

Abstract
Major bottlenecks in development of therapeutic post transcriptional gene silencing (PTGS) agents (e.g. ribozymes, RNA interference, antisense) include the challenge of mapping rare accessible regions of the mRNA target that are open for annealing and cleavage, testing and optimization of agents in human cells to identify lead agents, testing for cellular toxicity, and preclinical evaluation in appropriate animal models of disease. Methods for rapid and reliable cellular testing of PTGS agents are needed to identify potent lead candidates for optimization. Our goal was to develop a means of rapid assessment of many RNA agents to identify a lead candidate for a given mRNA associated with a disease state. We developed a rapid human cell-based screening platform to test efficacy of hammerhead ribozyme (hhRz) or RNA interference (RNAi) constructs, using a model retinal degeneration target, human rod opsin (RHO) mRNA. The focus is on RNA Drug Discovery for diverse retinal degeneration targets.
To validate the approach, candidate hhRzs were tested against NUH↓ cleavage sites (N=G,C,A,U; H=C,A,U) within the target mRNA of secreted alkaline phosphatase (SEAP), a model gene expression reporter, based upon in silico predictions of mRNA accessibility. HhRzs were embedded in a larger stable adenoviral VAI RNA scaffold for high cellular expression, cytoplasmic trafficking, and stability. Most hhRz expression plasmids exerted statistically significant knockdown of extracellular SEAP enzyme activity when readily assayed by a fluorescence enzyme assay intended for high throughput screening (HTS). Kinetics of PTGS knockdown of cellular targets is measureable in live cells with the SEAP reporter. The validated SEAP HTS platform was transposed to identify lead PTGS agents against a model hereditary retinal degeneration target, RHO mRNA. Two approaches were used to physically fuse the model retinal gene target mRNA to the SEAP reporter mRNA. The most expedient way to evaluate a large set of potential VAI-hhRz expression plasmids against diverse NUH↓ cleavage sites uses cultured human HEK293S cells stably expressing a dicistronic Target-IRES-SEAP target fusion mRNA. Broad utility of this rational RNA drug discovery approach is feasible for any ophthalmological disease-relevant mRNA targets and any disease mRNA targets in general. The approach will permit rank ordering of PTGS agents based on potency to identify a lead therapeutic compound for further optimization.
Keywords: high throughput screening, gene therapy, hammerhead ribozyme, RNAi, siRNA, shRNA, DNAzyme, RNA Drug Discovery, hereditary retinal degenerations, autosomal dominant retinitis pigmentosa
1. Introduction
Development of therapeutic nucleic-acid based PTGS agents is a challenging task as evidenced by the fact that only two agents have been FDA-approved for human clinical use during the last three decades of academic and corporate research (Anderson et al., 1996; Jabs et al., 2002; Merki et al., 2008). The dense secondary and tertiary structures of the target mRNA, RNA-protein association, and expected molecular dynamics severely restrict the regions that are accessible to essential second-order annealing reactions with smaller PTGS ligands in trans (Sullivan et al., 2008). Additional biocomplexity results from cellular compartmentalization of the target mRNA, on both gross and fine scales, which promotes spatial and temporal distributions of target mRNAs within the cell. To be effective the PTGS agent must journey through the same cellular locales and have residence, stability, and kinetic performance that overlaps with the target mRNA lifetime in each spatial compartment. However, these challenges due to biocomplexity can be addressed by new approaches that attack bottlenecks in RNA drug development (Sullivan et al., 2008, 2012). Here we developed a human cell-based screening platforms to rapidly and reliably identify lead hhRz or RNAi candidate agents (“hits”) from substantial sets of potential agents.
Both hhRz and RNAi catalyze the sequence specific cleavage of target mRNAs. HhRzs are small RNA sequences capable of enzymatic cleavage of polyribonucleotides (Vaish et al., 1998; Amarzguioui and Pyrdz, 1998; Hauswirth and Lewin, 2000; Lewin and Hauswirth, 2001; Sullivan et al., 2011). Originally discovered as intramolecular self-cleaving (cis) sequences in self-replicating plant viroids (Flores et al. 2012), the hhRz consists of three helices surrounding an evolutionarily conserved catalytic core. Trans-cleaving hhRzs are readily constructed by embedding the hhRz core enzyme into a target annealing sequence which gives the unimolecular RNA the capacity for both molecular recognition and enzymatic cleavage of an independent target mRNA. The target molecular recognition arms of the trans hhRz are designed to provide antisense complementarity (Watson-Crick) to a defined accessible region of an independent target mRNA (Uhlenbeck, 1987). After 2nd-order collision-based interaction and kissing complex formation, full annealing with the target may occur over the antisense spans to form a complete hhRz: target hybrid structure. This hybrid undergoes conformational changes to align specific bases within the RNA enzyme core that mediate proton transfer chemistry and accelerate target mRNA cleavage at a specific phosphodiester bond. The trans design strategy allows for potential realization of hhRzs that possess potent sequence-specific endoribonuclease activity against any given target RNA (Haseloff and Gerlach, 1988). HhRz target motifs are NUH↓ triplets, where N is any nucleotide (nt), U is a uridine, and H can be any nucleotide except guanosine (Perriman et al., 1992; Ruffner et al., 1990; Zoumadakis and Tabler, 1995; Birikh et al., 1997). Given this versatility, any moderately sized mRNA target has numerous potential NUH↓ cleavage sites. For example, in SEAP mRNA (1777 nt) there are a total of 180 NUH↓ cleavage sites and in the dominant polyadenylated form of human RHO mRNA there are 236 potential NUH↓ cleavage sites. Similarly, RNAi agents can be designed for cellular expression as short hairpin RNAs (shRNA). These are processed intracellularly by Drosher and Dicer into short-interfering RNAs (siRNA) that associate as guide sequences within the RNA-induced silencing complex (RISC), built upon the endonuclease Ago2, which anneal with the target mRNA and drive cleavage by protein-mediated catalysis (Brummelkamp et al., 2002; Rossi, 2008). While this might seem to make PTGS therapeutics a straightforward endeavor, in live cells most potential NUH↓ cleavage sites and RNAi target sites within any mRNA are inaccessible due to strong secondary and tertiary structures and protein binding (Amarzguioui et al., 2000; Brown et al., 2005; Lima et al., 1992; Patzel and Sczakiel, 1998; Patzel et al., 1999; Scherr and Rossi, 1998; Scherr et al., 2000). Identifying the optimum site for targeting is a daunting task, yet critical for successful RNA drug discovery.
