

Abstract
Extracting histones from cells is the first step in studies that aim to characterize histones and their post‐translational modifications (hPTMs) with MS. In the last decade, label‐free quantification is more frequently being used for MS‐based histone characterization. However, many histone extraction protocols were not specifically designed for label‐free MS. While label‐free quantification has its advantages, it is also very susceptible to technical variation. Here, we adjust an established histone extraction protocol according to general label‐free MS guidelines with a specific focus on minimizing sample handling. These protocols are first evaluated using SDS‐PAGE. Hereafter, a selection of extraction protocols was used in a complete histone workflow for label‐free MS. All protocols display nearly identical relative quantification of hPTMs. We thus show that, depending on the cell type under investigation and at the cost of some additional contaminating proteins, minimizing sample handling can be done during histone isolation. This allows analyzing bigger sample batches, leads to reduced technical variation and minimizes the chance of in vitro alterations to the hPTM snapshot. Overall, these results allow researchers to determine the best protocol depending on the resources and goal of their specific study. Data are available via ProteomeXchange with identifier PXD002885.
Keywords: Comparative study, Epigenetics, Extraction protocol, Histone, Label‐free MS quantification, Technology
Abbreviations
- DDA
data‐dependent acquisition
- hESC
human embryonic stem cells
- hPTMs
histone post‐translational modifications
- QC
quality control
- RA
relative abundance
- RT
room temperature
- SDC
sodium deoxycholate
1. Introduction
Chromatin, the heritable material of eukaryotic cells, consists of highly structured DNA wrapped around nucleosomes. Each nucleosome comprises four different, evolutionarily well‐conserved core histone proteins (H2A, H2B, H3, and H4) that play a key role in the regulation of DNA‐templated biological processes. Histones encode epigenetic information, which constitutes a complex regulatory level on top of the genome sequence, through the expression of variant histone forms as well as through the presence of an extensive array of histone post‐translational modifications (hPTMs) 1, 2.
Significance of the study
Label‐free quantitative MS is increasingly being used in the histone community to relatively quantify dynamic changes of histone post‐translational modifications (hPTMs) across different samples. Because many histone extraction protocols have been established before this trend and increasingly different biological systems are being studied with the label‐free approach, validation of repeatable protocols for accurate and precise results is needed. The data provided here make a useful starting point for the chromatin community: if the cell type allows for it, reducing the protocol can be done with no impact on relative abundance (RA) of hPTMs, while saving time and means, reducing technical variation and avoiding the need for specific inhibitors of epigenetic writers and erasers. Because the choice of a protocol when starting an experiment is a crucial but sometimes difficult task, we provide an overview that allows researchers to make a conscious choice.
With increasing insight into the complexity of this histone code, an experimental shift from targeted, antibody‐based techniques to using LC–MS as a powerful, unbiased method for discovering, screening, and quantifying global hPTMs has been observed 3. Although identification of (novel) hPTMs remains a major goal in the field, mapping dynamic changes of histone marks across samples is increasingly of interest to the histone community, for example a comparison between undifferentiated and differentiated stem cells or among samples from different patients 4, 5, 6.
Relative quantification of hPTMs is increasingly being done by label‐free quantitative MS approaches because of their cost‐effectiveness, straightforward nature of sample preparation and flexibility in experimental design, both in the type and number of samples being analyzed 4, 7, 8. If not targeted, such as in MRM, shotgun approaches comprise data‐dependent (DDA) and data‐independent acquisition strategies. Contrary to label‐based workflows however, samples in label‐free approaches are not combined at the level of sample preparation, but in silico post‐data acquisition. Because all the systematic and nonsystematic variations between experiments are thus easily reflected in the obtained data, the number of sample preparation steps should be kept to a minimum and every effort should be made to control reproducibility at each step 7, 9.
Extraction is the first step in the histone label‐free MS workflow, followed, in bottom‐up analysis, by propionylation to block lysine residues, digestion of proteins into peptides using trypsin, a second round of propionylation, LC separation, and tandem MS acquisition 4. However, current histone extraction protocols were not specifically designed for label‐free MS 10, 11, 12, 13, 14, 15, 16, 17, 18. Because errors accumulate throughout the protocol, verification and adaption of histone extraction protocols for the specific purpose of label‐free quantification thus is needed. Here, we focus on: (i) avoiding non‐MS compatible components that need to be washed away or removed downstream (such as salts and detergents) or the application of (unautomated) mechanical lysis, (ii) minimizing sample handling, to reduce technical variation, and to allow bigger sample batches to be processed, and (iii) verifying the impact of freezing, which is often required during, for example time‐lapse experiments, because all samples are preferably extracted, processed, and measured in a single batch. Additionally, these latter measures also minimize the time frame wherein residual activity of epigenetic mediators could change the histone fingerprint because, to the best of our knowledge, no inhibitor cocktail for all epigenetic readers and writers has been described to date.
