

Summary
Background
ONO‐2952 is a novel and selective inhibitor of translocator protein 18 kDa that reduces stress‐induced defecation and visceral hyperalgesia in rat models.
Aim
To evaluate the efficacy and safety of ONO‐2952 in females with irritable bowel syndrome with diarrhoea in an exploratory proof‐of‐concept study.
Methods
A randomised, double‐blind, placebo‐controlled study was conducted at 49 US centres. Two hundred subjects with irritable bowel syndrome with diarrhoea (Rome III criteria) were randomised to ONO‐2952 20 mg, or 60 mg, or placebo. Subjects recorded irritable bowel syndrome symptoms daily during a 2‐week baseline period, the 4‐week treatment period and for 4 weeks post‐treatment. The co‐primary endpoints were change from baseline to week 4 in abdominal pain, stool consistency and stool frequency.
Results
Improvements in irritable bowel syndrome symptoms were seen with ONO‐2952 over placebo in per‐protocol analyses for all three co‐primary endpoints, but these did not reach statistical significance at the 5% level. The largest improvement was seen with ONO‐2952 60 mg. ONO‐2952 was well tolerated with a safety profile similar to that of placebo. Most adverse events were mild or moderate in severity and not treatment related.
Conclusion
ONO‐2952 showed evidence of clinical efficacy in reducing irritable bowel syndrome‐related symptoms in female subjects with irritable bowel syndrome with diarrhoea, and further evaluation is, therefore, warranted to assess its potential as a treatment for irritable bowel syndrome with diarrhoea (NCT01844180).
Introduction
Irritable bowel syndrome (IBS) is a common chronic functional gastrointestinal disorder that affects approximately 11% of the population worldwide.1 It is characterised by abdominal pain or discomfort associated with altered bowel habits.2, 3, 4 Three main IBS subtypes are recognised, determined by stool consistency pattern: IBS with diarrhoea (IBS‐D), IBS with constipation (IBS‐C) and IBS with mixed constipation and diarrhoea (IBS‐M).2 IBS significantly impacts on health‐related quality of life (QoL)5 and is a substantial socioeconomic burden due to the high healthcare costs, lost work days and reduced productivity6, 7 associated with the condition.
The pathophysiology of IBS remains unclear; however, the most important mechanisms include visceral sensitivity, abnormal gut motility and autonomic nervous system dysfunction.8 Genetics, the gut microbiome, immune activation, altered intestinal permeability and brain–gut interactions are also thought to play a role.9
The management of IBS is challenging due to the complex nature of the disease. Management options include dietary and lifestyle modifications and psychological and pharmacological therapies.10 There are currently only three approved pharmacological treatments for IBS‐D, which have been shown to improve both abdominal pain and diarrhoea.11 The 5‐HT3 antagonist alosetron was initially approved in 2000, but due to serious gastrointestinal adverse effects, its use is now limited to women with severe IBS‐D symptoms that are refractory to other treatments. Eluxadoline (a mixed μ‐opioid receptor agonist and δ‐opioid receptor antagonist) and rifaximin (a broad‐spectrum, non‐absorbable, gut‐specific antibiotic) were both approved in 2015.11, 12 Although these new treatments have expanded the treatment options available for IBS‐D, there remains a need for further effective and well‐tolerated therapies.11
Translocator protein 18 kDa (TSPO) is a five‐domain transmembrane protein that is highly expressed in steroid‐producing tissues, including the glial cells within the brain.13, 14, 15, 16 TSPO ligands can modulate the synthesis of neurosteroids that act as allosteric modulators of excitatory and/or inhibitory neurotransmitter receptors.17, 18, 19, 20 ONO‐2952 is a novel and selective antagonist that binds with high affinity to TSPO in rat brain and human tumour cell‐line membrane preparations. The antagonism of TSPO by ONO‐2952 has been shown to reduce stress‐induced defecation and visceral hyperalgesia in rat models.21, 22 It is hypothesised that IBS symptoms and symptoms of stress feed into one another, so that a reduction in stress leads to a reduction in IBS symptoms. ONO‐2952 has been granted Fast Track status for IBS‐D by the United States Food and Drug Administration (FDA). Phase 1 studies in healthy adults have shown ONO‐2952 to be safe and well tolerated.23, 24
This Phase 2, exploratory study evaluated the efficacy, safety and tolerability of orally administered ONO‐2952 (20 and 60 mg once daily for 4 weeks) vs. placebo in female subjects with IBS‐D. The aim of this study was to identify signals of efficacy over a relatively short treatment period prior to larger scale clinical evaluation. The primary objective of the study was to investigate the efficacy of ONO‐2952 in abdominal pain and stool symptoms. Secondary objectives included assessment of effects on stool urgency, abdominal discomfort, adequate relief of IBS symptoms or IBS‐related pain, QoL and evaluation of safety and tolerability.
Materials and methods
Study design
This double‐blind, randomised, placebo‐controlled study enrolled subjects from April 2013 to July 2014 at 49 US study centres. The trial was designed, conducted and reported in compliance with US federal and local regulations and the ethical principles of the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice guidelines. An institutional review board‐approved informed consent form was signed by all subjects before their participation in the trial (ClinicalTrials.gov identifier: NCT01844180).
The study consisted of a screening period of up to 30 days, a 4‐week, on‐treatment period, then a 4‐week, treatment‐free follow‐up period (Figure 1a). Subjects completed an electronic diary (e‐diary) to record IBS‐related symptoms, adequate symptom relief, dates of onset and termination of menstrual bleeding, and rescue medication use. At visit 3, eligible subjects were randomly assigned (1:1:1) to treatment with oral placebo, ONO‐2952 20 mg or ONO‐2952 60 mg once daily for 4 weeks. Subjects were assigned to treatment groups by site personnel using an Interactive Web Response System and central randomisation scheme (Cenduit Interactive Response Technology).
Figure 1.
