan TH, Wong KY. PLoS One. 2014;9:e97514. doi: 10.1371/journal.pone.0097514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parhi A, Lu S, Kelley C, Kaul M, Pilch DS, LaVoie E. J Bioorg Med Chem Lett. 2012;22:6962. doi: 10.1016/j.bmcl.2012.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kornika LG. Cell Mol Biol. 2007;53:15. [Google Scholar]
- 58.Puupponen-Pimiä R, Nohynek L, Meier C, Kähkönen M, Heinonen M, Hopia A, Oksman-Caldentey KM. J App Microbiol. 2001;90:494. doi: 10.1046/j.1365-2672.2001.01271.x. [DOI] [PubMed] [Google Scholar]
- 59.Hemaiswarya S, Soudaminikkutty R, Narasumani ML, Doble M. J Med Microbiol. 2011;60:1317. doi: 10.1099/jmm.0.030536-0. [DOI] [PubMed] [Google Scholar]
- 60.Ali SM, Khan AA, Ahmed I, Musaddiq M, Ahmed KS, Polasa H, Rao LV, Habibullah CM, Sechi LA, Ahmed N. Ann Clin Microbiol Antimicrob. 2005;4:20. doi: 10.1186/1476-0711-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Domadia P, Swarup S, Bhunia A, Sivaraman J, Dasgupta D. Biochem Pharmacol. 2007;74:831. doi: 10.1016/j.bcp.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 62.Li X, Sheng J, Huang G, Ma R, Yin F, Song D, Zhao C, Ma S. Eur J Med Chem. 2015;97:32. doi: 10.1016/j.ejmech.2015.04.048. [DOI] [PubMed] [Google Scholar]
- 63.Ohashi Y, Chijiiwa Y, Suzuki K, Takahashi K, Nanamiya H, Sato T, Hosoya Y, Ochi K, Kawamura aF. J Bacteriol. 1998;181:1348. doi: 10.1128/jb.181.4.1348-1351.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Czaplewski LG, Collins I, Boyd EA, Brown D, East SP, Gardiner M, Fletcher R, Haydon DJ, Henstock V, Ingram P, Jones C, Noula C, Kennison L, Rockley C, Rose V, Thomaides-Brears HB, Ure R, Whittaker M, Stokes NR. Bioorg Med Chem Lett. 2009;19:524. doi: 10.1016/j.bmcl.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 65.Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. Science. 2008;321:1673. doi: 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- 66.Andreu JM, Schaffner-Barbero C, Huecas S, Alonso D, Lopez-Rodriguez ML, Ruiz-Avila LB, Nunez-Ramirez R, Llorca O, Martin-Galiano AJ. J Biol Chem. 2010;285:14239. doi: 10.1074/jbc.M109.094722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elsen NL, Lu J, Parthasarathy G, Reid JC, Sharma S, Soisson SM, Lumb KJ. J Am Chem Soc. 2012;134:12342. doi: 10.1021/ja303564a. [DOI] [PubMed] [Google Scholar]
- 68.Stokes NR, Baker N, Bennett JM, Chauhan PK, Collins I, Davies DT, Gavade M, Kumar D, Lancett P, Macdonald R, Macleod L, Mahajan A, Mitchell JP, Nayal N, Nayal YN, Pitt GR, Singh M, Yadav A, Srivastava A, Czaplewski LG, Haydon D. J Bioorg Med Chem Lett. 2014;24:353. doi: 10.1016/j.bmcl.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 69.Kaul M, Mark L, Zhang Y, Parhi AK, LaVoie EJ, Pilch DS. Biochem Pharmacol. 2013;86:1699. doi: 10.1016/j.bcp.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 70.Kaul M, Mark L, Zhang Y, Parhi AK, Lyu YL, Pawlak J, Saravolatz S, Saravolatz LD, Weinstein MP, LaVoie EJ, Pilch DS. Antimicrob Agents Chemother. 2015;59:4845. doi: 10.1128/AAC.00708-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun J, Li M-H, Wang X-Y, Zhang Y, Yuan R-J, Liu H-Y, Zhu H-L. Med Chem Res. 2013;23:2985. [Google Scholar]
- 72.Cerrutti P, Alzamora SM, Vidales SI. J Food Sci. 1997;62:608. [Google Scholar]
- 73.Ruiz-Avila LB, Huecas S, Artola M, Vergonos A, Ramirez-Aportela E, Cercenado E, Barasoain I, Vazquez-Villa H, Martin-Fontecha M, Chacon P, Lopez-Rodriguez ML, Andreu JM. ACS Chem Biol. 2013;8:2072. doi: 10.1021/cb400208z. [DOI] [PubMed] [Google Scholar]