We employ a mutation-independent approach to hhRz development for RNA Drug Discovery for autosomal dominant retinal degenerations (Montgomery and Dietz, 1997; Millington-Ward et al., 1997; Sullivan et al., 2002; Farrar et al., 2002; Gorbatyuk et al., 2005, 2007; Sullivan et al., 2011). In this approach one works to identify the most potent hhRz or shRNA that can maximally suppress a given disease target mRNA and protein. In the context of a genetic disease such as an autosomal dominant retinitis pigmentosa, a mutation independent hhRz will suppress not only the mutant mRNA but also the WT mRNA. Such a single therapeutic agent would be expected to be useful for treatment of most if not all of the known mutations in a given gene as the optimum targeting location within the mRNA is likely to harbor relatively few, if any, random mutations. Prevention of haploinsufficiency due to suppression of the intrinsic WT mRNA is achieved in a combined gene therapy paradigm in which the knockdown hhRz agent is expressed in concert with an allelic variant of the WT target which transcribes a “hardened” WT mRNA which cannot be cleaved by the potent therapeutic agent yet encodes the WT protein (Sullivan et al., 2011; Millington-Ward et al., 1997; Gorbatyuk et al., 2007).
The rationale for this study is that efficient and timely realization of potent lead candidates in the RNA drug discovery process requires initial approaches to identify regions of accessibility in a target mRNA, and the means to rapidly screen the efficacy (potency) of sets of agents designed against accessible regions and (control) inaccessible regions in live cells in order to identify the lead candidate on the basis of rank-ordered activity. In this study we addressed both issues and exploited the SEAP reporter protein to establish a platform for rapid and reliable assessment of the efficacy of trans-cleaving hhRz and shRNA constructs. The stable SEAP reporter protein is secreted into culture medium in proportion to its steady-state intracellular mRNA levels, which makes it an ideal “model” target mRNA to assay the immediate and long term kinetic impact of PTGS agents on gene expression in live cell cultures (Berger et al., 1988). We first developed a HTS fluorescence plate assay for secreted SEAP enzyme and used computational RNA folding algorithms to map SEAP mRNA accessibility to guide hhRz design at regions expected to be accessible or inaccessible. Accessibility predictions led to successful identification of lead hhRz and shRNA expression constructs that knockdown SEAP mRNA and protein. PTGS lead optimization is also feasible on the SEAP HTS platform. We demonstrated that the SEAP reporter in a HTS screening platform can be used for PTGS development against arbitrary mRNAs by embedding the SEAP cDNA into two expression constructs that contain full or partial elements of a validated retinal disease target mRNA (human RHO). In this report we describe in detail the methodological approach used to conduct the HTS for ribozyme or shRNA agents. We demonstrate the identity of a new strong lead hhRz agent against human RHO mRNA (725 GUC↓) that is now being subjected to rational optimizations, and a potent shRNA against RHO against this same accessible region of RHO mRNA. In a subsequent report we will demonstrate how this HTS approach was used to screen human RHO and the early efforts to optimize these candidate gene therapeutics. Critically, the SEAP-based HTS approach is modular and useful for lead PTGS agent identification and optimization to any disease mRNA target.
2. Materials
2.1 Oligonucleotides
Oligodeoxynucleotides for hhRz or shRNA cDNAs were synthesized by Sigma GenoSys (Houston, TX, USA) or Integrated DNA Technologies (Coralville, IA, USA), annealed, and phosphorylated using T4 polynucleotide kinase (New England Biolabs, Ipswich, MA, USA) prior to T4 ligase-mediated ligation into linearized vector by a highly optimized positive selection approach (Abdelmaksoud et al., 2009). All constructs achieved were proven by DNA sequence determination.