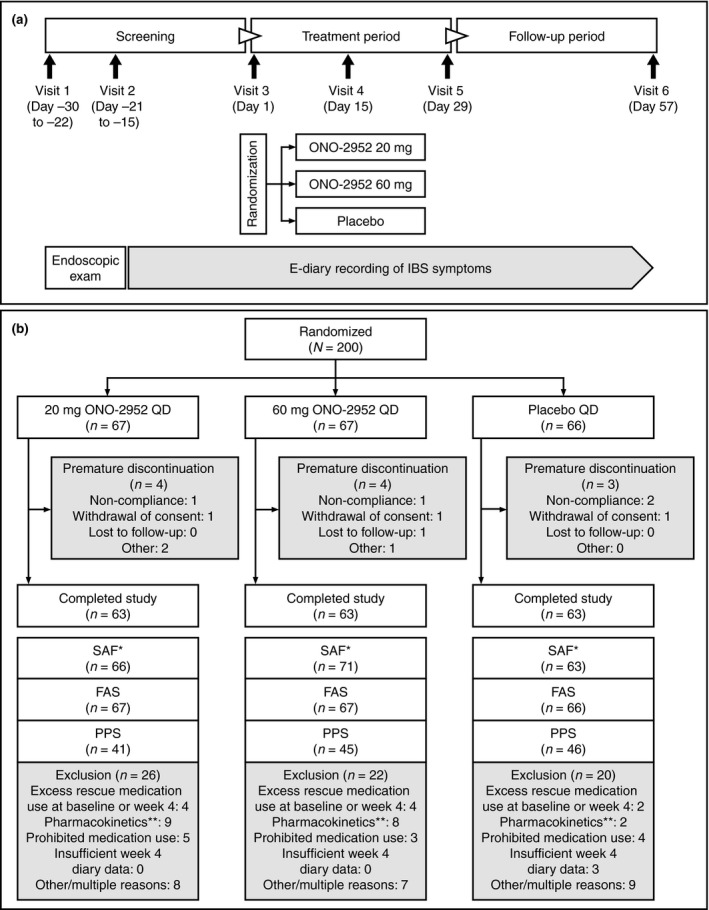
(a) Study design and (b) subject disposition. *Based on actual treatment received. **Pharmacokinetic concentrations on day 29 that were below the limit of quantification and so were consistent with noncompliance with the study protocol. FAS, full analysis set; IBS, irritable bowel syndrome; PPS, per‐protocol set; q.d., once daily; SAF, safety analysis set.
Study population
Eligible subjects were adult females aged 18–65 years who met the Rome III criteria for IBS‐D and had a diagnosis of IBS‐D (defined as loose/watery stools in ≥25% and hard/lumpy stools in <25% of defecations) confirmed by the pre‐treatment (days −14 to −1) IBS‐D symptom diary and physician evaluation. Other inclusion criteria were an average daily score of ≥3.0 (on a 0‐ to 10‐point numerical rating scale) for IBS‐related worst abdominal pain during the 2‐week baseline window (days −14 to −1); ≥1 stool with a consistency of type 6 or 7 on ≥2 days/week and ≤1 stool with a consistency of type 1 or 2 on <2 days/week using the Bristol Stool Scale (BSS)25 during the 2‐week baseline window; and completed self‐assessment of IBS symptoms using a subject e‐diary on ≥5 days/week during the 2‐week baseline. Subjects were also required to have undergone a flexible sigmoidoscopy or colonoscopy within the past 5 years or after the onset and/or significant worsening of IBS symptoms, with a normal result based on clinical findings. Pregnant women were excluded and eligible patients had to meet one of the following criteria: (i) surgically sterilised or post‐menopausal, (ii) females of child‐bearing potential who are nonlactating agreed to use a double barrier method of contraception for the study, and (iii) negative β‐hCG test for pregnancy at both visit 1 and visit 3. There was no requirement for subjects to have a specific Hamilton Anxiety Rating Scale (HAM‐A), Hamilton Depression Rating Scale (HAM‐D) or Perceived Stress Scale (PSS) value on entry into the study.
Key exclusion criteria were as follows: structural abnormalities of the gastrointestinal tract other than oesophagitis or gastritis; clinically significant biochemical abnormality within the past 6 months; diagnosis with a major psychiatric disorder that required hospitalisation within the past 2 years; faecal occult blood test indicating an inflammatory gastrointestinal disorder; upper gastrointestinal symptoms that would impact on IBS symptoms or their assessments; history of abdominal surgery or Crohn's disease, ulcerative colitis, diabetes mellitus, lactose malabsorption, malabsorption syndromes or coeliac sprue; or any other medical conditions that might interfere with assessment of safety or efficacy of the study drug.
Medications likely to interfere with the assessment of ONO‐2952 had to be discontinued at least 14 days prior to randomisation.1 Laxatives, enemas, colonics and hydrotherapy were prohibited from 7 days prior to randomisation. The use of cytochrome P450 (CYP) inhibitors, inducers and substrates was prohibited during randomisation and the on‐treatment period of the study. Subjects agreed to remain on a stable diet and maintain their usual level of physical activity.
Analysis sets defined prior to study unblinding included the following: the safety analysis set (SAF; all subjects who received ≥1 dose of study medication); the full analysis set [FAS; all subjects in the SAF who had ≥1 post‐baseline efficacy assessment – i.e. ≥5 valid diary entries within a week period (2‐week period during the baseline period)]; and the per‐protocol set (PPS). Strict criteria were applied to the PPS to obtain a population of ONO‐2952‐treated subjects to provide the best possible chance of observing a signal of efficacy in this exploratory study. The PPS comprised all subjects in the FAS except for those with violations of inclusion/exclusion criteria that might affect the evaluation of the co‐primary efficacy endpoints (IBS‐D symptoms), subjects who used concomitant medications that could have impacted efficacy assessments, and subjects who were inadequately treated with study medication. Subjects who were insufficiently exposed to ONO‐2952 (compliance <80%, n = 2) or those with excess exposure (compliance >120%, n = 1) were excluded from analysis.
Rescue medication
Subjects who had severe IBS symptoms for >3 consecutive days during the period from 14 days prior to randomisation could take loperamide (up to 4 mg/day). Subjects who had severe IBS‐related abdominal pain could take acetylsalicylic acid (up to 650 mg/day) or paracetamol (up to 1000 mg/day). Daily doses of acetylsalicylic acid or paracetamol were also permitted for IBS‐unrelated symptoms (e.g. fever or headache), and restricted to a maximum of twice during the 14 days prior to randomisation until the end of the on‐treatment period, and four times during the 4‐week follow‐up period.
Study outcomes
Given this was the first assessment of efficacy in IBS patients, co‐primary endpoints as recommended in FDA Guidance26 were selected to assess the effect of treatment on two major IBS signs and symptoms: abnormal defecation (both frequency and consistency) and abdominal pain. The co‐primary endpoints consisted of the following three variables measured on continuous scales: (i) daily ratings of worst abdominal pain experienced during the past 24 h (measured on a 0–10 numeric rating scale), (ii) daily ratings of stool consistency on the 7‐point BSS and (iii) frequency of stools per week measured via the daily e‐diary. For each outcome, the primary analyses were based on the mean change from baseline to week 4. In this exploratory study, the primary outcomes were continuous variables rather than binary measures of treatment responsiveness because continuous measures are more sensitive to between‐group comparisons. However, secondary analyses included binary measures of response to treatment.
A daily responder in abdominal pain intensity was defined as a subject who experienced a decrease of ≥30% compared with the baseline average in the score recorded for worst abdominal pain in the past 24 h. A weekly responder in abdominal pain intensity was defined as a subject who experienced a decrease of ≥30% compared with baseline in the weekly average of worst abdominal pain in the past 24 h.
A daily stool consistency responder was defined as a subject who experienced BSS classifications of <5 for all bowel movements of the day or who had no bowel movement for that day. A weekly stool consistency responder was defined as a subject who experienced a ≥50% reduction in the number of days per week with at least one stool that has a BSS classification of type 6 or 7 compared with baseline.