- 74.Artola M, Ruiz-Avila LB, Vergonos A, Huecas S, Araujo-Bazan L, Martin-Fontecha M, Vazquez-Villa H, Turrado C, Ramirez-Aportela E, Hoegl A, Nodwell M, Barasoain I, Chacon P, Sieber SA, Andreu JM, Lopez-Rodriguez ML. ACS Chem Biol. 2015;10:834. doi: 10.1021/cb500974d. [DOI] [PubMed] [Google Scholar]
- 75.Beuria TK, Singh P, Surolia A, Panda D. Biochem J. 2009;423:61. doi: 10.1042/BJ20090817. [DOI] [PubMed] [Google Scholar]
- 76.Singh P, Jindal B, Surolia A, Panda D. Biochemistry. 2012;51:5434. doi: 10.1021/bi201813u. [DOI] [PubMed] [Google Scholar]
- 77.Nepomuceno GM, Chan KM, Huynh V, Martin KS, Moore JT, O’Brien TE, Pollo LA, Sarabia FJ, Tadeus C, Yao Z, Anderson DE, Ames JB, Shaw JT. ACS Med Chem Lett. 2015;6:308. doi: 10.1021/ml500497s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parhi AK, Zhang Y, Saionz KW, Pradhan P, Kaul M, Trivedi K, Pilch DS, LaVoie E. J Bioorg Med Chem Lett. 2013;23:4968. doi: 10.1016/j.bmcl.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chan FY, Sun N, Neves MA, Lam PC, Chung WH, Wong LK, Chow HY, Ma DL, Chan PH, Leung YC, Chan TH, Abagyan R, Wong KY. J Chem Inf Model. 2013;53:2131. doi: 10.1021/ci400203f. [DOI] [PubMed] [Google Scholar]
- 80.Mathew B, Srivastava S, Ross LJ, Suling WJ, White EL, Woolhiser LK, Lenaerts AJ, Reynolds RC. Bioorg Med Chem. 2011;19:7120. doi: 10.1016/j.bmc.2011.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang Q, Kirikae F, Kirikae T, Pepe A, Amin A, Respicio L, Slayden RA, Tonge PJ, Ojima I. J Med Chem. 2006;49:463. doi: 10.1021/jm050920y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taraboletti G, Micheletti G, Rieppi M, Poli M, Turatto M, Rossi C, Borsotti P, Roccabianca P, Scanziani E, Nicoletti MI, Bombardelli E, Morazzoni P, Riva A, Giavazzi R. Clin Cancer Res. 2002;8:1182. [PubMed] [Google Scholar]
- 83.Appendino G, Danieli B, Jakupovic J, Belloro E, Scambia G, Bombardelli E. Tetrahedron Lett. 1997;38:4273. [Google Scholar]
- 84.Huang Q, Tonge PJ, Slayden RA, Kirikae T, Ojima I. Curr Top Med Chem. 2007:527. doi: 10.2174/156802607780059790. [DOI] [PubMed] [Google Scholar]
- 85.Ojima I, Kumar K, Awasthi D, Vineberg JG. Bioorg Med Chem. 2014;22:5060. doi: 10.1016/j.bmc.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh D, Bhattacharya A, Rai A, Dhaked HP, Awasthi D, Ojima I, Panda D. Biochemistry. 2014;53:2979. doi: 10.1021/bi401356y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reynolds RC, Srivastava S, Ross LJ, Suling WJ, White EL. Bioorg Med Chem Lett. 2004;14:3161. doi: 10.1016/j.bmcl.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 88.White EL, Suling WJ, Ross LJ, Seitz LE, Reynolds RC. J Antimicrob Chemother. 2002;50:111. doi: 10.1093/jac/dkf075. [DOI] [PubMed] [Google Scholar]
- 89.Sarcina M, Mullineaux CW. FEMS Microbiol Lett. 2000;191:25. doi: 10.1111/j.1574-6968.2000.tb09314.x. [DOI] [PubMed] [Google Scholar]
- 90.Ojima I, Lee S-y. The Research Foundation of State University of New York; United States: US 8,232,410 United States patent. 2012