3. Detailed Methods
3.1 Computational methods of RNA Secondary Structure Prediction
The secondary structure of the full-length SEAP mRNA transcribed from the SV40 early promoter and enhancer in the pSEAP2-control plasmid (Clontech Laboratories Inc., Mountain View, CA, USA) was analyzed, using both free energy minimization (MFold algorithm) (Zuker, 2003) (http://mfold.rna.albany.edu/?=mfold/download-mfold) and a Boltzmann-weighted sampling of all sub-structures (SFold algorithm) (Ding et al., 2004) (http://sfold.wadsworth.org/cgi-bin/index.pl). The use of multiple algorithms is expected to allow more robust identification of accessible single-stranded regions in target RNA structures (Sullivan et al., 2008; Abdelmaksoud et al., 2009; Shao et al., 2007). Using MFold, SEAP target mRNA was folded in 200 nucleotide windows with 100 nucleotide overlapping steps, and a range of structures (maximum = 20) not exceeding 20% deviation from the minimal free energy structure were generated for each window (Scherr and Rossi, 1998; Scherr et al., 2000). The total ensemble of structures was analyzed visually in the printed pictorial output for single-stranded bulges or loops greater than or equal to 7 nt that also contained potential hhRz target sites (NUH↓). The frequencies of such single-stranded structures were calculated. Mfold first identifies the minimum free energy (MFE) structure and then inclusively displays and analyzes a set of less stable structures within a certain user-specified energetic and structural difference range (neighborhood) of the MFE structure. Statistical estimates of accessibility in such an ensemble sampling an important but only local neighborhood of folding are therefore biased but are, nonetheless, a proven useful approach to identify regions able to support hhRz knockdown (e.g. Abdelmaksoud et al., 2009). For MFold we use a “frequency of occurrence” to estimate accessibility, which is not a true probability of occurrence, however. SEAP mRNA was also analyzed with SFold which, in contrast to MFold, samples the entire conformational space (astronomical in size and proportional to 4N where N is the number of nts). SFold output is therefore an unbiased estimate of the probability of being single stranded along the mRNA. In addition to the individual outputs from these two algorithms, we also averaged the single-stranded probability maps around each chosen attack site across obtained from both computational platforms. We obtained a range of estimated access probabilities along the SEAP target mRNA for the proof-of-principle design and testing of hhRzs against this target. Finally, we folded the SEAP mRNA with RNA-Structure (http://rna.urmc.rochester.edu/RNAstructureWeb/) and then used OligoWalk to obtain a map of the local folding energy along the transcript which was then normalized into an additional probability estimator of accessibility (Mathews et al., 2004; Mathews, 2006).
3.2. Plasmid Constructions and Cloning
The construction of hhRz expression plasmid, pUC-VaL, the construction of the SEAP-RHO fusion RNA plasmid, the construction of the bicistronic RHO-IRES-SEAP expression plasmid, and the construction of shRNA expression plasmids all occurred by standard T4 ligase-mediated molecular approaches. Details are provided in the Supplementary Materials. Plasmid maps were generated with Clone Manager (vers 9) (Scientific and Educational Software, Denver CO). pUC-VaL expresses an engineered form of human adenoviral 2 VAI RNA (Fig. 1) in which the central domain is replaced by a stabilizing stem and engineered large stable single stranded loop (ribozyme harbor) into which the hhRz elements are ligated. pUC-VaL is a second generation form of pG-VaL which expresses an engineered adenoviral VAI scaffold RNA for embedding of hhRzs (Lieber and Strauss, 1995, Abdelmaksoud et al. 2009). The VAI-hhRz expression vectors harbor both an intrinsic RNA pol-III promoter (part of VAI gene), for cellular expression, and upstream T7 promoters for in vitro transcription. The pBlueScript plasmid (pShort-RHO) that is used to transcribe an element of the human RHO mRNA for in vitro hhRz cleavage assays has been previously described (Sullivan et al., 2002). In this construct a restriction fragment (450 bp) of the WT human RHO cDNA is cloned downstream of the T7 promoter in pBlueScriptII-SK and transcribes a 510 nt RNA (containing some peripheral vector sequences 5′ and 3′). Details on the construction of the RHO RNA fusion plasmids are described in Supplementary Methods. The in cellulo expression plasmid for full length human RHO mRNA (pRHO-fix5UT) placed the human RHO cDNA (true transcription start to 32 nt beyond first dominant polyA signal) immediately downstream of the CMV promoter transcription start site in an engineered pCDNA3.1(+)-Hyg plasmid (InVitrogen) that ensures cellular transcription of a native human full length human RHO mRNA with only 6 nt of excess vector sequence at the 5′ end (Bgl II ligation site at CMV transcription start), which is not expected to affect mRNA folding.
Figure 1. Secondary Structures of Chimeric VAI RNA and Ribozymes.
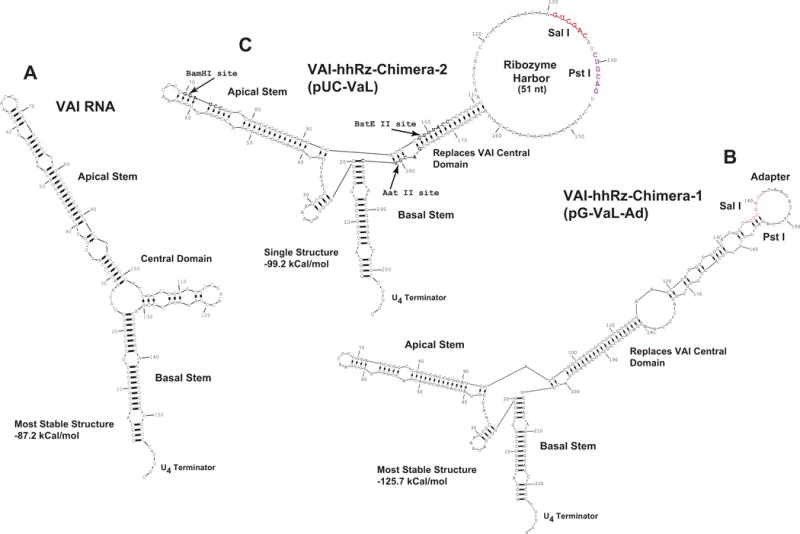
(A) The secondary structure of wild-type adenoviral VAI RNA in the most stable predicted state. (B) Expected secondary structure of the VAI-hhRz chimera as designed by Lieber and Strauss (1995). VAI RNA is transcribed in high levels (by RNA pol III) from an intragenic promoter (boxes A and B, not shown) and exported into the cytoplasm in mammalian cells. High transcription rate, export to cytoplasm, and long half-life make the VAI RNA an ideal chimeric carrier for ribozymes. Wild type adenoviral VAI RNA normally binds to and inhibits PKR, a cellular protein involved in the interferon response, but the inhibition is obviated by engineering of the central domain. In the VAI-hhRz chimera, the natural central domain of VAI is interrupted by a large stem-loop structure to inhibit the action of VAI on PKR (Lieber and Strauss, 1995). The expected most stable secondary structure of the VAI–Ad construct is shown into which hhRz cDNA constructs are ligated between the Sal I and Pst I restriction sites. (C) In the RNA of the pUC-VaL construct hhRz cDNAs are ligated to put the hhRz within the large loop (hhRz harbor) between the Sal I and Pst I restriction sites. The hhRz harbor is designed with the intent to allow the hhRz to flexibly interact with accessible regions of its target mRNA without interfering secondary structural interactions with the main body of the VAI scaffold RNA.
