

Abstract
Rheumatoid arthritis (RA) patients with early elevations of antibodies against collagen type II (CII) have a distinct acute onset phenotype, associated with cytokine induction by surface‐bound anti‐CII‐containing immune complexes (ICs) and high C‐reactive protein (CRP) and erythrocyte sedimentation rate (ESR). Polymorphonuclear granulocytes (PMNs) and peripheral blood mononuclear cells (PBMCs) are abundant in the vicinity of CII in RA joints, and both PMN and PBMC reactivity against anti‐CII IC individually relate to early joint destruction and early elevation of CRP and ESR in RA. We searched for CII‐dependent mechanisms that might attract PMNs and PBMCs to RA joints. Human PBMCs and PMNs were stimulated with anti‐CII ICs and control ICs, either individually or in cocultures. Cocultured PMNs and PBMCs stimulated with anti‐CII ICs synergistically augmented production of the chemokines CXCL8, RANTES and MCP‐1, whereas downregulation was seen with control IC. This upregulation was unique to chemokines, as TNF‐α, IL‐1β, and GM‐CSF were downregulated in anti‐CII IC‐stimulated cocultures. The coculture‐associated chemokine upregulation depended on endogenous TLR4 ligand(s) and functionally active PMN enzymes, and was partially mediated by GM‐CSF. As anti‐CII levels peak around the time of RA diagnosis, this mechanism can attract inflammatory cells to joints in early RA and intensify the anti‐CII‐associated acute onset RA phenotype.
Keywords: Anticollagen antibodies ⋅ Chemokine ⋅ Collagen type II ⋅ Immune complexes⋅ Polymorphonuclear granulocytes ⋅ Rheumatoid arthritis ⋅ TLR4
Introduction
Rheumatoid arthritis (RA) is a criterion‐based disease, clinically subgrouped according to the occurrence of autoantibodies. The most common RA phenotype is characterized by presence of antibodies against citrullinated proteins (ACPAs) 1. RA patients with ACPA have long‐term bad prognosis with radiological erosions 2, 3. Type II collagen (CII) make up the major protein content in joint cartilage, and between 3 and 27% of RA patients have elevated levels of anti‐CII 4, 5. Anti‐CIIs are produced locally in synovial tissue and synovial fluid, but not in the circulation 6, 7. Serum levels of anti‐CII are highest around the time of RA diagnosis, and thereafter decline 8, 9, 10. We have described a distinct anti‐CII‐associated RA phenotype with early elevation of acute phase reactants and early joint erosions, in contrast to the ACPA‐positive phenotype associated with late occurrence of more inflammation and erosions in the same RA cohort 10, 11. As CII is a joint‐specific autoantigen, conditions are met for local production of anti‐CII ICs, and we have previously proposed a mechanism driving this early RA phenotype, whereby anti‐CII ICs stimulate mononuclear cells to produce proinflammatory cytokines from monocytes/macrophages via FcγRIIa 12.
In RA synovium there is a significant increase in leucocyte trafficking compared with the normal synovium 13. Polymorphonuclear granulocytes (PMNs) are important in acute inflammation and are abundant in inflamed synovial fluid 14, 15. PMNs are also found in the pannus tissue and thus in the vicinity of CII and locally formed anti‐CII IC 16. We recently showed that PMN reactivity against anti‐CII IC is more closely associated with joint erosions than are peripheral blood mononuclear cell (PBMC) cytokine responses 17. CXCL8 is a major factor in the recruitment of neutrophils to the joint. CXCL8 levels are elevated in the joints of RA patients, and we have previously shown that CXCL8 is produced by anti‐CII IC‐stimulated PBMC 18, 19. Other chemokines such as CCL5 (RANTES), CCL2 (MCP‐1), CCL3 (MIP‐1α), and CXCL2 (GRO‐α) attract monocytes, memory T cells, and dendritic cells (DCs) 20. However, the mechanism behind the augmentation of chemokine levels in RA joints, responsible for leucocyte recruitment is not well known. In this paper, we describe a mechanism whereby joint‐specific anti‐CII ICs stimulate PMNs and PBMCs in synergy to increase chemokine production, thereby attracting inflammatory cells to the joints of patients with early RA.
Results
Selective enhancement of chemokines in anti‐CII IC‐stimulated cocultures
Anti‐CII IC stimulated PBMC+ PMN cocultures showed enhanced CXCL8 and diminished TNF‐α levels compared with anti‐CII IC stimulated PBMC cultures. This effect was specific for anti‐CII IC, as CXCL8 production was downregulated in cocultures stimulated with two control surface‐bound ICs, plate‐bound IgG and tetanus toxoid (TT) IC, compared with the corresponding PBMC cultures (Fig. 1A–D). Plate‐bound IgG followed by aftercoating with increasing concentrations of CII universally yielded downregulation of both CXCL8 and TNF‐α for all low CII concentrations, and a universal blocking of IgG‐mediated effects at higher CII concentrations (data not shown). TNF‐α downregulation was uniformly observed in cocultures irrespective of how cells were stimulated. Cell cultures with PMNs only generally produced insignificant amounts of cytokines.
Figure 1.
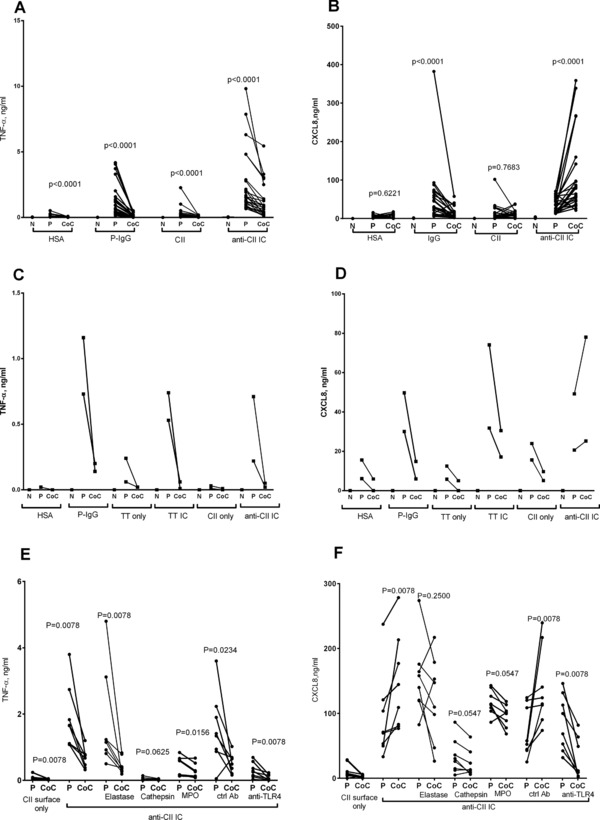
Selective upregulation of CXCL8 by anti‐CII IC cocultures is suppressed by anti‐TLR4 and blocking of PMN enzymes. (A and B) PMN and PBMC were stimulated with plate‐bound IgG (P‐IgG), with HSA as control, or IC containing CII and anti‐CII antibodies (anti‐CII IC), with CII as control. PMN (N), PBMC (P), and PMN + PBMC cocultures (CoC) were investigated in parallel in 12 experiments with two donors each (total 24 donors). (A and B) Production of (A) TNF‐α and (B) CXCL8. TNF‐a (C) and CXCL8 (D) are shown for two more donors that were also stimulated with TT or anti‐TT IC. They were investigated in parallel to directly coated IgG and anti‐CII IC. The effect on (E) TNF‐α and (F) CXCL8 of TLR4 blockade (anti‐TLR4) with control antibody (ctrl ab) and the inhibition of elastase, cathepsins L and S (cathepsin), and MPO on PBMC (P) and PBMC + PMN cocultures (CoC) were investigated in four independent experiments with two donors each (total eight donors). Paired investigations were made with the Wilcoxon test.