Shechter et al. describe a standard extraction protocol using a hypotonic lysis buffer for isolation of nuclei prior to acid extraction of histones that is easily amenable to label‐free MS owing to the lack of detergents in the lysis buffer 14. We here refer to it as Protocol A. Next, we evaluated the impact of modifications to this protocol especially outlined for label‐free MS. First, in Protocol B, the nuclei isolation step was omitted and the cells were directly placed in dilute acid 14, 15. Second, the impact of snap freezing of the cells directly after harvest was assessed (suffix—fresh vs. frozen). Because extraction efficiency proved to be dependent on cell type and state, we also assessed the suitability of the detergent sodium deoxycholate (SDC) as a component of nuclei isolation buffers for subsequent histone extraction (Protocol C) 19, 20. Because it precipitates in acid, the “MS compatible” detergent SDC is easily implemented in the histone extraction protocol as it does not demand additional washing steps 21.
Using SDS‐PAGE we first compared the general purity of the six different protocols (A/B/C, fresh, and frozen) on two cell lines. Based on this purity evaluation, we selected four protocols for a final analysis by label‐free DDA MS to assess their drawbacks and benefits in histone research in which we focus on repeatability and quality assessment of the extracts.
2. Materials and methods
2.1. Cell culture and sample collection
The human embryonic stem cells (hESC) WA01 Oct4‐eGFP knock‐in reporter cell line (WiCell) was cultured (5% O2 and 5% CO2 at 37°C) in a feeder‐free manner on Vitronectin XF coated 6‐well plates (coating concentration 0.5 μg/cm²; Primorigen Biosciences) in Essential 8 Medium (Life Technologies). Every 3–4 days, cultures were split using 0.5 mM EDTA according to the manufacturer's protocol (Life Technologies). Jurkat cells were grown in culture flasks in RPMI 1640 medium supplemented with 10% FBS, 2 mM l‐glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin, and were maintained at a density of 0.5–1 × 106 cells/mL in a humidified atmosphere containing 5% CO2 at 37°C. Both cultures were verified to be free of mycoplasma contamination.
After a washing step with dPBS, hESC were detached with 750 μL 0.25% trypsin‐EDTA per well for 6 min at 37°C and harvested as single cells. Subsequently, an equal amount of 0.25% trypsin‐inhibitor from Glycine max (Sigma‐Aldrich) was added and hESC were centrifuged for 5 min at 100 g at room temperature (RT). Jurkat cells were harvested by centrifugation (10 min, 250 g, RT). Afterwards, cells were washed three times using PBS, counted and distributed evenly in Eppendorfs. Cell pellets were either snap‐frozen in liquid nitrogen as a dry pellet (snap‐frozen cells) or processed for histone extraction directly (fresh cells). Prior to histone extraction, pellets of snap‐frozen cells were resuspended in ice‐cold PBS, aliquoted in 1.5 mL Eppendorfs according to the required cell number, and spun.
2.2. Histone extraction
Differences between Protocols A, B, and C are situated in the first part of the protocol, that is before the acid extraction of histones. In Protocol A, the cell pellet was resuspended in hypotonic lysis buffer (10 mM Tris‐HCl pH 8.0, 1 mM KCl, 1.5 mM MgCl2) supplemented with 1 mM DTT, 1 mM PMSF, complete protease inhibitor cocktail EDTA‐free (1183617001, Roche Diagnostics, 1 tablet for 50 mL of buffer) and phosphatase inhibitor cocktails II and III (P5726 and P0044, Sigma‐Aldrich, 1 mL of cocktail for 100 mL of buffer) at a cell density of 5 × 106 cells/mL, and incubated for 30 min on a rotator at 4°C 14. For Protocol B, no cell lysis buffer was used. Acid was added directly to fresh or snap‐frozen cells 14, 15. In Protocol C, the cell pellet was lysed with a 10:1 v/v ratio of nuclear isolation buffer‐250 (15 mM Tris‐HCl pH 7.5, 15 mM NaCl, 60 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose) supplemented with inhibitors (1 mM DTT, 0.5 mM AEBSF, 5 nM microcystin‐LR (Enzo Life Sciences) and 10 mM sodium butyrate) and 0.1% SDC 13, 21. Homogenized cells were incubated on ice for 10 min and washed three times with 10:1 v/v nuclear isolation buffer‐250 + inhibitors (without SDC). For all protocols, acid extraction was performed using 0.2 N HCl at a cell density of 8 × 103 cells/μL, after which samples were incubated on a rotator at 4°C for 30 min. After spinning (16 000× g, 10 min, 4°C), equal amounts of supernatant were transferred to fresh 0.5 mL Eppendorfs (Eppendorf Protein LoBind microcentrifuge tubes, Eppendorf). A final concentration of 25% trichloroacetic acid was added slowly to the histone solution, after which the Eppendorfs were inverted several times and incubated on ice for 30 min. Pelleted (16 000 × g, 10 min, 4°C) histones were washed twice with ice‐cold acetone and air‐dried in a fume hood for about 30 min at RT. Histone pellets were dissolved in milliQ water (Merck Millipore), centrifuged (16 000 × g, 10 min, 4°C) to remove the remaining insoluble pellet and transferred into fresh 0.5 mL Protein LoBind Eppendorfs.