Our highly efficient evolutionary selection approach to ligate small hhRz (or shRNA) cDNAs into plasmids is also described in Supplementary Materials. Ribozyme cDNA constructs were directionally ligated between the Sal I and Pst I restriction sites in pUC-VAL, which places hhRz sequences within the engineered large central loop harbor of the VAI scaffold. Antisense substrate-binding arms were designed to direct hhRzs to target annealing sites in SEAP mRNA or in human RHO mRNA (Table 1). The expected secondary structures of the hhRzs used in this study are shown (Fig. 2) (NUH↓ sites are shown italicized and the number of the site refers to the nt (“H”) preceding the cleaved phosphodiester bond, as indicated by a vertical arrow (↓).
Table 1. Ribozyme Targeting sites in SEAP Human RHO mRNA for hhRzs and shRNAs.
The targeting sequences for hhRzs used in this study against SEAP and human RHO mRNAs are shown. The NUH↓ site in the target mRNA is bolded in all cases.
|
SEAP hhRz Target Sequences
| |
|---|---|
| Site | Target sequence in SEAP mRNA |
| 150 | 5′-UCCCAGUUGAGGAGG-3′ |
| 246 | 5′-AGAACCUCAUCAUCU-3′ |
| 800 | 5′-GAUGACUACAGCCAA-3′ |
| 965 | 5′-AUGAAAUACGAGAUC-3′ |
| 1260 | 5′-GCUCCAUCUUCGGGC-3′ |
| 1654 | 5′-AUAAGAUACAUUGAU-3′ |
|
RHO hhRz Target Sequences
| |
|---|---|
| Site | Target sequence in RHO mRNA |
| 266 | 5′-ACUUCCUCACGCUCU-3′ |
| 725 | 5′-UCGUGGUCCACUUCA-3′ |
Figure 2. hhRz Secondary Structures and Lead RHO hhRz cDNA and RNA.
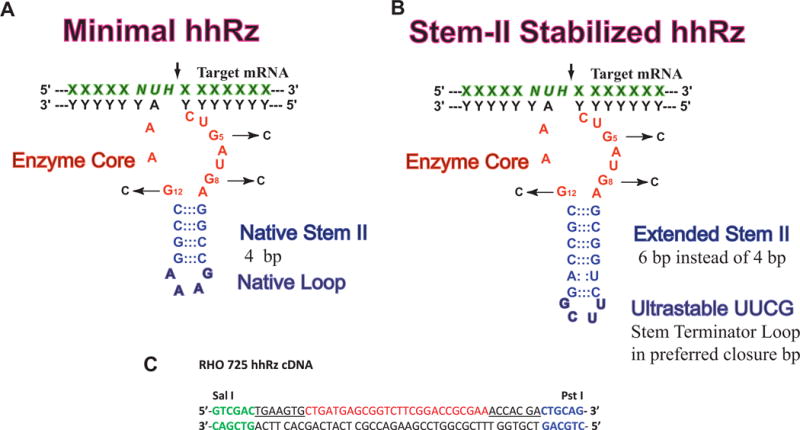
(A) Expected secondary structure of the 4 bp Stem II hhRzs tested in this study design. The antisense flanks anneal to a target region by Watson Crick base pairing to position an NUH↓ cleavage site relative to the enzyme core. Mutations in the enzyme core that inhibit catalysis are shown (G5C, G8C, G12C). (B) The expected secondary structure of the Stem II extended hhRz with two additional bp (total of 6 bp) to stabilize hhRz folding and is capped by an ultrastable UUCG terminator loop. (C) The cDNA for a stabilized hhRz targeting the 725 GUC↓ motif in human RHO mRNA is shown. The Sal I and Pst I recognition sequences are shown. The antisense flanks for the hhRz are underlined. The enzyme core of the hhRz is in red.
3.3. Cell Culture and Transfection
The HEK293S cell line was obtained directly from Dr. Bruce Stillman (Stillman and Gluzman, 1985), initially expanded then frozen in liquid nitrogen storage, and used in experiments in early passage number expansions (~10). These cells are used only for heterologous expression of the target mRNA and proteins within the housekeeping machinery of an easily transfected human transformed cell line in a study that does not ascribe to or require any particular differentiated functions, for example, the simulation of retinal or photoreceptor tissue. HEK293S-SEAP cells were generated by stable co-transfection of pSEAP2-Control and pTK-Hyg (Clontech, #631750) plasmids into suspension-adapted human embryonic kidney cells (HEK293S) (Stillman and Gluzman, 1985; Sullivan and Satchwell, 2000), followed by selection in hygromycin (250 μg/ml). Clonal picks were screened with the SEAP assay (see below) and cell lines with different levels of stable SEAP expression and secretion were identified. HEK293S cells were also engineered to stably express pRHO-IRES-SEAP (HEK293S-RHO-IRES-SEAP) by transfection of pRHO-IRES-SEAP (contains a neomycin resistance construct for cellular selection) followed by selection in G418 (500 μg/mL).