Secondary efficacy endpoints (analysed or summarised using the PPS unless otherwise indicated) were based on the FDA definitions of specific weekly and daily responders for pain and stool parameters.26 Secondary endpoints included the overall proportion of weekly and daily responders for abdominal pain intensity, stool consistency and the composite of abdominal pain and stool consistency. The daily composite endpoint required that the worst abdominal pain rating be ≥30% below the baseline average daily worst abdominal pain and no bowel movement with a BSS classification of ≥5 for the same day. Other secondary endpoints were the proportion of subjects reporting adequate symptom relief over time and overall weekly adequate relief rate; change from baseline over time in the ‘number of pain‐free days’ and ‘days without any urgency’ per week; and total scores and changes from baseline over time in the IBS‐QoL scale and IBS Symptom Severity Scale (IBS‐SSS; compared using the FAS),26, 27, 28, 29 Exploratory endpoints (compared using the FAS) included changes from baseline over the treatment period for the planned psychiatric endpoints of the HAM‐A, HAM‐D and the PSS.
Safety monitoring consisted of physical examinations, vital signs, 12‐lead electrocardiography, clinical laboratory tests and surveillance for adverse events (AEs). Blood samples were collected pre‐dose and at ~3 h post‐dose on day 29 to measure the concentration of ONO‐2952 in plasma.
Statistical analyses
Determination of sample size
As this was a Phase 2 exploratory study, the sample size was determined by the ability to detect a signal of efficacy in IBS‐D subjects with ONO‐2952. It was planned to analyse 150 subjects (50 per treatment arm). A 25% dropout rate was estimated, and thus, approximately 195 female subjects with IBS‐D were to be randomised.
Co‐primary endpoints
ONO‐2952 20 and 60 mg doses were compared with placebo using a general linear model, with additional factors added as appropriate, using the PPS. A separate model was applied for each endpoint, giving three models in all. In each model, the dependent variable was the endpoint in question. The model included the baseline value (of the corresponding primary efficacy measure) and treatment as explanatory variables. In addition, the following explanatory factors and covariates known to affect IBS symptoms were included: race dichotomised into white/nonwhite, body mass index (BMI; as continuous covariate); and menstrual bleeding. All comparisons used a two‐sided test at the alpha = 0.05 level of significance, uncorrected for multiple comparisons, with P values presented with point estimates and 95% confidence intervals (CIs). Adjusted least squares means for the treatment effects and 95% CIs were also presented.
Sensitivity analyses
In addition to using the FDA‐recommended threshold for defining daily and weekly responders for abdominal pain intensity (≥30% reduction from baseline),26 pre‐planned sensitivity analyses were undertaken for both the abdominal pain intensity endpoint and abdominal pain and stool consistency composite endpoints at week 4 using thresholds of ≥40% and ≥50% reduction in abdominal pain intensity from baseline.
Exploratory analyses
To permit exploration of the time course of the co‐primary endpoints from baseline through week 4 and to assess the impact of excluding subjects in the FAS from the PPS while accommodating missing data, mixed‐model repeated‐measures models were applied to each of the three co‐primary efficacy measures, initially using the PPS and then the FAS. To identify covariates that may have influenced outcomes, additional analyses were undertaken using the PPS, considering factors/covariates such as age, ethnicity, BMI, menstrual bleeding, mean stool consistency, baseline PSS score, mean weekly score of the worst abdominal pain experienced during the past 24 h at baseline, number of days per week with ≥1 stool having a BSS classification of type 6 or 7 at baseline and number of stools per week at baseline.
Results
Participants
Two hundred female subjects were randomised; 189 (94.5%) completed the study and 11 discontinued treatment prematurely (Figure 1b). Reasons for discontinuation were noncompliance with the study protocol (n = 4), withdrawal of consent (n = 3), loss to follow‐up (n = 1) and other reasons (n = 3). No subject discontinued due to an AE. All subjects were included in the SAF and FAS and 132 (66.0%) of these met the stringent criteria for the PPS (Figure 1b).
Demographics and baseline characteristics were comparable between the three treatment arms (Table 1), except for a higher percentage of subjects with baseline BMI ≥30 kg/m2 in the ONO‐2952 60 mg group (49.3% vs. 36.4% and 42.9% in the ONO‐2952 20 mg and placebo groups, respectively). All had baseline PSS scores indicative of low stress (i.e. scores <14) and psychiatric scores (HAM‐A and HAM‐D) indicative of a normal population. Subjects in all treatment arms averaged ~25 bowel movements per week and had a mean BSS stool consistency of approximately 6. Treatment compliance (assessed by counting tablets, not with a dosing calendar) was 99.6%, 99.5% and 98.7% in the ONO‐2952 60 mg, ONO‐2952 20 mg and placebo groups, respectively.
Table 1.
Subject demographics and disease characteristics (PPS)
| Parameter | Placebo q.d. (n = 46) | ONO‐2952 20 mg q.d. (n = 41) | ONO‐2952 60 mg q.d. (n = 45) |
|---|---|---|---|
| Age, years, mean (s.d.) | 47.8 (12.5) | 46.8 (12.5) | 46.4 (12.1) |
| BMI, kg/m2, mean (s.d.) | 29.73 (6.55) | 29.24 (7.01) | 32.12 (7.25) |
| Ethnicity, n (%) | |||
| American Indian/Alaskan native | 1 (2.2) | 0 (0) | 1 (2.2) |
| Asian | 0 (0) | 0 (0) | 1 (2.2) |
| Black or African American | 5 (10.9) | 4 (9.8) | 5 (11.1) |
| Native Hawaiian or other Pacific Islander | 1 (2.2) | 0 (0) | 0 (0) |
| White | 38 (82.6) | 37 (90.2) | 38 (84.4) |
| Mixed | 1 (2.2) | 0 (0) | 0 (0) |
| Menstrual bleeding – yes, n (%) | |||
| At any time | 19 (41.3) | 15 (36.6) | 20 (44.4) |
| During the baseline period | 12 (26.1) | 6 (14.6) | 16 (35.6) |
| During week 4 | 8 (17.4) | 3 (7.3) | 11 (24.4) |
| Abdominal pain during baseline, mean (s.d.) | 6.36 (1.50) | 6.24 (1.36) | 6.01 (1.71) |
| No. of bowel movements/week during baseline, mean (s.d.) | 25.71 (11.96) | 23.25 (14.27) | 27.20 (14.76) |
| No. of days per week with ≥1 stool classed as BSS type 6/7 during baseline, mean (s.d.) | 6.08 (1.36) | 5.76 (1.40) | 5.89 (1.50) |
| Stool consistency (BSS classification) during baseline, mean (s.d.) | 6.11 (0.57) | 5.92 (0.56) | 5.89 (0.54) |
| PSS score during baseline, mean (s.d.) | 9.20 (5.35) | 9.66 (5.25) | 9.31 (6.21) |
| HAM‐A score during baseline, LS, mean (s.d.)a | 5.2 (4.55) | 5.4 (4.6) | 5.7 (5.34) |
| HAM‐D score during baseline, LS, mean (s.d.)a | 2.6 (3.79) | 2.8 (3.61) | 2.7 (4.29) |
BMI, body mass index; BSS, Bristol Stool Score; PSS, Perceived Stress Scale; QD, once daily; s.d., standard deviation.