Similar to CXCL8, RANTES and MCP‐1 were upregulated in anti‐CII IC‐stimulated cocultures, whereas GM‐CSF and IL‐1β followed the TNF‐α pattern with downregulation in cocultures. MIP1‐α and GRO‐α partly followed the other chemokines, as median levels were similar or higher in anti‐CII IC‐stimulated cocultures as in parallel PBMC cultures. All cytokines and chemokines were downregulated in cocultures stimulated with plate‐bound IgG (Fig. 2B and D). Levels of IL‐10, IP‐10, and fractalkine were generally below the measurement range in all cell cultures (data not shown).
Figure 2.
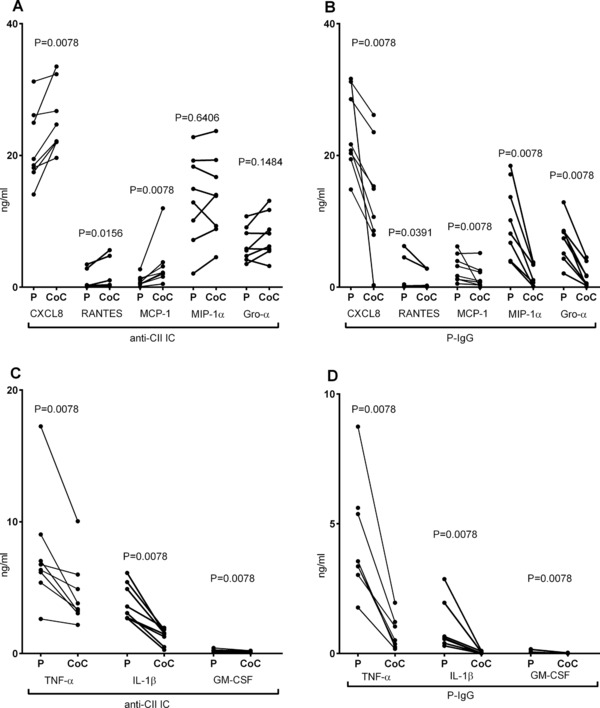
Selective upregulation of chemokines in anti‐CII IC‐stimulated cocultures. (A and B) Production of chemokines CXCL8, RANTES, MCP‐1, MIP‐1α, and GRO‐α and (C and D) cytokines TNF‐α, IL‐1β, and GM‐CSF after stimulation of PBMC (P) and PBMC + PMN cocultures (CoC) with (A and C) IC containing CII and anti‐CII (anti‐CII IC) and (B and D) plate bound IgG (P‐IgG). Data are pooled from four independent experiments with two donors each (total eight donors), but with ALBIA measurements performed in parallel. Paired investigations were made with the Wilcoxon test.
Anti‐CII IC‐induced CXCL8 in cocultures depend on TLR4 and functionally active PMN enzymes
After blocking TLR4, the augmentation of CXCL8 in anti‐CII IC‐stimulated cocultures was reversed for a downregulation as compared with PBMC cultures, whereas a control antibody had no effect (Fig. 1E and F). Both PBMC and coculture responses against anti‐CII ICs depended on TLR4, as anti‐TLR4 downregulated TNF‐α in both PBMC and PBMC + PMN cocultures, whereas only cocultures showed diminished CXCL8 levels after TLR4 blockade (Fig. 3A and B). When PBMCs and PMNs were individually treated with anti‐TLR4 before being merged in cocultures, TLR4 blockade was effective when treating PBMCs, but had no effect if only PMNs were treated (Fig. 4). TLR 4 blockade was only effective in anti‐CII IC‐stimulated cultures; no effect was noted in any cell cultures stimulated with surface‐bound IgG (Fig. 3C and D).
Figure 3.
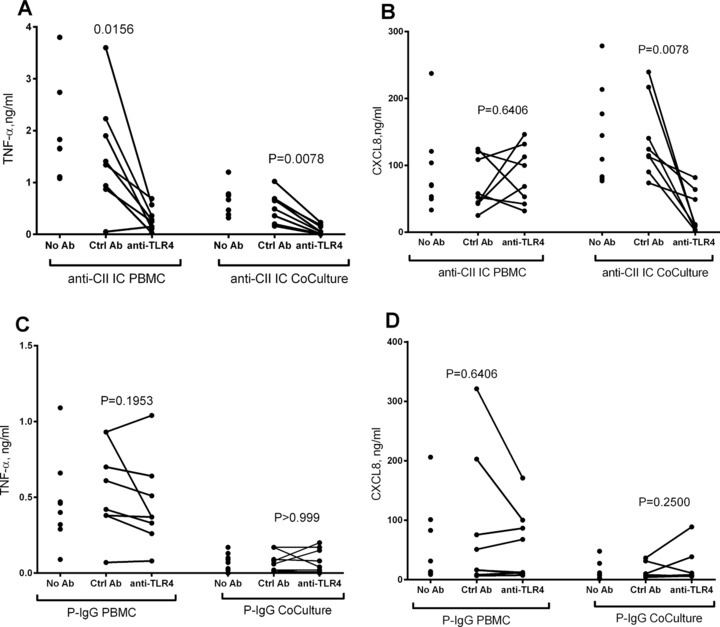
Both PBMC and PBMC/PMN cocultures respond to anti‐CII IC but not to control IC in a TLR4‐dependent manner. (A and B) Effect of TLR4 blockade on the production of (A) TNF‐α and (B) CXCL8 from PBMC and PBMC + PMN cocultures (CoCulture) stimulated with anti‐CII IC. (C and D) Effect of TLR4 blockade on the production of (C) TNF‐α and (D) CXCL8 from PBMC and PBMC + PMN cocultures (CoCulture) stimulated with plate bound IgG (P‐IgG). Significance levels are shown between cell cultures treated with anti‐TLR4 antibodies or with control rabbit IgG (ctrl Ab). Data are from the eight donors also depicted in Figure 1C and D. Data were obtained in four experiments with two donors each (total eight donors). Paired investigations were made with the Wilcoxon test.
Figure 4.