2.3. Gel electrophoresis and image analysis
Histones extracted from 106 cells were vacuum‐dried, dissolved in 2× Laemmli buffer (4% SDS, 20% glycerol, and 10% 2‐mercaptoethanol in 50 mM Tris‐HCl pH 6.8) and separated on 15% Tris‐HCl gels (Bio‐Rad Laboratories). Gels were stained with the fluorescent Sypro Ruby gel stain after fixation and scanned using a Versadoc imaging system (Bio‐Rad Laboratories). The Quantity One software (Bio‐Rad Laboratories) was used for purity analysis.
2.4. MS sample preparation
Histone extracts from 106 cells performed in independent technical replicates were subjected to a double round of propionylation pre‐ and postdigestion as described before (Meert et al., Method A.2) 22. Proteolysis was performed overnight at 37°C using trypsin (1:20, trypsin:protein w/w, Promega) in 50 mM ammonium bicarbonate buffer supplemented with 1 mM CaCl2 and 5% acetonitrile 23. Propionylated histones were dissolved in 0.1% formic acid in HPLC grade water (buffer A), sonicated and centrifuged to remove insoluble aggregates prior to loading 4 μL containing 800 ng per injection onto the LC‐MS system. Equal fractions of all samples were pooled to generate quality control (QC) samples, which were run in fixed intervals in between the other samples. All samples were spiked with digested beta‐galactosidase (20 fmole on column; Sciex) to monitor chromatographic quality and variation between LC‐MS runs.
2.5. LC‐MS method
LC was performed using a nanoACQUITY UPLC system (Waters). First, samples were delivered to a trap column (180 μm × 20 mm nanoACQUITY UPLC 2G‐V/MTrap 5 μm Symmetry C18, Waters) at a flow rate of 8 μL/min for 2 min in 99.5% buffer A. Subsequently, peptides were transferred to an analytical column (100 μm × 100 mm nanoACQUITY UPLC 1.7 μm Peptide BEH, Waters) and separated at a flow rate of 300 nL/min using a gradient of 60 min going from 1 to 40% buffer B (0.1% formic acid in acetonitrile). MS data acquisition parameters were set according to Helm et al., with minor adaptations 24. A Q‐TOF SYNAPT G2‐Si instrument (Waters) was operated in positive mode for High Definition‐DDA, using a nano‐ESI source, acquiring full scan MS and MS/MS spectra (m/z 50–5000) in resolution mode. Survey MS scans were acquired using a fixed scan time of 400 ms. Tandem mass spectra of up to eight precursors with charge state 2+ to 5+ were generated using CID in the trapping region with intensity threshold set at 2000 cps, using a collision energy ramp from 6/9 V (low mass, start/end) up to 147/183 V (high mass, start/end). MS/MS scan time was set to 100 ms with an accumulated ion count “TIC stop parameter” of 350 000 cps allowing a maximum accumulation time of 200 ms. Dynamic exclusion of fragmented precursor ions was set to 10 s. Ion mobility spectrometry wave velocity was ramped from 2500 to 400 m/s. Wideband enhancement was used to obtain a near 100% duty cycle on singly charged fragment ions. LockSpray of Glufibrinopeptide‐B (m/z 785.8427) was acquired at a scan frequency of 60 s.
2.6. Data analysis
Progenesis QI for Proteomics (Progenesis QIP 2.0, Nonlinear Dynamics, Waters) was used to process the raw LC–MS data. One QC run served as alignment template. MS precursors were filtered based on charge state (2+ to 6+) and the data were normalized to all MS precursors prior to calculations of fold enrichment and statistics. A multivariate statistical analysis was performed on all 6539 retained MS precursors, without any prior peptide identification. Using MS precursor abundance levels across runs, PCA in Progenesis QIP determines the principle axes of abundance variation, and transforms and plots the abundance data in the principle component space, separating the samples according to abundance variation. Based on the first principal component, sample B from Protocol A‐frozen and samples A and H from Protocol B‐frozen were deemed outliers (Supporting Information Fig. 1 for PCA and accompanying boxplot) and ignored in all subsequent analyses. The CV is reported as percentage and was calculated using peptide‐level abundance measurements (m/z 400–1250, RT 17–50 min (RT window 10–90 s), median intensity >1000). For comparison of the repeatability of the different protocols, instrument CV as calculated from the eight QC runs spread throughout the sample list was subtracted from each peptide (which we equated to zero when negative), resulting in a corrected CV. Only precursors detected in all replicates were used.
For the untargeted, first‐quantify‐then‐identify analysis at the peptide level in search for unanticipated effects induced by the protocols, the MS/MS spectra of the differential MS precursors significantly (q‐value < 10−6) most abundant in either Clusters 1 or 2 (highest mean) were exported as separate *.mgf peaklists. An error tolerant search against Swissprot Human (20 210 sequences, version 2014_7) supplemented with internal standards and prevalent contaminants was performed using a Mascot 2.5 in‐house server (Matrix Science), with propionylation of lysine and the N‐terminus as variable modifications. Mass error tolerance for the precursor ions was set at 15 ppm and for the fragment ions at 0.3 Da. Enzyme specificity was set to Arg‐C, allowing for up to two missed cleavages.