All HEK293S lines were maintained in Dulbecco’s Modified Eagle’s Medium/F12 nutrient/salts mix (DMEM/F12) with 10% (v/v) heat-inactivated calf serum and antibiotics (penicillin and streptomycin). For transfection of stable HEK293S-SEAP and HEK293S-RHO-IRES-SEAP lines, cells were grown in 10 cm plates (BD Falcon #35-3003), seeded into 96 well plates (BD Falcon Optilux black-walled #35-3220) and co-transfected using Lipofectamine 2000 (InVitrogen, Carlsbad, CA, USA) with 500 ng of VAI-Chimera control vector or VAI-hhRz constructs and 100 ng of pEGFP-N1 plasmid (Clontech, Mountain View, CA) per well (1.74 nM VAI constructs: 0.21 nM EGFP). The green fluorescent protein reporter plasmid, pEGFP-N1 (Clontech), was used to monitor and control for transfection efficiency. Each transfection condition was replicated in 8 wells in each experiment. Conditioned culture media was assayed for SEAP activity at 72 hours post-transfection for HEK293S-SEAP transfections. Enhanced green fluorescent protein (EGFP) fluorescence (488 max. excitation/507 max. emission) was measured on an Ascent Fluoroskan FL plate reader (Thermo Corp., Waltham, MA, USA) at 488 nm peak excitation/538 nm peak emission (filters selected: 488 ± 7 nm full width half maximum (FWHM)/538 ± 12.5 nm FWHM).
In transient transfections of plasmids expressing trans-acting PTGS agents, naïve HEK293S cells were grown in 10 cm plates and seeded into 24-well plates (BD Falcon #35-3047). Ribozyme or shRNA plasmids were co-transfected (Lipofectamine 2000) with pRHO-IRES-SEAP or pSEAP-STOP-L57RHO at a 2 μg (ribozyme or shRNA plasmid): 133 ng (target plasmid) ratio (1.4 nM: 0.04 nM). Each transfection was replicated in duplicate wells. Conditioned culture media was assayed for SEAP activity at 48 hours post-transfection, or at different time points as indicated in the legends.
3.4. SEAP Assay
SEAP is a secreted form (64 kDa) of human placental alkaline phosphatase (PLAP) (65 kDa) engineered by truncating the PLAP gene (Berger et al., 1988). SEAP has several properties that make it attractive for use as a reporter for cellular gene expression: 1) it is an engineered form (truncation of the C-terminal 24 amino acids which contains the membrane retention signal) of human PLAP that is efficiently (98.5%) secreted out of live cells (Berger et al., 1988); this configures a protein reporter assay of gene expression in live cells over time by simple sampling of culture media without cell extraction, 2) fixed time end point or time-resolved kinetic assays of SEAP expression can occur by simply sampling culture media without the need for cell extraction and while maintaining cell viability; and 3) critically, SEAP reporter protein is secreted into culture medium in proportion to its steady-state mRNA levels (Berger et al., 1988). These features make SEAP an ideal genetically encoded reporter of gene expression and intracellular mRNA dynamics, allowing for live cell measures, and in our case, assaying for the impact of PTGS agents on gene expression in live cell cultures. Alkaline phosphatase is widely used in many assays because of its high affinity for a range of substrates, a high substrate turnover number, and good enzymatic stability. A potential disadvantage of alkaline phosphatases as reporters of gene expression is endogenous cellular expression of phosphatases that create background noise in an assay. Fortunately, normally expressed only by human placental cells, PLAP possesses a number of features to decrease this background: 1) PLAP functions optimally at pH levels (pH 9.8) that inactivate other phosphatases, 2) PLAP is unaffected by the presence of 10 mM homoarginine, which efficiently inhibits other alkaline phosphatase isozymes, 3) PLAP is also not significantly affected by heating to 65°C for up to 30 minutes unlike other alkaline phosphatases (Berger et al., 1988; Stigbrand, 1984), and 4) it is highly stable in cell culture fluids. These properties allow SEAP protein secretion to stably reflect the temporal integration of SEAP mRNA transcription and translation in cells, in proportional to steady state levels. A potential downside, in stable cell lines, is that prior to transfection of PTGS expression plasmids, pre-transcribed SEAP mRNA will produce protein in translational trafficking streams before any hhRz RNA or shRNA can be made. This constrains the dynamic range of available PTGS knockdown of target to a level less than the total level of expressed target (we estimate maximum knockdown at around 50%). The loss of dynamic range is not a problem in studies used to identify lead candidates PTGS agents so long as the variance of measures is relatively low, as it is with the assays we developed. It is important to realize that the level of knockdown in such screening assays does not reflect the knockdown that can occur when naïve cells are simultaneously transfected with both PTGS and target plasmids de novo.