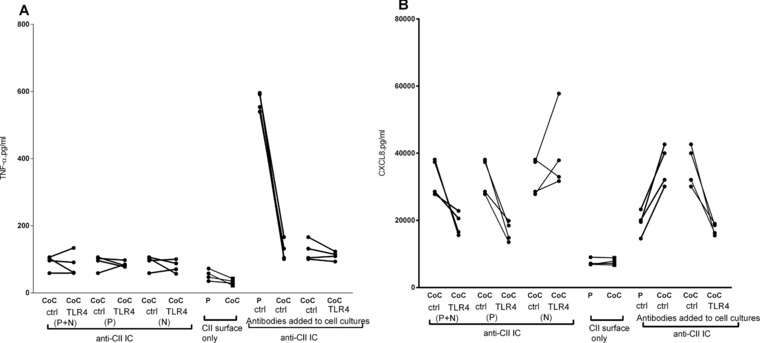
Upregulation of CXCL8 in anti‐CII IC‐stimulated cocultures is suppressed by selectively blocking anti‐TLR4 on PBMC (A and B) Effect on the production of (A) TNF‐a and (B) CXCL8 of selectively blocking TLR in the PBMC and PMN coculture compartments. PBMC and PMN were individually treated with anti‐TLR antibodies (TLR4) or control antibodies (ctrl) and thereafter washed before being merged into cocultures (CoC). In the first comparisons from the left both PBMC and PMN were treated with anti‐TLR4 (P + N), in the second only PBMC (P) and in the third only PMN (N). The following comparisons are controls, showing that the individual treatment/washing steps did not change the overall effect of anti‐TLR4 blockade in cocultures. Comparison was done also with PBMC cultures (P), to show that the experimental conditions yielded the awaited upregulation in anti‐CII IC‐stimulated cocultures. Data were obtained in two experiments with two donors each (total four donors).
Inhibitors targeting the PMN enzymes elastase, myeloperoxidase (MPO) and cathepsins L and S abolished the CXCL8 upregulation in cocultures, and led to lower median CXCL8 levels in cocultures than in PBMC cultures, but without the total reversal noted after addition of anti‐TLR4 (Fig. 1F). PMN enzyme inhibitors did not change the downregulation of TNF in anti‐CII IC‐stimulated cocultures (Fig. 1E). Enzyme inhibitors targeting MPO and especially cathepsins had a certain general suppressive effect on cytokine responses (Fig. 1E and F).
Anti‐CII IC‐induced CXCL8 upregulation is not mediated via LPS contamination
As we suspected that the TLR4‐mediated enhancement of chemokine levels in anti‐CII IC‐stimulated cocultures might be due to endotoxin contamination, we evaluated the endotoxin level in the commercially available CII preparation used. The undiluted commercial CII preparation used to prepare anti‐CII IC contained very low levels of endotoxin, 0.42 EU/mL, slightly above the level accepted in fluids uses to prepare hemofiltration fluids (0.25 EU/mL, data not shown). Anti‐CII IC‐stimulated cocultures with submaximal lipopolycaccharide (LPS) concentrations (100 pg/mL) did not show higher CXCL8 levels than PBMC cultures, neither if cultured on human serum albumin (HSA) or on CII surfaces without anti‐CII (i.e. without anti‐CII IC; Fig. 5A). The same result was also seen with 1000× higher LPS concentration (100 ng/mL, data not shown). Addition of LPS did not change the universal downregulation of TNF‐α in cocultures, although TNF levels were generally increased (Fig. 5A). LPS‐stimulated isolated cultures with PMNs only produced negligible cytokine levels (data not shown).
Figure 5.
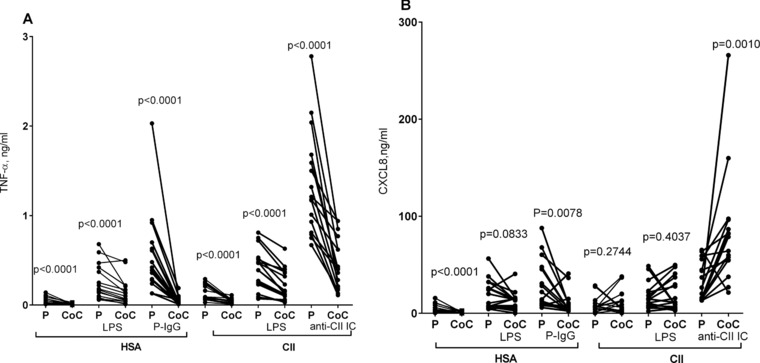
The anti‐CII IC‐induced upregulation of CXCL8 in PMN + PBMC cocultures can not be mimicked by LPS. (A and B) Production of (A) TNF‐α and (B) CXCL8 from PBMC (P) or PBMC + PMN cocultures (CoC). The effects of LPS on surfaces of HSA or collagen type II (CII) are compared with stimulation by plate‐bound IgG (P‐IgG) or anti‐CII IC. Data are pooled from four experiments with four donors each (total 16 donors). Paired investigations were made with the Wilcoxon test.
GM‐CSF partially mediate the anti‐CII IC‐induced CXCL8 enhancement in cocultures
GM‐CSF‐stimulated cocultures incubated on HSA surfaces or on CII surfaces (i.e. without anti‐CII) yielded increased CXCL8 levels compared with parallel PBMC cultures, whereas TNF‐α levels were generally increased but with maintained downregulation in cocultures (Fig. 6A and B). GM‐CSF‐stimulated PMNs alone produced negligible cytokine levels (data not shown). As we hypothesized that GM‐CSF might mediate the enhancement of CXCL8 production in anti‐CII‐IC‐stimulated cocultures, we performed neutralization experiments. A neutralizing antibody against GM‐CSF lowered the CXCL8 enhancement in anti‐CII IC‐stimulated cocultures, but did not totally abolish the increase in CXCL8 production (Fig. 6D), and had minimal effect on anti‐CII IC‐stimulated TNF‐α responses (Fig. 6C). When the same data were expressed as fold change in cocultures as compared with PBMC cultures, the effect of GM‐CSF neutralization was significant for CXCL8 but not for TNF‐α (Fig. 6E and F).
Figure 6.
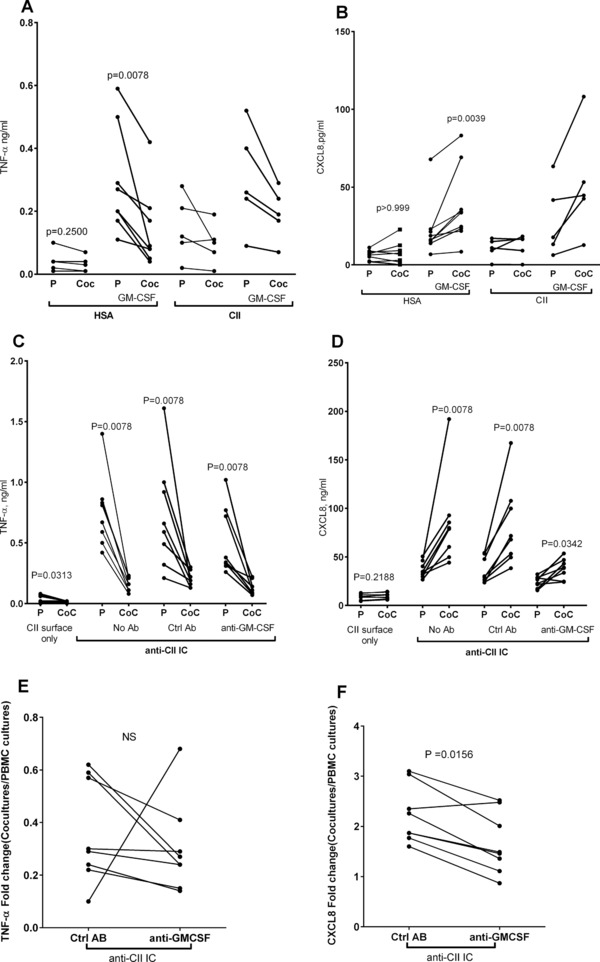
GM‐CSF mimics anti‐CII IC‐induced CXCL8 enhancement in cocultures. (A and B) When GM‐CSF was added to cell cultures on HSA and CII surfaces without IC (A) TNF‐a production was downregulated and (B) CXCL‐8 production was upregulated in PMN/PBMC cocultures (CoC) as compared with PBMC cultures (P). (C and D) Neutralization of GM‐CSF (anti‐GM‐CSF) as compared with control antibody (ctrl Ab) diminished the (D) CXCL8 enhancement in anti‐CII IC‐stimulated cocultures (CoC) as compared with PBMC cultures (P), with no effect on (C) TNF‐α. (E and F) When the same data were expressed as fold change between cocultures and PBMC cultures, GM‐CSF neutralization significantly diminished production of (F) CXCL8 but not of (E) TNF‐α. Data are pooled from four independent experiments with two donors each (total eight donors), except for the addition of GM‐CSF on CII surface where five donors were investigated in two experiments. Paired investigations were made with the Wilcoxon test. The fold change was calculated as the ratio between cytokine production in cocultures and in PBMC cultures, and this ratio was compared between cultures with anti‐GM‐CSF and with control antibody.