For the identification of contaminating proteins, histone variants and hPTMs, a search was performed in a reduced search space (detailed description in Supporting Information) with the following search parameters: enzyme specificity was set to Arg‐C because lysines are blocked by propionylation, tolerating up to two missed cleavages with a peptide tolerance of 15 ppm and an MS/MS tolerance of 0.3 Da. As variable modifications propionylation, butyrylation, acetylation, dimethylation, and trimethylation on lysine, monomethylation and dimethylation on arginine, acetylation on serine and on threonine were selected. Propionylation of the N‐terminus was set as a fixed modification. Monomethylation of lysine was searched as butyrylation, which equals the sum of the masses for propionylation and monomethylation, because the Ɛ‐amino group of monomethylated lysines will be propionylated. Propionylation and butyrylation were interpreted as unmodified residues and monomethylation, respectively, only if allowed by the snapshot by Huang et al. 25. Decoy search was set to randomized. Scores were adjusted by the Percolator algorithm 26, resulting in a protein FDR of <0.01.
A list of the proteins identified in the dataset together with their relative quantification based on unique peptides was exported from Progenesis QIP for evaluation of the contaminating, coextracted proteins (Supporting Information Table 1). In the radar charts, relative abundance (RA) is visualized. Averages are given of ≥6 independent technical replicates of each protocol. Scatter plots and bar graphs were generated using GraphPad Prism 5, the violin plot using R with the ggplot2 package and the other graphics using Progenesis QIP 2.0.
3. Results and discussion
Sample preparation is increasingly recognized to be of critical importance for the overall accuracy and precision of a label‐free MS quantification experiment 7, 27. Here, we evaluate different (adapted) histone extraction protocols (Fig. 1) for their quality and repeatability in that context. Protocol A uses a hypotonic lysis buffer for nuclei isolation prior to acid extraction, and is the reference protocol by Shechter et al. to which the other protocols are compared 14. Protocol B omits the nuclei isolation step and aims at a direct acid extraction of histones from the whole cell pellet, representing the shortest protocol 14, 15. This equally minimizes the time frame wherein residual activity of epigenetic mediators can change the histone fingerprint because, to the best of our knowledge, no cocktail exists to date that can simultaneously inhibit all writers and readers. Protocol C uses the acid‐precipitating SDC as detergent for nuclei isolation, followed by acid extraction 13, 19, 20, 21. For SDS‐PAGE analysis, all protocols were tested with and without snap freezing of the cell pellet after cell harvest (resulting in six protocols), on two different cell lines (Jurkat and hESC). For label‐free MS, a selection of protocols was evaluated in terms of quality and repeatability. Together, this leads to an overview of the protocols evaluated regarding cost, purity, yield, and repeatability.
Figure 1.
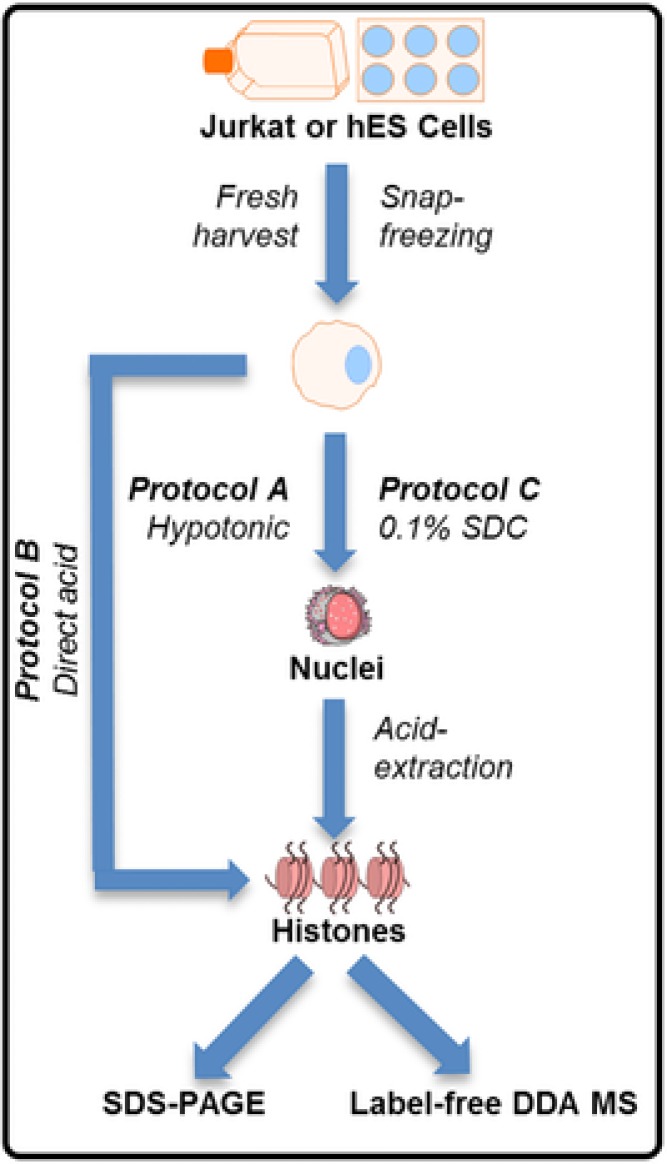
Overview of the different histone extraction protocols, highlighting the differences between Protocols A, B, and C.