Full analysis set for HAM‐A and HAM‐D.
Efficacy
Co‐primary endpoints
Over the 4‐week on‐treatment period, a greater decrease in IBS symptoms was evident in both ONO‐2952 groups, compared with placebo, for all three co‐primary endpoints (Table 2). The largest decreases in abdominal pain and stool frequency were observed in subjects receiving ONO‐2952 60 mg. However, subject numbers were not high enough to consistently achieve statistical significance with between‐group comparisons at the 5% level. For stool consistency, mean decreases in the number of days per week with ≥1 stool of BSS type 6 or 7 were similar in both ONO‐2952 groups. Neither were significantly different vs. placebo at week 4 (Table 2).
Table 2.
Change from baseline to week 4 in mean weekly scores for worst abdominal pain experienced during the past 24 h, number of days per week with ≥1 stool of BSS type 6 or 7, and weekly number of stools (on‐treatment period, per‐protocol set)
| Abdominal pain | Stool consistency | Stool frequency | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo QD (n = 46) | ONO‐2952 | Placebo q.d. (n = 46) | ONO‐2952 | Placebo q.d. (n = 46) | ONO‐2952 | ||||
| 20 mg q.d. (n = 41) | 60 mg q.d. (n = 45) | 20 mg q.d. (n = 41) | 60 mg q.d. (n = 45) | 20 mg q.d. (n = 41) | 60 mg q.d. (n = 45) | ||||
| Adjusted treatment mean | |||||||||
| LS mean estimate | −2.07 | −2.23 | −2.70 | −2.38 | −3.03 | −3.01 | −7.59 | −8.36 | −8.52 |
| 95% CI | −2.82 to −1.31 | −3.09 to −1.37 | −3.44 to −1.96 | −3.30 to −1.45 | −4.10 to −1.96 | −3.93 to −2.10 | −10.41 to −4.77 | −11.62 to −5.09 | −11.31 to −5.73 |
| Treatment comparison (ONO‐2952 vs. placebo) | |||||||||
| Difference | – | −0.16 | −0.63 | – | −0.65 | −0.64 | – | −0.77 | −0.93 |
| P value for zero difference | – | 0.7162 | 0.1519 | – | 0.2396 | 0.2406 | – | 0.6498 | 0.5733 |
| 95% CI | – | −1.05 to 0.72 | −1.50 to 0.24 | – | −1.75 to 0.44 | −1.71 to 0.43 | – | −4.10 to 2.57 | −4.18 to 2.33 |
CI, confidence interval; LS, least squares; q.d., once daily.
Mean weekly IBS symptoms were similar in each treatment group at baseline. At each time point on‐treatment and during follow‐up, the largest improvements from baseline in IBS symptoms, relative to placebo, were seen in the ONO‐2952 60 mg group (Figure 2). These improvements became evident at 1–2 weeks after dosing, reaching statistical significance at week 3 for abdominal pain (P < 0.05) but missing statistical significance at week 4. Stool consistency also showed improvement over 4 weeks compared with placebo, but was not statistically significant at the 5% level. No differences were observed in weekly stool frequency scores between groups. Following the end of treatment, there was a rapid return to baseline for abdominal pain and stool consistency scores in the ONO‐2952 60 mg group.
Figure 2.
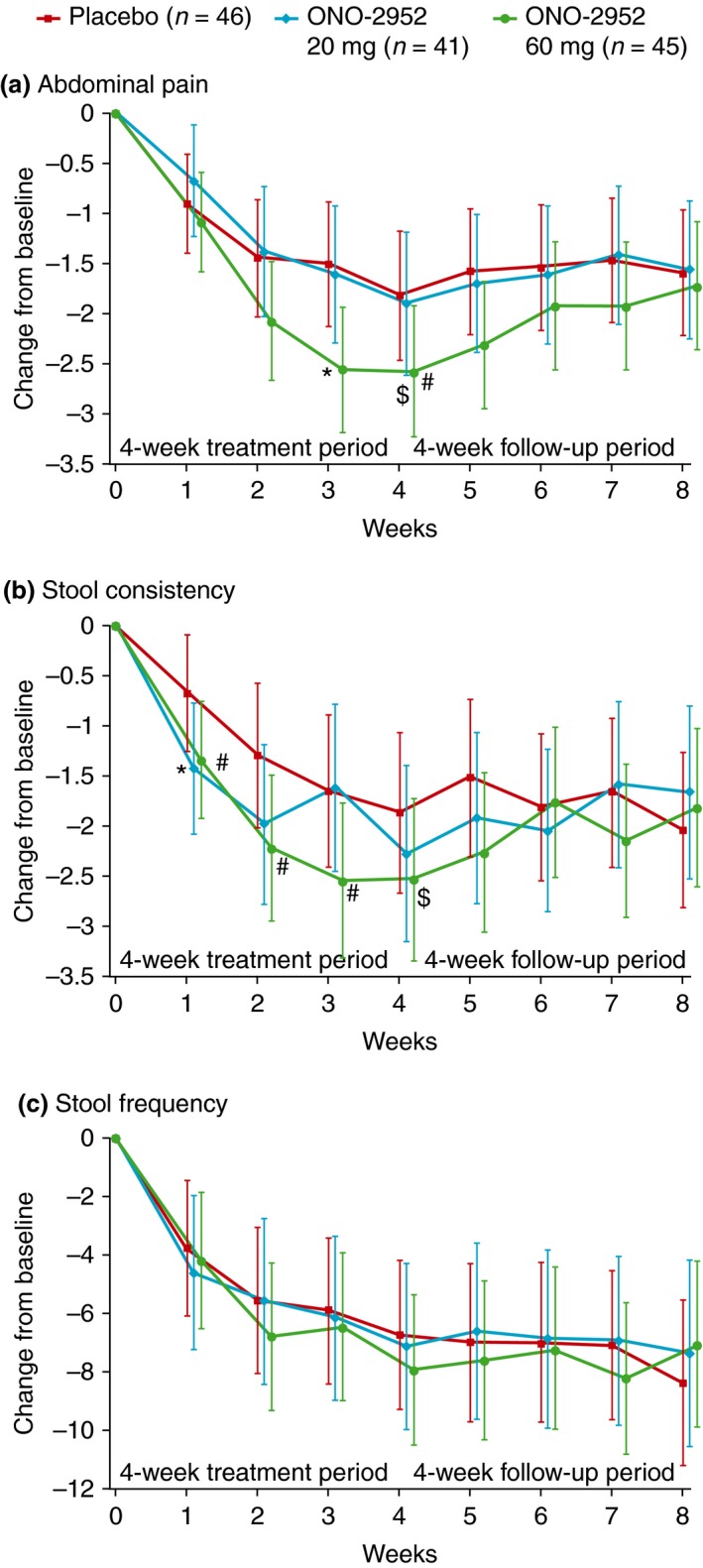
Least‐squares mean (95% CI) change from baseline in weekly scores for (a) worst abdominal pain experienced in the past 24 h, (b) number of days per week with ≥1 stool of BSS type 6 or 7, and (C) weekly number of stools (per‐protocol set). *P < .05; # P < 0.10 vs. placebo; $ P < 0.10 vs. placebo over weeks 1–4 (repeated measures analysis). BSS, Bristol Stool Score; CI, confidence interval.