A fluorescence enzyme assay was used to measure SEAP reporter activity. Conditioned cell culture media (50–100 μL) from cells stably or transiently transfected with SEAP encoding vectors was transferred to separate wells in black-walled 96-well plates (BD Falcon Optilux black-walled #35-3220) and incubated at 65°C for 30 min to inactivate irrelevant phosphatases (SEAP is stable under these conditions). After cooling to room temperature, an equal volume (50–100 μL) of diethanolamine assay buffer (1M diethanolamine, pH 9.8, 1 mM MgCl2, 1 mM L-homoarginine (inhibitor of nonspecific phosphatases)) was added per well, followed by 5 μL of 4-methyl-umbelliferyl-phosphate (4-MUP) fluorescent substrate (Fluka 69607, from Sigma-Aldrich, St. Louis, MO) to a final concentration of 50 μM per well. Hydrolysis of the phosphate group of 4-MUP by SEAP enzyme converts 4-MUP into the highly fluorescent 4-methylubelliferrone (Fluka 69580) (excitation maximum: 364 nm: emission maximum: 448 nm, at pH 10.3). SEAP enzyme reaction was routinely incubated at room temperature (22°C) for 1 hr before measuring fluorescence on an Ascent Fluoroskan FL plate reader (excitation: 355 ± 19 nm FWHM; emission: 460 ± 12 nm FWHM). The assay was validated using PLAP as a standard, for both concentration and time, such that the general 1 hr screen chosen was not limited by either substrate depletion or product accumulation (see Supplementary Results, Supplementary Fig. 1). Properties of the assay and statistical assessment are reported. Given the linearity of the assay we expect that the levels of measured SEAP activity are directly proportional to the levels of secreted SEAP protein. For 96-well transfection of stable cell lines, each well was independently assayed (transfections were replicated in 8 wells). For 24-well transient transfections, each well was assayed in duplicate (transfections were replicated in duplicate wells).
3.5. RT-PCR. Real-Time Quantitative RT-PCR
Total RNA was purified from cell cultures 48 hours post-transfection (RNeasy, Qiagen, Valencia, CA). RNA was treated with TURBO DNA-free™ DNase kit (also RNase negative) (Ambion Inc., Austin, TX, USA) for 30 min at 37°C to reduce the potential for contaminating genomic or plasmid DNA and purified a second time using the RNeasy kit. Total RNA was quantified by OD260 nm measures on a NanoDrop™ spectrophotometer instrument (ND-1000, NanoDrop Products, Wilmington, DE). cDNA synthesis was performed using 400 ng of total RNA with the AffinityScript™ Reverse Transcriptase system (Stratagene, La Jolla, CA, USA) using the supplied oligo(dT) primers. Quantitative PCR for RHO was performed in a Smart Cycler II thermocycler (Cepheid Inc., Sunnyvale, CA, USA). Primers that spanned adjacent exons and a probe primer containing a fluorescent dye (6-carboxyfluorescein or 6-FAM; absorbance maximum: 495 nm, emission maximum: 520 nm) at the 5′ end and a quenching dye (BHQ1) at the 3′ end were designed using Primer Quest software (Integrated DNA Technologies, Coralville, IA). RHO primers (5′-AATTTGGAGGGCTTCTTTGCCACC-3′, 5′-AGTTGCTCATGGGCTTACACACCA-3′ and probe primer (6-FAM-5′-AAATTGCCCTGTGGTCCTTGGTGGT-3′-BHQ1) were initially tested on plasmid DNA and genomic DNA to demonstrate their specificity and sensitivity. Quantitative PCR reactions were assembled by mixing equal volumes of stock PCR primers (0.5 μM) and stock probe primer (0.25 – 0.5 μM) with 2× Amplitaq Gold® PCR master mix (Applied Biosystems, Foster City, CA, USA), (final concentrations: 15 mM Tris HCl (pH 8.05), 2.5 mM MgCl2, 50 mM KCl, 200 μM dNTPs, 0.25 μM PCR primer, 0.125–0.25 μM probe primer, 0.025 units enzyme) dispensed into 25 μl reaction tubes (Cepheid) and adding 2 μl of the 1st strand cDNA sample or plasmid cDNA standard (control). Thermocycler conditions were 94°C for 6 min followed by 45 cycles at 94°C (30 sec), 58°C (15 sec), and 72°C for 30 sec. Fluorescent intensity was measured during the 72°C extension, which showed log-linear detection of the respective cDNA over a range from 10 ag (attograms) to 20 pg (data not shown). Standard samples were analyzed in quadruplicate and 1st strand cDNA samples were analyzed in duplicate or triplicate with the software provided with the Smart Cycler II instrument (Cepheid). Endpoint RT-PCR was performed following established protocol (Supplementary Methods).
3.6. Quantitative Analysis
Statistical tests were conducted with Origin (OriginLab Corp, Northampton MA) and SPSS Statistics (SPSS Inc., Chicago, IL). Data is presented as values of the mean ± Standard Error of the Mean (SEM). Transfection experiments evaluating hhRz and shRNA knockdown vs. control plasmid were subject to one-way Analysis-of-Variance (ANOVA). If the null hypothesis was refuted (all independent variables not equal, p < 0.05) then post-hoc parametric t-tests were used to evaluate differences between the means of samples and controls or between samples. Levene’s algorithm was used to test for homogeneity of variance. Tests of normality of the data distribution were determined by Kolmogorov-Smirnov and Shapiro-Wilk tests (if p < 0.05, the data is not normally distributed).
3.7. Results
3.71. Bioinformatics Analysis and Mapping of SEAP mRNA Accessibility
We chose SEAP mRNA as a model target for PTGS development because we could directly test hhRz or shRNA constructs for suppression of SEAP protein activity in live cells in a HTS 96-well fluorescent enzyme assay without need for cellular extraction. The first step in this development was to investigate if SEAP mRNA itself was a useful target for testing PTGS agents.