3.1. Purity evaluation of the different extraction protocols using SDS‐PAGE
Before comparison of the different protocols, a preliminary experiment was performed for optimization of the common part of the extraction protocol. Different incubation times of the acid extraction step (30 min, 2 h, 4 h, overnight) and of the trichloroacetic acid precipitation step (30 min versus overnight), as well as different acid concentrations (0.2, 0.8 N) different concentrations of cells (cells/μL), and acids utilized (HCl, H2SO4) were examined. We conclude that none of these considerably affect the quality or yield of the extracts (representative gel image in Supporting Information Fig. 2).
Next, histone extracts from 106 cells, performed in independently processed technical triplicates, were first visualized using SDS‐PAGE and their purity ratios calculated. Bovine histones, commercially available pure histone standards, were included as additional references for purity.
Figure 2A clearly indicates differences in purity between the protocols tested. These differences tend to be cell‐line dependent, although this cannot be concluded solely based on two cell lines. Yet, even changing the state of a single cell line induces considerable changes in the efficiency of histone extraction. For example, Protocol B‐frozen has a marked reduced efficiency when applied to stem cells after they have been made naïve over a 2‐week period (data not shown). Here, all protocols, except Protocol B‐fresh, display a comparable purity of about 70% for Jurkat cells, whereas for hESC, a larger difference can be noted: only protocols making use of the detergent SDC for fresh cells (Protocol C‐fresh) or protocols including a snap‐freezing step (or a combination thereof) display acceptable purity ratios. Thus, this suggests that extraction protocols need to compared for each individual experiment.
Figure 2.
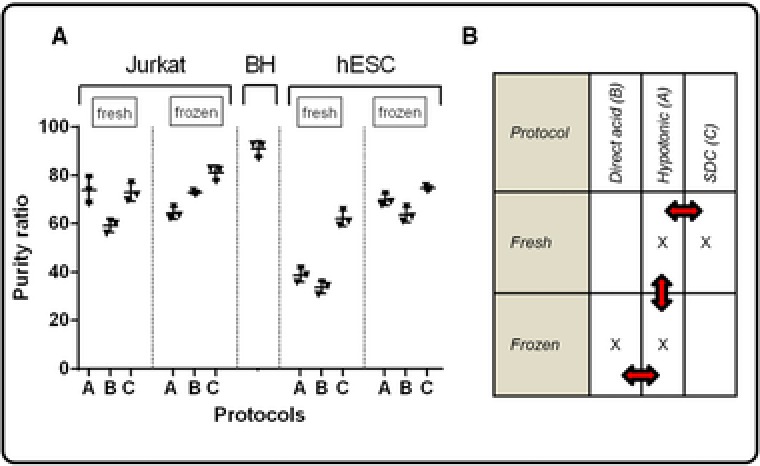
Extraction efficiency evaluation of the different protocols using SDS‐PAGE based purity ratios. (A) Purity of histone extracts prepared using Protocols A, B, and C on fresh and snap‐frozen cells for Jurkat cells and hESC, as compared to bovine histones, a commercial extract obtained after extensive fractionation. Independent technical replicates n = 3, mean ± SD. Replicates were prepared in different tubes at different time points by the same person. (B) Four protocols were selected for subsequent MS analysis and performed on Jurkat cells.
3.2. Evaluation of quality and repeatability using label‐free MS
Based on the SDS‐PAGE analysis, Protocols A‐fresh, C‐fresh, A‐frozen and B‐frozen (Fig. 2B) were repeated on Jurkat cells in eight independent technical replicates to verify whether these protocols yield comparable results during a label‐free MS analysis. All samples were measured in a randomized sample list interspersed by QC samples, which are pooled samples derived from all of the samples in the experiment. These fulfill a dual role in that they (i) serve as precursor alignment templates and (ii) can assess the overall variation of the data acquisition and data analysis platform (independently of the variation in extraction) as they are periodically run throughout the study. A CV of peptide and/or protein level variations is commonly calculated across all qualitatively identified proteins within the QC sample to assess system variability 7. The median CV of the identified peptides across the different QC injections was 9.7%, and 95.8% of the peptides had a CV <20% (technical replicates of injection, n = 8) 7. In total 6539 multiply charged (2+ to 6+) MS precursors were detected and each aligned for 90% against the QC (Progenesis QIP, Waters).