CXCL8 upregulation in cocultures is dependent on anti‐CII IC density
When anti‐CII‐positive sera from 13 RA patients were investigated in parallel, 8 of 13 sera induced CXCL8 enhancement in anti‐CII IC‐stimulated cocultures as compared with PBMC cultures (data not shown). We hypothesized that the CXCL8 enhancement in cocultures might depend on anti‐CII density in IC and performed titration experiments. At low anti‐CII densities, cocultures showed lower CXCL8 production than in corresponding PBMC cultures, but at anti‐CII densities corresponding to a coating concentration of 1 μg/mL or higher, the situation was reversed (Fig. 7B). For anti‐TT ICs and directly coated IgG, coculture CXCL8 levels were always lower than PBMC levels, even at antibody densities higher than those causing CXCL8 enhancement in anti‐CII IC‐stimulated cocultures (Fig. 7D and F). TNF‐α production was always higher in PBMC cultures than in cocultures for all antibody concentrations in all IC systems (Fig. 7A, C, and E).
Figure 7.
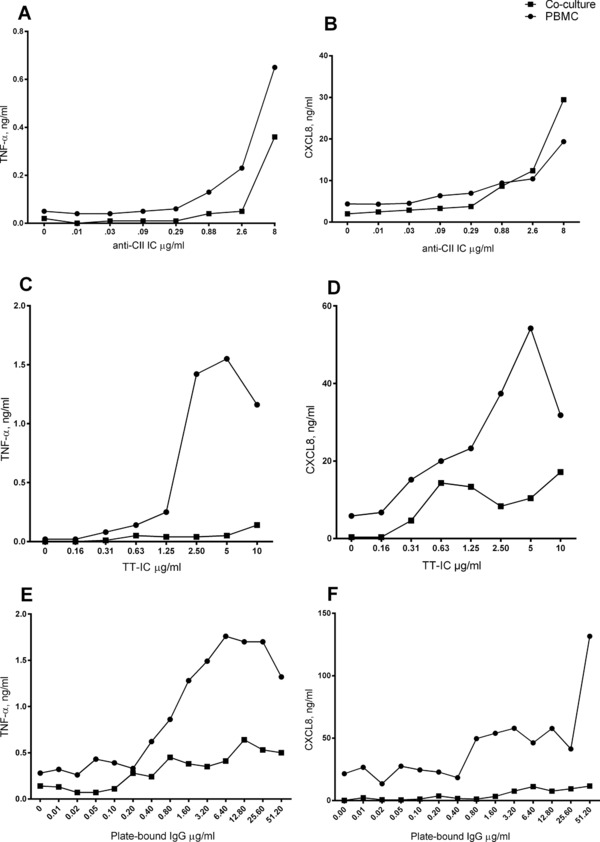
Upregulation of CXCL8 in anti‐CII IC‐stimulated cocultures depends on high anti‐CII antibody density and is not seen for other IC. Effect of antibody density on the difference between PBMC and PBMC + PMN cocultures (Co‐culture) in production of TNF‐α and CXCL8. (A and B) Stimulation was done with anti‐CII containing IC (anti‐CII IC) with increasing antibody concentrations and responses were evaluated as production of (A) TNF‐a and (B) CXCL8. (C and D) Stimulation was performed with surface‐bound IC made with tetanus toxoid and anti‐TT antibodies (TT‐IC) and evaluated as production of (C) TNF‐a and (D) CXCL8. (E and F) Stimulation was performed with directly coated IgG (plate‐bound IgG) and evaluated as production of (E) TNF‐a and (F) CXCL8. Antibody density in all ICs had been normalized against a standard curve consisting of plate‐bound IgG as described in 17. Eight micrograms per milliliter of anti‐CII represents the highest anti‐CII concentration available. The results are from one of two experiments with similar results.
Discussion
Surface‐bound ICs containing the joint‐specific autoantigen CII together with anti‐CII antibodies specifically upregulated the levels of the chemokines CXCL8, CCL5, and CCL2 in PBMC + PMN cocultures as compared with PBMC cultures, whereas the cytokines TNF‐α, IL‐1β, and GM‐CSF were downregulated. The effect depended on TLR4 ligation and functionally active granulocyte enzymes, and was partly mediated via GM‐CSF. The coculture‐dependent and TLR4‐dependent upregulation of chemokines was only found for anti‐CII ICs, and was absent using other surface‐bound ICs. When PBMC and PMN were individually treated with anti‐TLR4 before being merged in cocultures, only PBMC treatment was effective in downregulating CXCL8 production.
CII is an autoantigen with very narrow tissue distribution. Except for hyaline cartilage in joints, it is found in the ear, larynx, trachea, and vitreous of the eye 21. Anti‐CII are produced by synovial tissue and synovial fluid B lymphocytes from RA patients, but not in parallel blood samples 6, 7. Contrary to ACPA and rheumatoid factor serum levels that gradually increase during the years before RA onset 22, 23, anti‐CII levels are reported not to be elevated during the years before RA diagnosis 24. Such pre‐RA studies are however difficult to perform for infrequent RA‐associated autoantibodies such as anti‐CII, and the study by Möttönen et al investigated only 22 preillness sera, which probably is too low given the rarity of anti‐CII in early RA; in our previous study we found 8.8% positive, with 3.3% belonging to a very high outlier group 10. Anti‐CII levels peak around the time of RA diagnosis and thereafter decline 8, 9, 10, whereas ACPA levels change less after diagnosis. Thus both the specific autoantigen and autoantibodies are produced and present locally in the joints facilitating local IC formation, probably especially around the time of RA onset. Mononuclear cells and PMNs accumulate both in the synovial fluid and tissue, in close proximity to hyaline cartilage where anti‐CII ICs might form 14, 15, 16. Our findings therefore have a certain bearing on early RA patients where anti‐CII IC‐induced enhancement of CXCL8, RANTES, and CCL2 may attract both PMN, monocytes, memory T cells, and DCs and thereby amplify early joint inflammation 20. In Figure 8, we have summarized our view on how anti‐CII and ACPAs change during preillness years and during the first years after RA diagnosis based on own publications 3, 10, 22 and the paper by Möttönen et al 24. The figure also summarizes how we envisage that anti‐CII ICs stimulate PBMC and PMN to produce chemokines and thereby attract a wide array immunological cell types by stimulating the production of many different chemokines, especially during the time of RA onset when anti‐CII levels peak.