Secondary endpoints
FDA responder analyses
Compared with placebo, a greater proportion of subjects treated with ONO‐2952 60 mg met FDA responder criteria over the 4‐week treatment period in overall daily response for a composite endpoint of both pain intensity (using a ≥30% reduction in pain threshold) and stool consistency (20.0% vs. 6.5%) and single endpoints of abdominal pain (53.3% vs. 37.0%) or stool consistency (26.7% vs. 10.9%; Figure 3 and Table S1), although the differences did not reach statistical significance at the 5% level. A similar pattern was observed for overall weekly response (Figure 3b).
Figure 3.
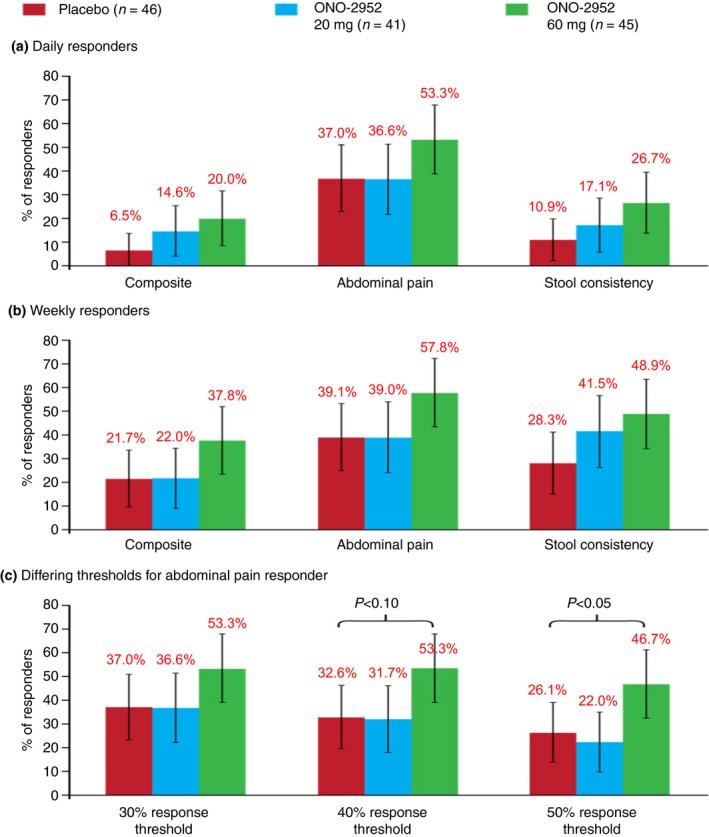
Responder analysis for (a) daily responders, (b) weekly responders and (c) daily responders according to differing thresholds for abdominal pain (on‐treatment period, per‐protocol set).
Pre‐planned sensitivity analyses were performed on the overall daily responders over the 4‐week treatment period for abdominal pain response as well as for the composite endpoint of both pain intensity and stool consistency using ≥40% and ≥50% change from baseline thresholds for abdominal pain response. Using both thresholds, there were higher proportions of responders for the composite endpoint in the ONO‐2952 60 mg group (20.0% and 13.3%, respectively), compared with the ONO‐2952 20 mg (12.2% and 9.8%, respectively) and placebo groups (6.5% and 6.5%, respectively; Table S1). For abdominal pain, a statistically significant difference in the odds ratio (OR) for percentage of responders was observed following ONO‐2952 60 mg treatment, compared with placebo, for change from baseline of ≥50% (OR: 2.89; 95% CI: 1.04–8.00; P = 0.04; Table S1).
Other responder analyses and quality‐of‐life and psychiatric assessments
During the treatment period, a higher proportion of responders in the ONO‐2952 60 mg group reported adequate relief from symptoms (68.9%), compared with the ONO‐2952 20 mg (56.1%) and placebo (56.5%) groups, which was most evident at weeks 2, 3 and 4. This difference between treatments across the 4‐week treatment period was not statistically significant. The proportion of responders then decreased in each treatment group during the follow‐up period. The biggest decrease in the proportion of subjects who reported adequate relief was seen in the 60 mg group (from 68.9% during treatment to 31.1% during follow‐up), while in the placebo group 45.7% reported adequate relief in the follow‐up phase. This difference between groups was not statistically significant (Figure S1). The number of days without pain or urgency vs. baseline was also higher in the ONO‐2952 60 mg group, compared with the ONO‐2952 20 mg and placebo groups, during the treatment and follow‐up periods (Figure S2).
Mean baseline values were similar for all treatment groups for IBS‐QoL and IBS‐SSS total scores, HAM‐A, HAM‐D and PSS total scores (Table S2). At the end of the on‐treatment period (day 29), improvements from baseline were evident in each treatment group for IBS‐QoL and IBS‐SSS and for the three psychiatric assessments.
Rescue medication use (paracetamol, acetylsalicylic acid or loperamide) as permitted by the protocol was low (<12%) in all treatment groups during the treatment period (Table S3).
Safety
The incidence of treatment‐emergent AEs (TEAEs) was similar for subjects in the ONO‐2952 60 mg and placebo groups (47.9% and 47.6%, respectively) and slightly lower for those in the ONO‐2952 20 mg group (39.4%) (Table 3). Most TEAEs were mild or moderate in severity and considered unrelated to study treatment. No subjects discontinued treatment due to TEAEs. The most frequently reported TEAEs (by system organ class) were gastrointestinal disorders (placebo: 23.8%, ONO‐2952 20 mg: 12.1%, ONO‐2952 60 mg: 16.9%), infections and infestations (placebo: 15.9%, ONO‐2952 20 mg: 10.6%, ONO‐2952 60 mg: 14.1%), and nervous system disorders (placebo: 6.3%, ONO‐2952 20 mg: 10.6%, ONO‐2952 60 mg: 4.2%) (Table 3). None of the differences between treatment groups were considered clinically relevant. There were no clinically significant CNS‐related AEs.
Table 3.