First, we performed an untargeted, first‐quantify‐then‐identify analysis to verify clustering (and thus repeatability) of the experimental conditions and to check for outliers without prior knowledge of these features’ identity. This was done under the form of an “MS‐precursor intensity‐based” PCA (Progenesis QIP) 22. Using this PCA three of 32 extractions appeared to have failed and these outliers were removed. Figure 3A shows that the multiply‐charged MS precursors aggregate the four protocols into two clusters along PC 1, explaining 18% of the variation: detergent‐based (Protocol C‐fresh, Cluster 1) versus detergent‐free (Protocols A‐fresh/frozen and B‐frozen, Cluster 2). Because samples were run in a randomized fashion and no major differences in total sample load (and yield) were found (normalization factors of 0.8 to 1.3 throughout the experiment), we extracted the MS/MS spectra of the differential (q‐value < 10−6) features that cause the experiments to “cluster” in the PCA. After assuring that these features are not multiply charged “nonpeptide features” (chemical noise, PEG) (data not shown), we performed an error tolerant search in Mascot that allows to determine if unanticipated modifications on the peptides introduced by the protocols actually cluster the experiments at the feature level, because this would not show up in a differential protein analysis. No considerable difference in modification prevalence was apparent from the Modification Statistics (Mascot). The difference between Clusters 1 and 2 could thus only be explained by the fact that different proteins are being extracted, giving rise to differential peptides. Therefore, we next identified the coextracted, contaminating proteins using a conventional search. This indicated that, for example ribosomal proteins are enriched in protocols that do not use SDC, probably due to a more efficient nuclear isolation in Protocol C‐fresh (for a list of all proteins identified and quantified in each protocol, see Supporting Information Table 1). Note however, that direct acid extraction and hypotonic nuclear isolation do cluster, implying that nuclear isolation can be omitted in this case.
Figure 3.
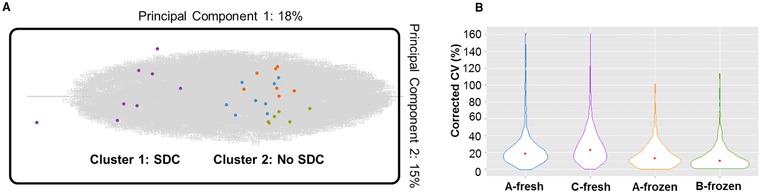
Feature‐level based analysis of the different protocols. (A) Based on MS precursor intensities (gray cloud), a PCA was performed resulting in the clustering of Protocol C‐fresh (Cluster 1: SDC) versus Protocols A‐fresh, A‐frozen and B‐frozen (Cluster 2: No SDC) along PC 1. Color code: A‐fresh (blue), C‐fresh (purple), A‐frozen (orange), B‐frozen (green). (B) Distribution of corrected CV values obtained for independent technical replicates (n = 6–8) shown as violin plots. The median CV is depicted by a red dot.
Next, we determined the variance (after subtraction of the system variability defined above) within the protocols by calculating the CV across the signal intensities of the identified peptides in the protocol replicates. The results are given in a frequency violin plot in Fig. 3B, showing that the frozen protocols are less variable. This is in line with the PCA, where the dispersion of the independent technical replicates of Protocols C and A‐fresh is wider.
3.3. Label‐free quantification of histones and their PTMs in the different protocols
We evaluated whether the protocols differentially deplete or enrich certain histone variants or hPTMs, either physically, by unanticipated chemical reaction or because of interference with other, co‐extracted peptides.
Overall, based on total ion intensity of histone‐derived peptides, a small but insignificant increased yield of histones could be detected in Protocol C‐fresh and A‐fresh (data not shown). At the protein level 26 histone variants could be quantified based on unique peptides only. Eight variants were significantly different between the selected protocols at the 0.01 significance level (ANOVA q‐value), but only two showed a maximum fold enrichment >2, Histone H1.1 and macroH2A (Fig. 4A). These results indicate subtle yet statistically significant changes in extraction efficiencies of the various histone variants, especially in those histones that are more difficult to extract: H1 is only quantitatively extracted using perchloric acid, and macroH2A has a very different and larger sequence than most histones and is also less acid extractable.
Figure 4.