Computational analysis of the full-length SEAP mRNA transcript revealed a highly ordered and dense secondary structure, where large single-stranded regions were absent (Fig. 3). This initial outcome indicated that SEAP mRNA was a challenging target for PTGS mediated suppression. Local structure was rigorously examined with MFold (see Methods) as previously described (Patzel and Sczakiel, 1998; Patzel et al., 1999; Abdelmaksoud et al., 2009). The frequency of occurrence of single stranded regions (≥ 7nt) in each ensemble (two to three) was averaged over all windows that included each candidate target region. SEAP target accessibility was also analyzed with SFold to calculate true probabilities of being single-stranded at each nt. The frequencies or actual probabilities of being single-stranded in each region from the MFold, SFold, and OligoWalk analyses, respectively, were tabulated individually (Table 2). The outcomes were also averaged between the three algorithm outputs and we also took the product which we call the multiparameter prediction of RNA accessibility (mppRNA) (Abdelmaksoud et al., 2009). We found correlation between accessibility estimates by MFold and SFold for several targets (data not shown), including SEAP and RHO, even though the algorithms sample overlapping but distinct conformational spaces. Therefore, averaging these outcomes appears reasonable. There was general consistency between predictions of accessibility by MFold and SFold. Predicted stable single-stranded accessible regions of SEAP were then examined for potential hhRz target sites (NUH↓), and a rank order of predicted accessible target sites was determined. Regions around the cleavage sites predicted by full MFold outcome survey are displayed, and the SFold maps of access probability surrounding the NUH↓ sites are shown (Fig. 4).
Figure 3. Minimal Folding Energy Structure of SEAP mRNA.
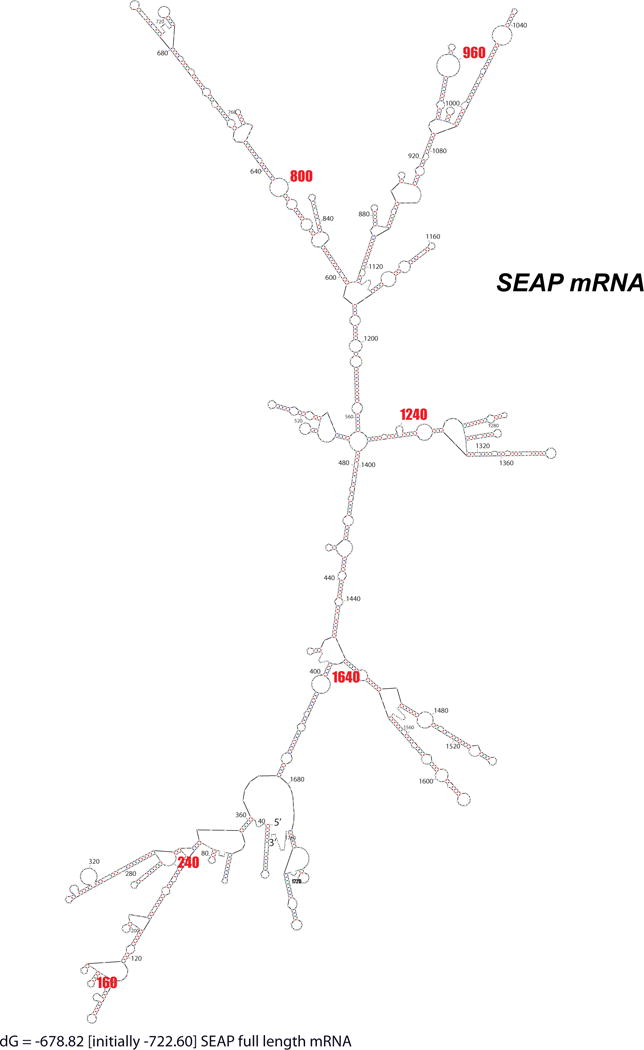
The minimal folding energy (most stable) structure of SEAP mRNA is shown. The mRNA is densely folded with few single stranded platforms available for annealing. Note this is only the most stable structure out of a huge set of potential conformations.
Table 2. Rank ordering of predicted SEAP mRNA accessibility sites.
Target hhRz sites (in bold) were chosen based on accessibility determined by rank-ordering according to average regional single-stranded frequencies and probabilities determined by MFold, SFold, and OligoWalk analyses, respectively. Four sites were chosen for their high predicted accessibility (AUA↓ 965, AUA↓ 1654, AUC↓ 1260, CUA↓ 800) and the GUU↓ 150 site chosen for its lower predicted accessibility. In addition, a site targeted by a previous study was also chosen (CUC↓ 246) (Zakharchuk et al., 1995), which we predicted to be poorly accessible. Three of the four predicted accessible sites with average probability over 50% supported hhRz knockdown. The two low probability sites (GUU↓ 150 and CUC↓ 246) did not support hhRz knockdown.
| Target Site | Single-stranded loop size (nt) | MFold Probability (%) | SFold Probability (%) | OligoWalk Probability (%) | Average Probability (%) | mppRNA Probability (%) |
|---|---|---|---|---|---|---|
| AUA 965 | 8 | 97.50 | 63.40 | 80.0 | 80.3 | 49.45 |
| AUA 1654 | 16 | 80.00 | 72.90 | 68.57 | 73.82 | 40.00 |
| CUA 800 | 8 | 50.00 | 72.40 | 100.0 | 74.13 | 36.2 |
| UUA 1707 | 7 | 30.00 | 71.90 | 82.08 | 61.33 | 17.70 |
| AUC 1260 | 9 | 80.00 | 53.50 | 37.4 | 56.97 | 16.01 |
| AUU 1709 | 7 | 30.00 | 64.90 | 62.08 | 52.33 | 12.09 |
| GUA 755 | 7 | 42.50 | 35.00 | 89.87 | 55.79 | 13.37 |
| AUU 726 | 8 | 17.50 | 55.10 | 83.38 | 51.99 | 8.04 |
| CUC 713 | 8 | 20.00 | 45.80 | 54.29 | 40.03 | 4.97 |
| GUU 150 | 7 | 10.00 | 47.90 | 79.74 | 45.88 | 3.82 |
| CUC 246 | 7 | 10.00 | 36.90 | 77.14 | 41.35 | 2.85 |
| CUU 458 | 12 | 10.00 | 32.40 | 75.58 | 39.33 | 2.45 |
Figure 4. Computational Analysis of Predicted Accessible Target Sites in SEAP mRNA.