Summary of most frequently reported (by >1 subject in any treatment group) treatment‐emergent adverse events by MedDRA Preferred Term version 16.1 (safety analysis set)
| n (%) | Placebo q.d. (n = 63) | ONO‐2952 | |
|---|---|---|---|
| 20 mg q.d. (n = 66) | 60 mg q.d. (n = 71) | ||
| Number of subjects with TEAEs | 30 (47.6) | 26 (39.4) | 34 (47.9) |
| Abdominal pain | 3 (4.8) | 0 (0) | 1 (1.4) |
| Abdominal tenderness | 2 (3.2) | 0 (0) | 0 (0) |
| Diarrhoea | 0 (0) | 2 (3.0) | 0 (0) |
| Flatulence | 2 (3.2) | 0 (0) | 1 (1.4) |
| Irritable bowel syndrome | 1 (1.6) | 2 (3.0) | 2 (2.8) |
| Nausea | 2 (3.2) | 1 (1.5) | 3 (4.2) |
| Vomiting | 0 (0) | 2 (3.0) | 1 (1.4) |
| Influenza | 2 (3.2) | 1 (1.5) | 0 (0) |
| Nasopharyngitis | 4 (6.3) | 1 (1.5) | 4 (5.6) |
| Upper respiratory tract infection | 1 (1.6) | 1 (1.5) | 2 (2.8) |
| Urinary tract infection | 1 (1.6) | 2 (3.0) | 0 (0) |
| Myalgia | 0 (0) | 0 (0) | 2 (2.8) |
| Headache | 3 (4.8) | 3 (4.5) | 3 (4.2) |
| Pollakiuriaa | 2 (3.2) | 0 (0) | 0 (0) |
| Cold sweat | 0 (0) | 2 (3.0) | 0 (0) |
MedDRA, Medical Dictionary for Regulatory Activities; q.d., once daily; TEAE, treatment‐emergent adverse event.
Frequent daytime urination. Adverse events were collected from time of dosing and were all considered to be TEAEs. The table is based on actual treatment received. Three subjects were randomised to placebo but had plasma concentrations of ONO‐2952 greater than the limit of quantification, so were assigned to the ONO‐2952 60 mg treatment group. Two subjects were issued with incorrect kits in error, so were assigned to their first dispensed treatment (ONO‐2952 60 mg).
Eleven subjects in each treatment group experienced TEAEs that were considered related to study treatment. The most frequent of these were abdominal pain (placebo: n = 2, ONO‐2952 60 mg: n = 1) and nausea (placebo: n = 1, ONO‐2952 20 mg: n = 1, ONO‐2952 60 mg group: n = 3). One subject receiving ONO‐2952 60 mg experienced moderate constipation considered probably related to study treatment, which resolved spontaneously. This subject was also taking oral bupropion 150 mg once daily, which has been reported to be associated with constipation.30 No other subject reported constipation.
Seven subjects experienced severe TEAEs [placebo: n = 1 (1.6%), ONO‐2952 20 mg: n = 4 (6.1%), ONO‐2952 60 mg: n = 2 (2.8%)], all of which were gastrointestinal disorders [placebo: abdominal pain (n = 1); ONO‐2952 20 mg: exacerbation of IBS (n = 2), vomiting (n = 1); ONO‐2952 60 mg: exacerbation of IBS (n = 2)], except for one subject receiving ONO‐2952 20 mg (urinary tract infection). One subject each in the placebo and ONO‐2952 60 mg group experienced serious AEs (pneumonia and dehydration, and acute cholecystitis, respectively); none were considered related to study drug. No deaths were reported.
Discussion
This exploratory study of a TSPO antagonist, ONO‐2952, in female subjects with IBS‐D was designed to identify signals of IBS symptom improvement. Although the co‐primary endpoints were not met in this study, effects on some endpoints did show the potential of ONO‐2952 as a treatment for IBS patients. In this study, subjects treated with ONO‐2952 60 mg generally showed improvements in IBS symptoms (abdominal pain, stool consistency and stool frequency) over 4 weeks compared with placebo‐treated subjects. A statistically significant difference in abdominal pain compared with placebo was observed at week 3 (P < 0.05). Differences were also seen for stool consistency compared with placebo over weeks 1–4. These improvements disappeared during the treatment‐free follow‐up period, most rapidly for the 60 mg dose, with values becoming similar again to those of the placebo group. This rapid return to baseline after stopping of treatment is another indication that ONO‐2952 60 mg may be an effective treatment for IBS‐D (Figure 2).
In addition, a greater percentage of subjects treated with ONO‐2952 60 mg vs. placebo met FDA responder criteria over the 4‐week treatment period and met responder criteria for abdominal pain at thresholds of ≥40% and ≥50% decrease from baseline. A statistically significant improvement in abdominal pain with ONO‐2952 60 mg vs. placebo was observed for the ≥50% threshold (P < 0.05). Improvements were also evident in a number of secondary endpoints including adequate symptom relief, number of pain‐free days, and number of days without urgency following treatment with ONO‐2952 60 mg, compared with placebo providing additional signals for efficacy. There was no apparent difference in the changes from baseline for IBS‐QoL and IBS‐SSS; however, given the scale of the study, these measures were highly variable, with low likelihood of detecting any differences.
A dose–response relationship for ONO‐2952 was also evident for both primary and secondary efficacy measures, with greater improvements in IBS symptoms generally seen following administration of the 60 mg dose compared with the 20 mg dose. This observation is consistent with results from a Phase 1, single‐dose, positron emission tomography study in healthy male and female subjects that demonstrated a higher whole‐brain TSPO occupancy for ONO‐2952 60 mg (77.4%) compared with the 20 mg dose (61.3%).24 The higher whole brain TSPO occupancy for ONO‐2952 60 mg provides a plausible explanation for the observed dose–response effect; however, further studies are required to determine whether doses of ONO‐2952 above 60 mg are effective.
Thus, albeit in a relatively small‐scale clinical study, the potential impact on abnormal defecation and abdominal pain demonstrated by ONO‐2952 is considered highly relevant given that these are the cardinal symptoms of IBS patients. Indeed, abdominal pain and abnormal defecation management in IBS currently lacks one unique effective pharmacological remedy. Although the mechanism by which stool consistency is altered by ONO‐2952 is not fully understood, preclinical models showed a similar effect; ONO‐2952 prevented restraint stress‐induced defecation in rats at doses corresponding to >50% TSPO occupancy in the brain.22 In addition, questions are raised as to whether the effects of ONO‐2952 are limited to the central nervous system or may also be peripherally mediated. Due to the widespread expression of TSPO throughout the body (TSPO is found in many regions of the body including the heart, liver, adrenal and testis, as well as hemopoietic and lymphatic cells and human iris/ciliary body), it is possible that the effects of ONO‐2952 may not be limited to the CNS.
ONO‐2952 was generally well tolerated by female IBS‐D subjects in this study. The majority of AEs were mild in intensity and were considered to be unrelated to the study drug. Gastrointestinal disorders were the most commonly reported AEs, and there were no clinically relevant AEs involving the CNS. Interestingly, there was no apparent signal to suggest that constipation was observed in subjects treated with ONO‐2952. Constipation is the most common AE associated with both eluxadoline and alosetron in subjects with IBS‐D and was one of the reasons the use of alosetron is now limited to women with severe refractory IBS‐D.7, 31, 32, 33, 34, 35
There were limitations to this study. The antagonism of TSPO by ONO‐2952 has been shown to reduce stress‐induced defecation in rat models, which provided a rationale for testing the compound in IBS‐D patients where stress has been suggested to play an influential role in symptom generation. However, the subjects who participated in this study did not appear to be anxious or stressed. The assessment of several psychiatric parameters including the PSS, HAM‐A and HAM‐D was included in the study, but baseline values across the treatment groups were all in line with those of a healthy adult population. No attempt was made to enrol IBS‐D patients with a specific ‘stress’ phenotype as this would have impacted recruitment substantially. All patients had baseline PSS scores indicative of low stress (i.e. scores <14). HAM‐A total scores in a population of patients with generalised anxiety disorder were reported as between 20 and 30, while baseline HAM‐A scores in this study for all treatment groups were from 5.2 to 5.7.36 For the HAM‐D scale, a score of 0–7 is generally accepted to be within the normal range and scores of 20 or higher indicate moderate, severe or very severe depression and are usually required for entry into a clinical trial in depression. Baseline HAM‐D scores in this study for all treatment groups were between 2.6 and 2.8. There was, therefore, limited scope for improvement in any of these scales and post hoc analysis could not confirm any clear correlation between individual improvements in IBS symptoms such as abdominal pain and changes in psychiatric parameters.