Figure 8.
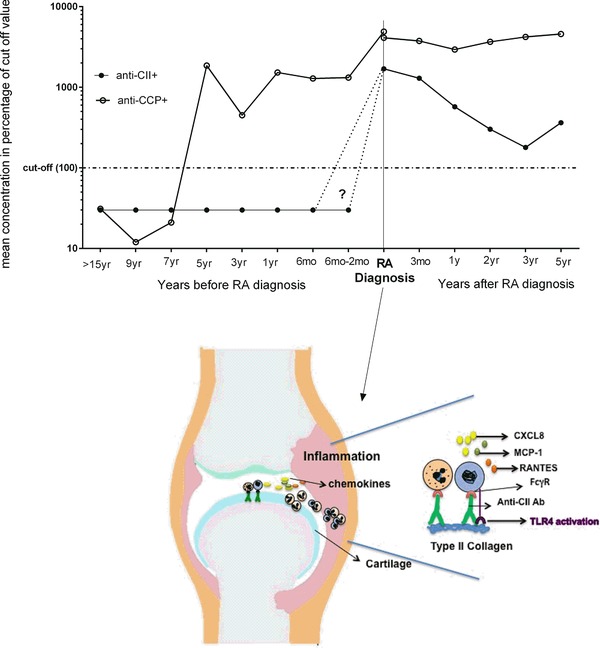
Proposed model of how PBMC and PMN interact to produce chemokines around the time of RA onset. Schematic figure showing how levels of anti‐CII change over time and the proposed model of how PBMC and PMN interact to produce CXCL8, RANTES, and MCP‐1 when stimulated with anti‐CII containing IC around the time of RA onset. Anti‐CII levels before RA diagnosis are based on 24, and after RA diagnosis on the mean of all anti‐CII‐positive individuals in 10. As comparison ACPA (anti‐CCP) levels before RA diagnosis are based on 22 and after RA diagnosis show the mean of all ACPA‐positive individuals in 3, with the omission of one patient with extremely high and undulating ACPA values. The question mark and dotted lines denotes that the anti‐CII preillness study 24 was based on very few individuals (n = 22) and data on anti‐CII before RA diagnosis therefore are uncertain.
RANTES and CCL2 operate in concert in RA joints and enhance the production of CXCL8 25. RANTES and CCL2 also activate osteoclast differentiation and can enhance bone resorbtion 26. As we find both RANTES and CCL2 to be increased in anti‐CII IC‐stimulated cocultures, this might explain the association between elevated anti‐CII levels in early RA and anti‐CII IC‐stimulated PMN activation on one hand and joint erosions in anti‐CII‐positive early RA patients on the other, as we have shown previously 11, 17.
The CXCL8 enhancement in anti‐CII IC‐stimulated cocultures strictly depended on TLR4 ligation. Limulus tests showed that the CII preparation contained very low endotoxin levels, and addition of LPS could not induce CXCL8 enhancement in IC‐free systems, neither on HSA nor on CII surfaces. Thus, contaminating endotoxin is an unlikely TLR4 ligand in this case, implicating an endogenous TLR4 ligand in CII or in anti‐CII IC.
Prerequisites for TLR4 ligation in conjunction to IC exist in RA joints as TLR4 is upregulated in RA synovial tissue, cross‐talk between TLR4 and FcγRIII has been described both in macrophages and PMN, and TLR4 ligation increases FcγRIIa expression in PMN 27, 28, 29. Animal studies confirm the importance of TLR4 in IC‐induced arthritis 30.
The fact that only anti‐CII ICs but not two other surface‐bound control ICs mediated increase in CXCL8 levels in cocultures made us ask whether the combination of non‐CII‐specific surface bound antibodies in conjunction to surface‐bound CII would create similar results. This was not the case: the response against surface‐bound IgG was gradually blocked by increasing concentrations of CII added, with complete blocking of IgG‐mediated stimulation at the highest CII concentrations, but always with higher CXCL8 responses in PBMC cultures than in cocultures. Therefore, we cannot rule out the possibility that specific properties of anti‐CII in RA sera contribute to the enhancement of chemokine levels in anti‐CII IC‐stimulated cocultures. Such factors might, for example, be differences in glycosylation patterns, IgG subclass distribution, or even anti‐CII of other isotypes than IgG in the used serum samples, as the control used contained essentially only IgG. Physical characterization including, for example, surface plasmon resonance or quartz crystal microbalance with dissipation monitoring might also shed light on unique properties of anti‐CII IC as compared with other surface‐bound IC, but have not yet been performed. Due to shortage of high level anti‐CII sera, we have not been able to perform analyses with affinity‐purified anti‐CII IgG.
The universal TNF‐α downregulation in cocultures is probably due to degradation of monocyte‐derived TNF‐α by PMN enzymes, including elastase and cathepsin G, as previously shown 31, 32, 33. These studies solely focused on TNF degradation, and we are not aware of any literature investigating whether there are similar effects on CXCL8 or on other chemokines. It is anyhow intriguing that CXCL8 enhancement in anti‐CII IC‐stimulated cocultures is abrogated by PMN enzyme inhibition. Our data are however in agreement with a previous paper reporting that neutrophil elastase upregulated CXCL8 via TLR4, and that elastase‐induced CXCL8 production could be blocked with anti‐TLR4 antibodies 34. N‐terminal processing of chemokines by, for example, elastase and cathepsin G is also known to increase chemokine receptor affinity 35. We hypothesize that similar functions might be found also for cathepsins S and L and for MPO, where enzyme inhibition abrogated and almost reversed the anti‐CII IC‐induced CXCL8 enhancement in cocultures. It is however important to note that in our system, the chemokine upregulation was found only in PMN + PBMC cocultures; we never found any significant cytokine or chemokine production in any isolated PMN cultures.
Addition of GM‐CSF to cocultures without anti‐CII IC mimicked the CXCL8‐inducing effect of anti‐CII ICs, and neutralization of GM‐CSF in the anti‐CII IC system lead to a significantly decreased fold induction of CXCL8 in cocultures compared with PBMC cultures. We hypothesize that CXCL8 enhancement in cocultures is at least partly mediated via GM‐CSF, and interpret the lower GM‐CSF levels cocultures (Fig. 2D) as GM‐CSF consumption by PMNs.
Sokolove et al. showed that ACPA‐containing IC stimulated macrophage TNF‐α production via dual engagement of FcγR and TLR4 36. In our system, TLR4 blockade downregulated TNF‐α production in both PBMC and cocultures, whereas CXCL8 levels diminished in cocultures but not in PBMC cultures (Fig. 3A and B). Thus TLR4 blockade had different effects on TNF‐α and CXCL8 production induced by anti‐CII IC. TLR4 blockade had no effect on cytokine production induced by irrelevant ICs, again showing the uniqueness of anti‐CII ICs. Due to short survival of PMNs, we have so far not been able to perform cytokine staining of cells in anti‐CII IC‐stimulated cocultures. Given the universally low production of cytokines in our PMN cultures, our hypothesis is that monocytes are the main cytokine producers in our anti‐CII IC‐stimulated cocultures 19. Our findings that only TLR4 blockade of PBMCs but not of PMNs suppressed the enhancement of CLXL8 production in cocultures (Fig. 4) is also an indirect argument that the major chemokine producing cells reside in PBMCs, and we have previously shown that monocytes are the main CXCL8 producers in anti‐CII IC‐stimulated PBMCs 19.