- 91.Kumar K, Awasthi D, Lee SY, Zanardi I, Ruzsicska B, Knudson S, Tonge PJ, Slayden RA, Ojima I. J Med Chem. 2011;54:374. doi: 10.1021/jm1012006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Awasthi D, Kumar K, Knudson SE, Slayden RA, Ojima I. J Med Chem. 2013;56:9756. doi: 10.1021/jm401468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Park B, Awasthi D, Chowdhury SR, Melief EH, Kumar K, Knudson SE, Slayden RA, Ojima I. Bioorg Med Chem. 2014;22:2602. doi: 10.1016/j.bmc.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Knudson SE, Awasthi D, Kumar K, Carreau A, Goullieux L, Lagrange S, Vermet H, Ojima I, Slayden RA. PLoS One. 2014;9:e93953. doi: 10.1371/journal.pone.0093953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Knudson SE, Kumar K, Awasthi D, Ojima I, Slayden RA. Tuberculosis. 2014;94:271. doi: 10.1016/j.tube.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Knudson SE, Awasthi D, Kumar K, Carreau A, Goullieux L, Lagrange S, Vermet H, Ojima I, Slayden RA. J Antimicrob Chemother. 2015;70:3070. doi: 10.1093/jac/dkv226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kumar K, Awasthi D, Lee S-Y, Cummings JE, Knudson SE, Slayden RA, Ojima I. Bioorg Med Chem. 2013;21:3318. doi: 10.1016/j.bmc.2013.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ray S, Jindal B, Kunal K, Surolia A, Panda D. FEBS J. 2015;282:4015. doi: 10.1111/febs.13403. [DOI] [PubMed] [Google Scholar]
- 99.Gallo RL, Kim KJ, Bernfield M, Kozak Ca, Zanetti M, Merluzzi L, Gennaro R. J Biol Chem. 1997;272:13088. doi: 10.1074/jbc.272.20.13088. [DOI] [PubMed] [Google Scholar]
- 100.Bergman P, Johansson L, Wan H, Jones A, Gallo RL, Gudmundsson GH, Hökfelt T, Jonsson A-B, Agerberth B. Infect Immun. 2006;74:6982. doi: 10.1128/IAI.01043-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ray S, Dhaked HP, Panda D. Biochemistry. 2014;53:6426. doi: 10.1021/bi501115p. [DOI] [PubMed] [Google Scholar]
- 102.Hettinger TP, Craig LC. Biochemistry. 1970;9:1224. doi: 10.1021/bi00807a025. [DOI] [PubMed] [Google Scholar]
- 103.Shimotohno KW, Kawamura F, Natori Y, Nanamiya H, Magae J, Ogata H, Endo T, Suzuki T, Yamaki H. Biol Pharm Bull. 2010;33:568. doi: 10.1248/bpb.33.568. [DOI] [PubMed] [Google Scholar]
- 104.Kurylo-Borowska Z. Springer Berlin Heidelberg; Berlin, Heidelberg: 1967. p. 342. [Google Scholar]
- 105.Kurylo-Borowska Z, Szer W. Biochim Biophys Acta. 1972;287:236. doi: 10.1016/0005-2787(72)90373-5. [DOI] [PubMed] [Google Scholar]
- 106.Kurylo-Borowska Z, Szer W. Biochim Biophys Acta. 1972;259:357. doi: 10.1016/0005-2787(72)90310-3. [DOI] [PubMed] [Google Scholar]
- 107.Nielsen PE, Egholm M. Curr Issues Mol Biol. 1999;1:89. [PubMed] [Google Scholar]
- 108.Paulasova P, Pellestor F. Ann Genet. 2004;47:349. doi: 10.1016/j.anngen.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 109.Liang S, He Y, Xia Y, Wang H, Wang L, Gao R, Zhang M. Int J Infect Dis. 2014;30C:1. doi: 10.1016/j.ijid.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 110.Gruegelsiepe H, Brandt O, Hartmann RK. J Biol Chem. 2006;281:30613. doi: 10.1074/jbc.M603346200. [DOI] [PubMed] [Google Scholar]
- 111.Meng J, Da F, Ma X, Wang N, Wang Y, Zhang H, Li M, Zhou Y, Xue X, Hou Z, Jia M, Luo X. Antimicrob Agents Chemother. 2014;59:914. doi: 10.1128/AAC.03781-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baell JB. J Nat Prod. 2016;79:616. doi: 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- 113.Ingólfsson HI, Thakur P, Herold KF, Hobart EA, Ramsey NB, Periole X, de Jong DH, Zwama M, Yilmaz D, Hall K, Maretzky T, Hemmings HC, Blobel C, Marrink SJ, Koçer A, Sack JT, Andersen OS. ACS Chem Biol. 2014;9:1788. doi: 10.1021/cb500086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Baell JB, Holloway GA. J Med Chem. 2010;53:2719. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]