In addition, although the recommended treatment duration for studies in IBS‐D to formally assess efficacy is at least 8 weeks,26 the treatment duration in this trial was limited to 4 weeks, the longest period permitted by available pre‐clinical toxicology data at the time of study conduct. However, the 4‐week treatment period was considered appropriate to identify any signals of efficacy with ONO‐2952 and its novel mechanism of action. As this was an exploratory proof‐of‐concept study, it was not formally powered and the analysis of study endpoints was conducted using an analysis population (PPS) defined to provide the best possible chance of observing a signal of efficacy. Given the stage of development of ONO‐2952, this was deemed appropriate. Lastly, the study population was limited to women as the overall prevalence of IBS in women is 67% higher than in men.37 Further studies would be required to determine whether ONO‐2952 demonstrates efficacy in men. It is thus acknowledged that additional clinical trials of larger scale, longer treatment duration and expanded population will be required to further explore the efficacy of ONO‐2952 in IBS‐D.
In conclusion, treatment with ONO‐2952 60 mg once daily for 4 weeks in this study showed promising signals of clinical efficacy in female subjects with IBS‐D, although the a priori endpoints did not achieve statistical significance. Further evaluation of ONO‐2952 is warranted to assess its potential as a treatment for IBS‐D.
Authorship
Guarantor of the article: W. E. Whitehead.
Author contributions: W.E. Whitehead was responsible for the study concept and design. W.E. Whitehead, M. Bruce, K. Duffy and J. Sharpe performed the analysis and interpretation of data. W.E. Whitehead, M. Bruce, K. Duffy, J. Sharpe and T. Nabata wrote and critically revised the manuscript. All authors approved the final version of the manuscript.
Supporting information
Figure S1. Proportion of subjects (95% CI) reporting adequate relief from symptoms over time (per‐protocol set).
Figure S2. Least‐squares mean (s.d.) change from baseline in weekly scores for number of days free from (a) pain or (b) urgency (per‐protocol set).
Table S1. Sensitivity analysis of overall daily responder rate in abdominal pain intensity and the composite of pain intensity and stool consistency, when differing thresholds for abdominal pain were considered (on‐treatment period, per‐protocol set).
Table S2. Least squares mean (s.d.) and change from baseline in IBS‐QoL, IBS‐SSS, HAM‐A, HAM‐D and PSS scores (on‐treatment period, Per‐Protocol Set for IBS‐QoL and Full‐Analysis Set for IBS‐SSS, HAM‐A, HAM‐D, PSS).
Table S3. Number of subjects who recorded use of rescue medication (aspirin, paracetamol or loperamide) for abdominal pain/discomfort, urgency or diarrhoea in the e‐diary – safety analysis set.
Acknowledgements
We thank the subjects and personnel at each study site. Editorial support for the preparation of this manuscript, furnished by Jamie Ashman and James Reed, was provided by Prism Ideas and funded by Ono Pharmaceutical Co. Ltd. and Ono Pharma UK Ltd.
Declaration of personal interests: William E Whitehead has received consultancy fees from Ono Pharma US Inc. and Ono Pharma UK Ltd., research support from Salix Pharmaceuticals and the Rome Foundation. He received consultancy fees from Biomerica USA. Mark Bruce, John Sharpe, Toshiya Nabata and Kevin Duffy are employees of Ono Pharma UK Ltd.
Declaration of funding interests: This study and editorial support for the preparation of this manuscript were funded in full by Ono Pharmaceutical Co. Ltd. and Ono Pharma UK Ltd.
The Handling Editor for this article was Professor Alexander Ford, and it was accepted for publication after full peer‐review.
Note
Medications likely to interfere with the assessment of ONO‐2952: anxiolytics, anticonvulsants, alpha and beta blockers, calcium‐channel blockers, opioids, analgesics (except acetylsalicylic acid and paracetamol), 5‐HT3 antagonists, 5‐HT4 agonists, macrolides, antacids that contain Al or Mg, prostaglandins, sulfasalazine, infliximab, H2 blockers and proton‐pump inhibitors judged to impact on IBS symptoms or on symptom assessment, colchicine, leuprolide modafinil or armodafinil, iron supplements, otilonium bromide, pinaverium bromide, peppermint oil and antiobesity agents.