ACPA‐positive RA and anti‐CII‐positive RA represent two clinically distinct and partly reverse disease phenotypes. In three studies performed in the same RA cohort we have shown ACPA to be associated with increased signs of inflammation and radiological destruction rate after 1–5 years but not at RA onset. Anti‐CII‐positive patients on the other hand have increased laboratory signs of inflammation and more radiological destruction at the time of diagnosis, but not later. Anti‐CII‐positive patients also have shorter symptom duration at diagnosis, probably due to more intense disease onset 3, 10, 11. After RA diagnosis, serum anti‐CII levels and anti‐CII IC‐induced cytokine production decline in parallel to lowering of inflammatory markers, whereas ACPA levels remain rather stable or drop slightly over time in contrast to the parallel clinical worsening in ACPA‐positive patients examined in the same RA cohort 3, 10. Our data thereby tie temporal changes in anti‐CII autoantibody function to clinical RA phenotype in a unique way not seen for ACPA 10, 11, 17, 19. We believe that the collagen antibody induced arthritis (CAIA) rodent model where inflammation is detected already 5 days after anti‐CII injection, and that is aggravated by LPS, might be a counterpart to the acute onset pathology that we have described in anti‐CII‐positive RA patients 10, 37.
Although we do not know the exact TLR4 ligand in CII, it is most probably not a citrullinated epitope, as we have shown ACPA and anti‐CII to be inversely related, with many highly ACPA‐positive patients being anti‐CII negative 10.
In our in vitro system, the anti‐CII IC‐dependent enhancement of CXCL8 in cocultures depends on high anti‐CII density. Anti‐CII are produced by joint B cells as detected by ELISPOT on cells from synovial tissue and synovial fluid, whereas parallel investigations showed no anti‐CII production from peripheral B cells or nondetectable anti‐CII serum levels 6, 7. Therefore, we hypothesize that locally produced anti‐CII to a large extent are directly bound to CII epitopes exposed in joint cartilage, and that anti‐CII levels in serum represent the excess spilling over to the circulation. Therefore, high density of anti‐CII in IC might be acquired locally in joints without elevated serum anti‐CII levels, implying that our findings can be of clinical significance in far more arthritic individuals than in the small group of RA patients with elevated serum levels of anti‐CII 11. Our current data do however implicate anti‐CII in the early onset pathology only in a small subset of RA patients, and not to be involved in RA pathogenesis in general.
In a previous study, we investigated TNF production induced by polyethylene glycol‐precipitated ICs from RA synovial fluids 12. There was a nonsignificant (p = 0.06) trend for higher TNF production by PEG‐precipitated serum ICs from RA patients compared with controls, but significantly higher induction by ICs from RA synovial fluids, where TNF induction correlated to rheumatoid factor (but interestingly enough not to ACPA) levels, and to number of swollen and tender joints. Before establishing the currently used approach with surface‐bound CII‐containing ICs, we tried to make soluble CII‐containing ICs, but failed. However, we plan to investigate content of CII and anti‐CII in soluble ICs purified from RA synovial fluids. It might also be that many central findings in our surface‐bound IC systems in our present paper are not reproducible using soluble ICs, as they might depend on frustrated phagocytosis induced by the nonphagocytable ICs in our system.
We conclude that anti‐CII ICs induce a specific enhancement of levels of several chemokines in PBMC + PMN cocultures, via a mechanism dependent on TLR4, functionally active PMN enzymes, and GM‐CSF. As anti‐CII ICs are formed locally in the joints, this represents a mechanism that can attract a wide array of inflammatory cells to the inflamed joints and intensify the acute onset RA phenotype associated with anti‐CII. Targeting TLR4, PMN function and GM‐CSF might be used as means to suppress joint inflammation early in arthritis.
Materials and methods
Patients and cell donors
Sera from 13 anti‐CII‐positive RA sera from Karolinska University Hospital were used 11, 19. Most anti‐CII IC stimulations were done with an RA serum containing 8 μg/mL of anti‐CII as previously determined 17. Heparinized blood from healthy laboratory personnel and blood donors at Uppsala University hospital was used as responder cells. All subjects had given informed consent and the studies have been approved by the regional ethical boards in Uppsala and Stockholm.
Cell purification and IC stimulation
PBMC and PMN were immediately isolated using Ficoll (GE Healthcare, Uppsala, Sweden) as described previously 17. Purity of PBMCs and PMNs was checked by Türk's solution and was always >95%. Not more than 2% PMN were found in PBMC cultures, and not more than 2% PBMC were found in PMN cultures, remaining impurities representing erythrocytes. Viability (tryphan blue or flow cytometry using propidium iodide (PI) with comparable results) was ≥92% and ≥95% for PMNs and PBMCs, respectively. Surface‐bound anti‐CII ICs were prepared as previously described 17. Briefly, 50 μL of human native collagen type II (ELISA grade; Chondrex, Redmond, WA, USA), 10 μg/mL in PBS, was coated on Maxisorb ELISA plates (Nunc, Roskilde, Denmark) and incubated at +4°C overnight. After blocking the plates with 100 μL of 1% HSA (Alburex CSL Behring, Stockholm, Sweden) in PBS, 50 μL of an RA serum containing 8 μg/mL of anti‐CII antibodies was added, and incubated for 2 h at room temperature. Wells coated with CII and blocked with HSA but without any anti‐CII antibodies were used as control wells in these experiments. Two CII‐unrelated surface‐bound ICs were used as comparators: human polyclonal IgG, 8 μg/mL (Privigen; CSL Behring) directly bound to Maxisorb ELISA plates followed by HSA blocking, and with only HSA‐blocked wells as controls; and TT, obtained from Statens Biologiska Laboratorium, Stockholm, Sweden 5 flocking units, approximating 10 μg/mL coated to identical ELISA plates, followed by addition of an anti‐TT hyperimmune serum (Tetagam, CSL Behring). In the latter setup, TT‐coated and HSA‐blocked plates without any serum added were used as control, as almost all Swedes are tetanus vaccinated. To compare the effect of antibody density in different surface‐bound ICs, serially diluted IgG, anti‐CII and anti‐TT was added to empty ELISA wells, or wells coated with fixed concentrations of CII and TT, respectively, according to above, after adjusting levels of the specific antibodies to standard curves with directly coated plate‐bound IgG of known concentration, as previously described 17. Individual PMN and PBMC cultures contained 0.5 × 106 cells/mL, whereas cocultures contained 0.5 × 106 cells each of PBMC and PMN/mL. All cell cultures were stimulated for 18 h at 37°C in a cell incubator before collection of supernatants for cytokine and chemokine measurements.
TLR4 blocking and PMN enzyme inhibition experiments
PMN enzyme inhibition was performed by incubating cells with inhibitors against cathepsin S and cathepsin L (cathepsin1 inhibitor 100 nM), MPO (500 nM), and neutrophil elastase (100 nM) for 15 min before adding to the IC plates. All enzyme inhibitory peptides were bought from Merck Chemicals and Life Sciences, Stockholm, Sweden.