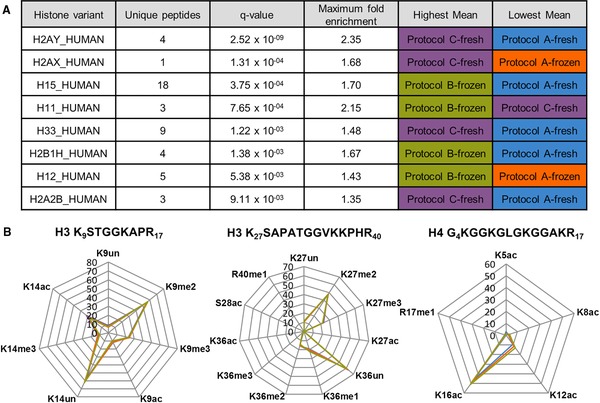
Label‐free quantification of histone variants and hPTMs. (A) Table depicting significantly different extracted histone variants (q‐value < 0.01) between the four protocols. Note that H2AX_HUMAN was only quantified by one unique peptide. (B) Radar charts representing the average RA of targeted hPTMs on two H3 peptides and one H4 peptide. Each targeted hPTM is located on one angle of the radar chart and each protocol is represented by a different color A‐fresh (blue), C‐fresh (purple), A‐frozen (orange), B‐frozen (green) whereby a RA of 0 is located in the center, ascending outwards. RAs and their SDs can be consulted in Supporting Information Table 2. K, lysine; R, arginine; S, serine; un, unmodified; me1, monomethylation; me2, dimethylation; me3, trimethylation; ac, acetylation; ph, phosphorylation
Then, we quantified the RA of 23 different hPTMs on three different peptides derived from H3 and H4 (Fig. 4B). RA is the standard measure for relatively quantifying changes of histone marks between two or more samples and is calculated as follows 28:
The radar charts in Fig. 4B show that the RA for all the modified forms of all the hPTMs of interest overlap well between the different protocols (numbers can be consulted in Supporting Information Table 2). This implies that: (i) all protocols are robust enough to mine biology by RA; (ii) none of the protocols chemically alters the biological fingerprint of the histones to a detectable extent, or they all do so to a similar degree; (iii) none of the protocols introduce any lasting chemical contamination that leads to ion suppression; (iv) none of the sample compositions seems to differ enough from the others in that chimericy increases to an extent reflected in RA measurements, by for example different amounts of coeluting peptides from other proteins. Thus, when using RA as a measure to map changes in the histone epigenetic signature, minimizing sample handling during histone extraction in Jurkat cells can be done and has several advantages. Still, because cell state (nucleus/cytoplasm ratio, cytoskeletal robustness, etc.) has a large impact on the outcome, Table 1 provides the reader with an overview of the main characteristics of each protocol as examined in this manuscript.
Table 1.
Comparison of the performance of the histone extraction protocols investigated by label‐free MS analysis
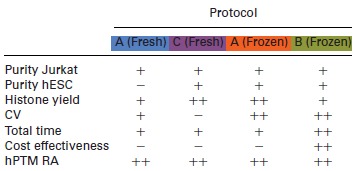
|
Timings range from 3 h (++) to 4–5 h (+).
4. Concluding remarks
Histone post‐translational modifications play a crucial role in chromatin remodeling. Dynamic changes of these histone marks are increasingly being quantified using label‐free MS, for which robust sample preparation is imperative, especially to study small biological changes. However, current histone extraction protocols, the first step in a label‐free MS workflow, are not specifically designed for label‐free MS.
Here, an established histone extraction technique and adjusted protocols with variations that focus on label‐free quantification were verified for label‐free MS applicability. Depending on the cell type under investigation, at the cost of some additional contaminating proteins, minimizing sample handling can be done during histone isolation. This results in reduced technical variation, minimizes the time frame wherein residual activity of epigenetic mediators can change the histone fingerprint and allows processing larger sample batches. In general, RA as a measure for hPTM quantification is robust enough to compensate for these small changes and based on the cell type under investigation and the sensitivity and peak capacity of the instrumentation, each lab should first select and compare the best suited protocol for maximum performance.
The authors have declared no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary Table 1. List of all identified proteins and their relative quantification for the different protocols.
Supplementary Table 2. Detailed data supporting Figure 4B.
Supporting Information
Acknowledgments
This study was mainly supported by BOF Mandate 01D23313, awarded to E.G. and by FWO Project grant G073112N. This study was further (partially) financed by GOA‐project 01G01112 (E.S.), IWT Mandates SB‐11179 (P.M.) and SB‐141209 (L.D.), and FWO grant G013916N and Mandate 12E9716N (M.D.). The authors are grateful to Sofie Vande Casteele and Yens Jackers for their excellent technical assistance.