References
- 1. Lovell RM, Ford AC. Global prevalence of and risk factors for irritable bowel syndrome: a meta‐analysis. Clin Gastroenterol Hepatol 2012; 10: 712–21. [DOI] [PubMed] [Google Scholar]
- 2. Makker J, Chilimuri S, Bella JN. Genetic epidemiology of irritable bowel syndrome. World J Gastroenterol 2015; 21: 11353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Longstreth GF, Thompson WG, Chey WD, et al Functional bowel disorders. Gastroenterology 2006; 130: 1480–91. [DOI] [PubMed] [Google Scholar]
- 4. Ford AC, Moayyedi P, Lacy BE, et al American College of Gastroenterology monograph on the management of irritable bowel syndrome and chronic idiopathic constipation. Am J Gastroenterol 2014; 109(Suppl. 1): S2–26. [DOI] [PubMed] [Google Scholar]
- 5. Zhu L, Huang D, Shi L, et al Intestinal symptoms and psychological factors jointly affect quality of life of patients with irritable bowel syndrome with diarrhea. Health Qual Life Outcomes 2015; 13: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ballou S, Bedell A, Keefer L. Psychosocial impact of irritable bowel syndrome: a brief review. World J Gastrointest Pathophysiol 2015; 6: 120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lembo T, Wright RA, Bagby B, et al Alosetron controls bowel urgency and provides global symptom improvement in women with diarrhea‐predominant irritable bowel syndrome. Am J Gastroenterol 2001; 96: 2662–70. [DOI] [PubMed] [Google Scholar]
- 8. Karantanos T, Markoutsaki T, Gazouli M, Anagnou NP, Karamanolis DG. Current insights in to the pathophysiology of irritable bowel syndrome. Gut Pathog 2010; 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lacy BE, Chey WD, Lembo AJ. New and emerging treatment options for irritable bowel syndrome. Gastroenterol Hepatol 2015; 11: 1–19. [PMC free article] [PubMed] [Google Scholar]
- 10. Trinkley KE, Nahata MC. Medication management of irritable bowel syndrome. Digestion 2014; 89: 253–67. [DOI] [PubMed] [Google Scholar]
- 11. Nee J, Zakari M, Lembo AJ. Novel therapies in IBS‐D treatment. Curr Treat Options Gastroenterol 2015; 13: 432–40. [DOI] [PubMed] [Google Scholar]
- 12. Cash BD, Lacy BE, Rao T, Earnest DL. Rifaximin and eluxadoline – newly approved treatments for diarrhea‐predominant irritable bowel syndrome: what is their role in clinical practice alongside alosetron? Expert Opin Pharmacother 2016; 17: 311–22. [DOI] [PubMed] [Google Scholar]
- 13. Batarseh A, Papadopoulos V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol Cell Endocrinol 2010; 327: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther 2008; 118: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papadopoulos V, Baraldi M, Guilarte TR, et al Translocator protein (18 kDa): new nomenclature for the peripheral‐type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 2006; 27: 402–9. [DOI] [PubMed] [Google Scholar]
- 16. Rupprecht R, Papadopoulos V, Rammes G, et al Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov 2010; 9: 971–88. [DOI] [PubMed] [Google Scholar]
- 17. Jasmin L, Rabkin SD, Granato A, Boudah A, Ohara PT. Analgesia and hyperalgesia from GABA‐mediated modulation of the cerebral cortex. Nature 2003; 424: 316–20. [DOI] [PubMed] [Google Scholar]
- 18. Ohara PT, Vit JP, Jasmin L. Cortical modulation of pain. Cell Mol Life Sci 2005; 62: 44–52. [DOI] [PubMed] [Google Scholar]
- 19. Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res 2010; 186: 113–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strous RD, Maayan R, Weizman A. The relevance of neurosteroids to clinical psychiatry: from the laboratory to the bedside. Eur Neuropsychopharmacol 2006; 16: 155–69. [DOI] [PubMed] [Google Scholar]
- 21. Mitsui K, Niwa T, Kawahara Y, et al Anti‐stress effects of ONO‐2952, a novel translocator protein 18 kDa antagonist, in rats. Neuropharmacology 2015; 99: 51–66. [DOI] [PubMed] [Google Scholar]
- 22. Mitsui K, Sasamura T, Katsumata S, et al Effects of Ono‐2952, a novel translocator protein 18 kDa antagonist, on stress‐induced rectal hyperalgesia and defecation in rats. Gastroenterology 2012; 142: S813–4. [Google Scholar]
- 23. Suto F, Wood A, Kobayashi M, et al Safety, tolerability and pharmacokinetics of the novel translocator protein 18 kDa antagonist ONO‐2952 in healthy subjects. Clin Ther 2015; 37: 2071–84. [DOI] [PubMed] [Google Scholar]
- 24. Frankle WG, Narendran R, Mason SN, et al A phase I PET study to evaluate brain translocator protein occupancy by ONO‐2952 in healthy adult subjects using 11C‐PBR28. Society for Neuroscience Annual Meeting 2014; Abstract BB21.
- 25. Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol 1997; 32: 920–4. [DOI] [PubMed] [Google Scholar]
- 26. U.S. Department of Health and Human Services, Food and Drug Administration , Center for Drug Evaluation and Research. Center for Drug Evaluation and Research, FDA, Silver Springs, MD, May 2012, Pp 1–14. [Google Scholar]
- 27. Drossman DA, Patrick DL, Whitehead WE, et al Further validation of the IBS‐QOL: a disease‐specific quality‐of‐life questionnaire. Am J Gastroenterol 2000; 95: 999–1007. [DOI] [PubMed] [Google Scholar]
- 28. Francis CY, Morris J, Whorwell PJ. The irritable bowel severity scoring system: a simple method of monitoring irritable bowel syndrome and its progress. Aliment Pharmacol Ther 1997; 11: 395–402. [DOI] [PubMed] [Google Scholar]
- 29. Mangel AW, Hahn BA, Heath AT, Northcutt AR, et al Adequate relief as an endpoint in clinical trials in irritable bowel syndrome. J Int Med Res 1998; 26: 76–81. [DOI] [PubMed] [Google Scholar]
- 30. Lounsbery JL, Medow MA, Green CG. Severe constipation associated with extended‐release bupropion therapy. Am J Health Syst Pharm 2008; 65: 1530–2. [DOI] [PubMed] [Google Scholar]
- 31. Lembo AJ, Lacy BE, Zuckerman MJ, et al Eluxadoline for irritable bowel syndrome with diarrhea. N Engl J Med 2016; 374: 242–53. [DOI] [PubMed] [Google Scholar]
- 32. Krause R, Ameen V, Gordon SH, et al A randomized, double‐blind, placebo‐controlled study to assess efficacy and safety of 0.5 mg and 1 mg alosetron in women with severe diarrhea‐predominant IBS. Am J Gastroenterol 2007; 102: 1709–19. [DOI] [PubMed] [Google Scholar]
- 33. Lembo AJ, Olden KW, Ameen VZ, et al Effect of alosetron on bowel urgency and global symptoms in women with severe, diarrhea‐predominant irritable bowel syndrome: analysis of two controlled trials. Clin Gastroenterol Hepatol 2004; 2: 675–82. [DOI] [PubMed] [Google Scholar]
- 34. Camilleri M, Chey WY, Mayer EA, et al A randomized controlled clinical trial of the serotonin type 3 receptor antagonist alosetron in women with diarrhea‐predominant irritable bowel syndrome. Arch Intern Med 2001; 161: 1733–40. [DOI] [PubMed] [Google Scholar]
- 35. Cremonini F, Nicandro JP, Atkinson V, et al Randomised clinical trial: alosetron improves quality of life and reduces restriction of daily activities in women with severe diarrhoea‐predominant IBS. Aliment Pharmacol Ther 2012; 36: 437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bandelow B. Defining response and remission in anxiety disorders: toward an integrated approach. CNS Spectr 2006; 11(10 Suppl. 12): 21–8. [DOI] [PubMed] [Google Scholar]
- 37. Canavan C, West J, Card T. The epidemiology of irritable bowel syndrome. Clin Epidemiol 2014; 6: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Proportion of subjects (95% CI) reporting adequate relief from symptoms over time (per‐protocol set).
Figure S2. Least‐squares mean (s.d.) change from baseline in weekly scores for number of days free from (a) pain or (b) urgency (per‐protocol set).
Table S1. Sensitivity analysis of overall daily responder rate in abdominal pain intensity and the composite of pain intensity and stool consistency, when differing thresholds for abdominal pain were considered (on‐treatment period, per‐protocol set).
Table S2. Least squares mean (s.d.) and change from baseline in IBS‐QoL, IBS‐SSS, HAM‐A, HAM‐D and PSS scores (on‐treatment period, Per‐Protocol Set for IBS‐QoL and Full‐Analysis Set for IBS‐SSS, HAM‐A, HAM‐D, PSS).
Table S3. Number of subjects who recorded use of rescue medication (aspirin, paracetamol or loperamide) for abdominal pain/discomfort, urgency or diarrhoea in the e‐diary – safety analysis set.