For TLR4 blockade, 10 μg/mL of a polyclonal rabbit anti‐TLR4 antibody or control rabbit IgG (both from Invivogen, Toulouse, France) was added to PBMCs and cocultures for 15 min before adding to the IC plates. In the experiments shown in Fig. 4, PBMCs, PMNs, or both were individually incubated with anti‐TLR4 or control antibody, thereafter washed and subsequently added in cocultures. The washing step per se did not change the effect of TLR4 blockade (Fig. 4).
LPS and GM‐CSF stimulation
Sub‐maximal TNF responses by PBMC had been titrated between 100 ng/mL and 0 ng/mL of LPS (Sigma Aldrich, Stockholm, Sweden), and the concentration 100 pg/mL was chosen and added to purified PMNs, PBMCs, or cocultures before addition to HSA or CII surfaces. Some experiments were also performed with a conventional 1000× higher LPS concentration (100 ng/mL). A kinetic chromogen limulus amebocyte lysate test (Endochrome‐K, Charles River, Wilmington, MA) was performed to evaluate endotoxin contamination in CII.
GM‐CSF (R&D Biosciences, Abingdon, UK; 10 ng/mL) was added to PMNs, PBMCs, and cocultures. For GM‐CSF neutralization, 0.5 μg/mL of polyclonal goat anti‐GM‐CSF or control normal goat IgG (both from R&D) was added to cell cultures. For both GM‐CSF stimulation and GM‐CSF neutralization, reagents were added directly after introduction of cell cultures on IC plates, and incubated for 18 h before harvesting.
Cytokine and chemokine assays
Cytokine ELISAs were performed as previously described for TNF‐α 10, 12. All antibodies and cytokine standards were purchased from R&D Biosciences. For CXCL8, the capture antibody was mouse monoclonal antibody MAB208 (2.0 μg/mL), and the detection antibody was biotinylated polyclonal goat antibody BAF208 (0.1 μg/mL). CXCL8, CCL5 (RANTES), CCL2 (MCP‐1), CCL3 (MIP‐1α), CXCL2 (GRO‐α), fractalkine, IP‐10, IL‐10, GM‐CSF, ΤΝF‐α, and IL‐1β were measured in parallel in cell cultures from eight donors with a customized chemokine‐focused addressable laser bead immunoassay (ALBIA; Millipore, Stockholm, Sweden) and analyzed on a Bio‐Plex Magpix Reader (Bio‐Rad, Berkeley, CA, USA).
Statistical analyses
As anti‐CII are dichotomously distributed in RA, we used the nonparametric Wilcoxon test for paired statistics. In GM‐CSF neutralization experiments, the fold change was calculated as the ratio between cytokine levels in cocultures and in PBMC cultures, and this ratio was compared between cultures with anti‐GM‐CSF and with control antibody. p‐values <0.05 were considered significant.
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Abbreviations
- ACPAs
antibodies against citrullinated proteins
- CII
collagen type II
- HSA
human serum albumin
- ICs
immune complexes
- MPO
myeloperoxidase
- RA
rheumatoid arthritis
- TT
tetanus toxoid
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Granulocyte‐augmented chemokine production induced by type II collagen‐containing immune complexes is mediated via TLR4 in rheumatoid arthritis patients
Acknowledgments
Financial support was obtained from the Swedish Research Council, the Swedish Rheumatism Association, Agnes Mac and Rudberg Foundation, King Gustav 5th 80‐year foundation, and the Signe and Reinhold Sund's Foundation for Rheumatological Research.
We would like to thank Eva Tano, Department of Clinical Microbiology, Uppsala University Hospital, for performing the Limulus test; Susanne Lindblom, Department of Immunology genetics and Pathology, Uppsala University, for helping in performing ALBIA. Amir Elshafie, Department of Immunology Genetics and Pathology, Uppsala University, critically read and commented on the final manuscript.
References
- 1. Schellekens, G. A. , Visser, H. , de Jong, B. A. , van den Hoogen, F. H. , Hazes, J. M. , Breedveld, F. C. and van Venrooij, W. J. , The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000. 43: 155–163. [DOI] [PubMed] [Google Scholar]
- 2. Kroot, E. J. , de Jong, B. A. , van Leeuwen, M. A. , Swinkels, H. , van den Hoogen, F. H. , van't Hof, M. , van de Putte, L. B. et al., The prognostic value of anti‐cyclic citrullinated peptide antibody in patients with recent‐onset rheumatoid arthritis. Arthritis Rheum. 2000. 43: 1831–1835. [DOI] [PubMed] [Google Scholar]
- 3. Rönnelid, J. , Wick, M. C. , Lampa, J. , Lindblad, S. , Nordmark, B. , Klareskog, L. and van Vollenhoven, R. F. , Longitudinal analysis of citrullinated protein/peptide antibodies (anti‐CP) during 5 year follow up in early rheumatoid arthritis: anti‐CP status predicts worse disease activity and greater radiological progression. Ann. Rheum. Dis. 2005. 64: 1744–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenbury, C. L. and Skingle, J. , Anti‐cartilage antibody. J. Clin. Pathol. 1979. 32: 826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cook, A. D. , Rowley, M. J. , Mackay, I. R. , Gough, A. and Emery, P. , Antibodies to type II collagen in early rheumatoid arthritis. Correlation with disease progression. Arthritis Rheum. 1996. 39: 1720‐1727. [DOI] [PubMed] [Google Scholar]
- 6. Tarkowski, A. , Klareskog, L. , Carlsten, H. , Herberts, P. and Koopman, W. J. , Secretion of antibodies to types I and II collagen by synovial tissue cells in patients with rheumatoid arthritis. Arthritis Rheum. 1989. 32: 1087–1092. [DOI] [PubMed] [Google Scholar]
- 7. Rönnelid, J. , Lysholm, J. , Engström‐Laurent, A. , Klareskog, L. and Heyman, B. , Local anti‐type II collagen antibody production in rheumatoid arthritis synovial fluid. Evidence for an HLA‐DR4‐restricted IgG response. Arthritis Rheum. 1994. 37: 1023–1029. [DOI] [PubMed] [Google Scholar]
- 8. Cook, A. D. , Rowley, M. J. , Stockman, A. , Muirden, K. D. and Mackay, I. R. , Specificity of antibodies to type II collagen in early rheumatoid arthritis. J. Rheumatol. 1994. 21: 1186–1191. [PubMed] [Google Scholar]
- 9. Pereira, R. S. , Black, C. M. , Duance, V. C. , Jones, V. E. , Jacoby, R. K. and Welsh, K. I. , Disappearing collagen antibodies in rheumatoid arthritis. Lancet 1985. 2: 501–502. [DOI] [PubMed] [Google Scholar]
- 10. Mullazehi, M. , Mathsson, L. , Lampa, J. and Rönnelid, J. , High anti‐collagen type‐II antibody levels and induction of proinflammatory cytokines by anti‐collagen antibody‐containing immune complexes in vitro characterise a distinct rheumatoid arthritis phenotype associated with acute inflammation at the time of disease onset. Ann. Rheum. Dis. 2007. 66: 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mullazehi, M. , Wick, M. C. , Klareskog, L. , van Vollenhoven, R. and Rönnelid, J. , Anti‐type II collagen antibodies are associated with early radiographic destruction in rheumatoid arthritis. Arthritis Res. Ther. 2012. 14: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mathsson, L. , Lampa, J. , Mullazehi, M. and Rönnelid, J. , Immune complexes from rheumatoid arthritis synovial fluid induce FcgammaRIIa dependent and rheumatoid factor correlated production of tumour necrosis factor‐alpha by peripheral blood mononuclear cells. Arthritis Res. Ther. 2006. 8: R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bartok, B. and Firestein, G. S. , Fibroblast‐like synoviocytes: key effector cells in rheumatoid arthritis. Immunol. Rev. 2010. 233: 233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pillinger, M. H. and Abramson, S. B. , The neutrophil in rheumatoid arthritis. Rheum. Dis. Clin. North Am. 1995. 21: 691–714. [PubMed] [Google Scholar]