5 References
- 1. Campos, E. I. , Reinberg, D. , Histones: annotating chromatin. Annu. Rev. Genet. 2009, 43, 559–599. [DOI] [PubMed] [Google Scholar]
- 2. Kouzarides, T. , Chromatin modifications and their function. Cell 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- 3. Britton, L. M. , Gonzales‐Cope, M. , Zee, B. M. , Garcia, B. A. , Breaking the histone code with quantitative mass spectrometry. Expert Rev. Proteomics 2011, 8, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang, H. , Lin, S. , Garcia, B. a. , Zhao, Y. , Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015, 115, 2376–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Soldi, M. , Cuomo, A. , Bremang, M. , Bonaldi, T. , Mass spectrometry‐based proteomics for the analysis of chromatin structure and dynamics. Int. J. Mol. Sci. 2013, 14, 5402–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sidoli, S. , Cheng, L. , Jensen, O.N. , Proteomics in chromatin biology and epigenetics: Elucidation of post‐translational modifications of histone proteins by mass spectrometry. J. Proteomics 2012, 75, 3419–3433. [DOI] [PubMed] [Google Scholar]
- 7. Soderblom, E. J. , Thompson, J. W. , Moseley, M. A. , in: Eyers C. E., Gaskell S. J. (Eds.), Quanitative. Proteomics, Vol. 1, The Royal Society of Chemistry, Cambridge, UK: 2014, pp. 131–153. [Google Scholar]
- 8. Bantscheff, M. , Lemeer, S. , Savitski, M. M. , Kuster, B. , Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [DOI] [PubMed] [Google Scholar]
- 9. Bantscheff, M. , Schirle, M. , Sweetman, G. , Rick, J. , Kuster, B. , Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [DOI] [PubMed] [Google Scholar]
- 10. Beck, H. C. , Nielsen, E. C. , Matthiesen, R. , Jensen, L. H. et al., Quantitative proteomic analysis of post‐translational modifications of human histones. Mol. Cell. Proteomics 2006, 5, 1314–1325. [DOI] [PubMed] [Google Scholar]
- 11. Bloom, K. S. , Anderson, J. N. , Fractionation and characterization of chromosomal proteins by the hydroxyapatite dissociation method. J. Biol. Chem. 1978, 253, 4446–4450. [PubMed] [Google Scholar]
- 12. Boyne, M. T. , Pesavento, J. J. , Mizzen, C. a. , Kelleher, N. L. , Precise characterization of human histories in the H2A gene family by top down mass spectrometry. J. Proteome Res. 2006, 5, 248–253. [DOI] [PubMed] [Google Scholar]
- 13. Lin, S. , Garcia, B. A. , Examining histone posttranslational modification patterns by high‐resolution mass spectrometry. Methods Enzymol. 2012, 512, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shechter, D. , Dormann, H. L. , Allis, C. D. , Hake, S. B. , Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [DOI] [PubMed] [Google Scholar]
- 15. Jufvas, A. , Stralfors, P. , Vener, A. V , Histone variants and their post‐translational modifications in primary human fat cells. PLoS One 2011, 6, e15960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodriguez‐Collazo, P. , Leuba, S. H. , Zlatanova, J. , Robust methods for purification of histones from cultured mammalian cells with the preservation of their native modifications. Nucleic Acids Res. 2009, 37, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vossaert, L. , Meert, P. , Scheerlinck, E. , Glibert, P. et al., Identification of histone H3 clipping activity in human embryonic stem cells. Stem Cell Res. 2014, 13, 123–134. [DOI] [PubMed] [Google Scholar]
- 18. Galasinski, S. C. , Resing, K. a. , Ahn, N. G. , Protein mass analysis of histones. Methods 2003, 31, 3–11. [DOI] [PubMed] [Google Scholar]
- 19. Griffin, M. J. , Cox, R. P. , Grujic, N. , A chemical method for the isolation of HeLa cell nuclei and the nuclear localization of HeLa cell alkaline phosphatase. J. Cell Biol. 1967, 33, 200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kirschner, R. H. , Rusli, M. , Martin, T. E. , Characterization of the nuclear envelope, pore complexes, and dense lamina of mouse liver nuclei by high resolution scanning electron microscopy. J. Cell Biol. 1977, 72, 118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin, Y. , Zhou, J. , Bi, D. , Chen, P. et al., Sodium‐deoxycholate‐assisted tryptic digestion and identification of proteolytically resistant proteins. Anal Biochem 2008, 377, 259–266. [DOI] [PubMed] [Google Scholar]
- 22. Meert, P. , Govaert, E. , Scheerlinck, E. , Dhaenens, M. , Deforce, D. , Pitfalls in histone propionylation during bottom‐up mass spectrometry analysis. Proteomics 2015, 15, 2966–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glibert, P. , Van Steendam, K. , Dhaenens, M. , Deforce, D. , iTRAQ as a method for optimization: enhancing peptide recovery after gel fractionation. Proteomics 2014, 14, 680–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Helm, D. , Vissers, J. P. , Hughes, C. J. , Hahne, H. et al., Ion mobility tandem mass spectrometry enhances performance of bottom‐up proteomics. Mol. Cell. Proteomics 2014, 13, 3709–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang, H. , Sabari, B. R. , Garcia, B. A. , Allis, C. D. , Zhao, Y. , SnapShot: Histone Modifications. Cell 2014, 159, 458–458.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brosch, M. , Yu, L. , Hubbard, T. , Choudhary, J. , Accurate and sensitive peptide identification with Mascot Percolator. J. Proteome Res. 2009, 8, 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scheerlinck, E. , Dhaenens, M. , Van Soom, A. , Peelman, L. et al., Minimizing technical variation during sample preparation prior to label‐free quantitative mass spectrometry. Anal. Biochem. 2015, 490, 14–19. [DOI] [PubMed] [Google Scholar]
- 28. Plazas‐Mayorca, M. D. , Zee, B. M. , Young, N. L. , Fingerman, I. M. et al., One‐pot shotgun quantitative mass spectrometry characterization of histones. J. Proteome Res. 2009, 8, 5367–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary Table 1. List of all identified proteins and their relative quantification for the different protocols.
Supplementary Table 2. Detailed data supporting Figure 4B.
Supporting Information