- 15. Edwards, S. W. and Hallett, M. B. , Seeing the wood for the trees: the forgotten role of neutrophils in rheumatoid arthritis. Immunol. Today 1997. 18: 320–324. [DOI] [PubMed] [Google Scholar]
- 16. Mohr, W. , Westerhellweg, H. and Wessinghage, D. , Polymorphonuclear granulocytes in rheumatic tissue destruction. III. an electron microscopic study of PMNs at the pannus‐cartilage junction in rheumatoid arthritis. Ann. Rheum. Dis. 1981. 40: 396–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manivel, V. A. , Sohrabian, A. , Wick, M. C. , Mullazehi, M. , Håkansson, L. D. and Rönnelid, J. , Anti‐type II collagen immune complex‐induced granulocyte reactivity is associated with joint erosions in RA patients with anti‐collagen antibodies. Arthritis Res. Ther. 2015. 17: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kraan, M. C. , Patel, D. D. , Haringman, J. J. , Smith, M. D. , Weedon, H. , Ahern, M. J. , Breedveld, F. C. et al., The development of clinical signs of rheumatoid synovial inflammation is associated with increased synthesis of the chemokine CXCL8 (interleukin‐8). Arthritis Res. 2001. 3: 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mullazehi, M. , Mathsson, L. , Lampa, J. and Rönnelid, J. , Surface‐bound anti‐type II collagen‐containing immune complexes induce production of tumor necrosis factor alpha, interleukin‐1beta, and interleukin‐8 from peripheral blood monocytes via Fc gamma receptor IIA: a potential pathophysiologic mechanism for humoral anti‐type II collagen immunity in arthritis. Arthritis Rheum. 2006. 54: 1759–1771. [DOI] [PubMed] [Google Scholar]
- 20. Charo, I. F. and Ransohoff, R. M. , The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006. 354: 610–621. [DOI] [PubMed] [Google Scholar]
- 21. Eyre, D. R. , The collagens of articular cartilage. Semin. Arthritis Rheum. 1991. 21: 2–11. [DOI] [PubMed] [Google Scholar]
- 22. Kokkonen, H. , Mullazehi, M. , Berglin, E. , Hallmans, G. , Wadell, G. , Rönnelid, J. and Rantapää‐Dahlqvist, S. , Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res. Ther. 2011. 13: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rantapää‐Dahlqvist, S. , de Jong, B. A. , Berglin, E. , Hallmans, G. , Wadell, G. , Stenlund, H. , Sundin, U. et al., Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003. 48: 2741–2749. [DOI] [PubMed] [Google Scholar]
- 24. Möttönen, T. , Hannonen, P. , Oka, M. , Rautiainen, J. , Jokinen, I. , Arvilommi, H. , Palosuo, T. et al., Antibodies against native type II collagen do not precede the clinical onset of rheumatoid arthritis. Arthritis Rheum. 1988. 31: 776–779. [DOI] [PubMed] [Google Scholar]
- 25. Koch, A. E. , Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum. 2005. 52: 710–721. [DOI] [PubMed] [Google Scholar]
- 26. Kim, M. S. , Day, C. J. and Morrison, N. A. , MCP‐1 is induced by receptor activator of nuclear factor‐{kappa}B ligand, promotes human osteoclast fusion, and rescues granulocyte macrophage colony‐stimulating factor suppression of osteoclast formation. J. Biol. Chem. 2005. 280: 16163–16169. [DOI] [PubMed] [Google Scholar]
- 27. Ospelt, C. , Brentano, F. , Rengel, Y. , Stanczyk, J. , Kolling, C. , Tak, P. P. , Gay, R. E. et al., Overexpression of toll‐like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll‐like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008. 58: 3684–3692. [DOI] [PubMed] [Google Scholar]
- 28. Rittirsch, D. , Flierl, M. A. , Day, D. E. , Nadeau, B. A. , Zetoune, F. S. , Sarma, J. V. , Werner, C. M. et al., Cross‐talk between TLR4 and FcgammaReceptorIII (CD16) pathways. PLoS Pathog. 2009. 5: e1000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krupa, A. , Fudala, R. , Florence, J. M. , Tucker, T. , Allen, T. C. , Standiford, T. J. , Luchowski, R. et al., Bruton's tyrosine kinase mediates FcgammaRIIa/Toll‐like receptor‐4 receptor crosstalk in human neutrophils. Am. J. Respir. Cell Mol Biol. 2013. 48: 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Lent, P. L. , Blom, A. B. , Grevers, L. , Sloetjes, A. and van den Berg, W. B. , Toll‐like receptor 4 induced FcgammaR expression potentiates early onset of joint inflammation and cartilage destruction during immune complex arthritis: Toll‐like receptor 4 largely regulates FcgammaR expression by interleukin 10. Ann. Rheum. Dis. 2007. 66: 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nortier, J. , Vandenabeele, P. , Noel, E. , Bosseloir, Y. , Goldman, M. and Deschodt‐Lanckman, M. , Enzymatic degradation of tumor necrosis factor by activated human neutrophils: role of elastase. Life Sci. 1991. 49: 1879–1886. [DOI] [PubMed] [Google Scholar]
- 32. Scuderi, P. , Nez, P. A. , Duerr, M. L. , Wong, B. J. and Valdez, C. M. , Cathepsin‐G and leukocyte elastase inactivate human tumor necrosis factor and lymphotoxin. Cell. Immunol. 1991. 135: 299–313. [DOI] [PubMed] [Google Scholar]
- 33. van Kessel, K. P. , van Strijp, J. A. and Verhoef, J. , Inactivation of recombinant human tumor necrosis factor‐alpha by proteolytic enzymes released from stimulated human neutrophils. J. Immunol. 1991. 147: 3862–3868. [PubMed] [Google Scholar]
- 34. Devaney, J. M. , Greene, C. M. , Taggart, C. C. , Carroll, T. P. , O'Neill, S. J. and McElvaney, N. G. , Neutrophil elastase up‐regulates interleukin‐8 via toll‐like receptor 4. FEBS Lett. 2003. 544: 129–132. [DOI] [PubMed] [Google Scholar]
- 35. Pham, C. T. , Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol. 2006. 6: 541–550. [DOI] [PubMed] [Google Scholar]
- 36. Sokolove, J. , Zhao, X. , Chandra, P. E. and Robinson, W. H. , Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll‐like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011. 63: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nandakumar, K. S. and Holmdahl, R. , Collagen antibody induced arthritis. Methods Mol. Med. 2007. 136: 215–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Granulocyte‐augmented chemokine production induced by type II collagen‐containing immune complexes is mediated via TLR4 in rheumatoid arthritis patients