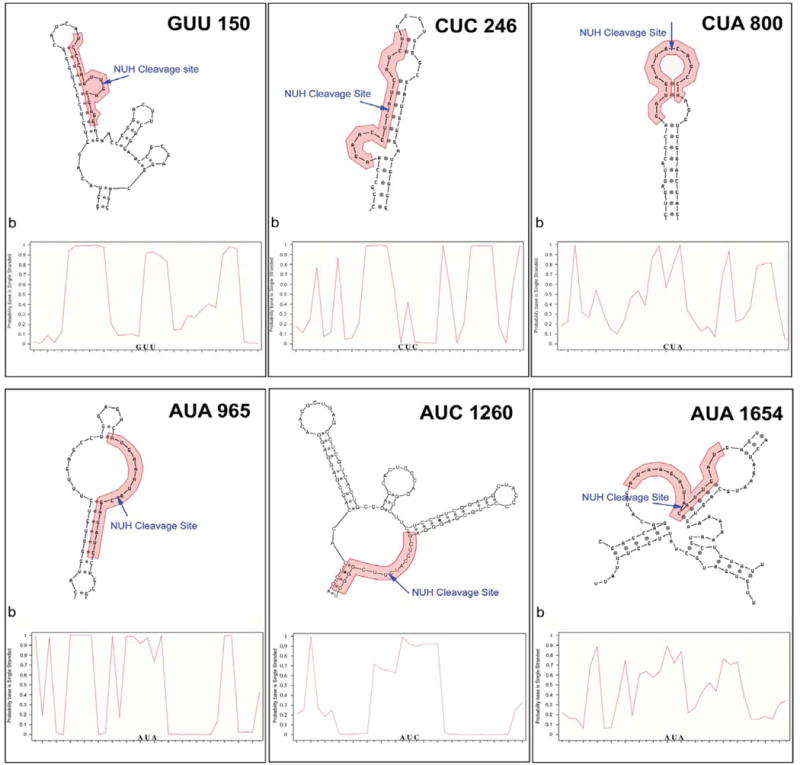
Predicted secondary structure based on the most prevalent Mfold structures observed is shown along with the SFold probability plot for each selected SEAP target site. Pictorial representation of the most commonly observed secondary structure in MFold analysis of local target regions is shown on the bottom of each panel, with hhRz target regions shown as red bases, and NUH↓ cleavage motifs shown in bold. In SFold probability plots on the top of each panel, the probability a base is single-stranded is plotted against base number. The relevant hhRz targeting region is shaded, and the corresponding mRNA base number is shown below the graph.
Six candidate target hhRz NUH↓ cleavage sites (out of a total of 180 NUH↓ sites) were chosen for hhRz targeting of SEAP mRNA. Four of the target NUH↓ hhRz cleavage sites were chosen because of higher predicted accessibility in regions in which they resided (Sites 800, 965, 1260, and 1654). One (Site 150) was chosen for its lower predicted accessibility, and a sixth (Site 246), also with a low predicted accessibility, was chosen based on its use in a previous hhRz study (Zakharchuk et al., 1995). Ribozyme cDNAs were designed with symmetrical 7 nt antisense flanks surrounding the catalytic consensus core and stabilized for proper secondary structure folding by an extended 6 bp stem II that was capped by an ultra-stable 4 nt 5′-UUCG-3′ loop (RzA6 design) (Abdelmaksoud et al., 2009; Sullivan et al., 2002; Koizumi et al., 1993; Tuerk et al., 1988; Homann et al., 1994) (Fig. 2). The hhRzs are embedded within a stable stem loop region (ribozyme harbor) engineered into the central domain of adenoviral VAI (expressed from pUC-VAL plasmid) (Fig. 1). VA1-hhRz constructs permit high level cellular transcription of chimeric RNA transcripts through an intragenic RNA polymerase-III promoter, and the stable (RNase-resistant) chimeric VAI-hhRz RNAs traffic abundantly to the cytoplasm, where SEAP mRNA, like most target mRNAs of interest to PTGS RNA drug discovery (e.g. RHO), has its greatest cellular compartmental lifetime. Intracellular co-localization of target mRNAs and PTGS agents is essential to achieve a high enzyme-target collision frequency that drives initial annealing of the antisense platforms of a hhRz or loaded RISC element (RNAi) to a single stranded regions in the target mRNA (Bertrand et al., 1997; Hormes et al., 1997; Thompson et al., 1995).
3.72. Cellular SEAP Screening for Lead Agents
To validate SEAP as a useful reporter for PTGS screening in disease targets, VA1-hhRz expressing plasmids that target candidate NUH↓ sites in SEAP mRNA (sites 150, 246, 800, 965, 1260, and 1654) were transiently transfected into stable HEK293S-SEAP cells in 96 well plates. Media from these cells were assayed 72 hours after transfection for SEAP enzyme activity. SEAP levels produced fell within the practical linear dynamic range of the assay. Statistically significant knockdown of SEAP protein levels was found for hhRzs targeting the 800, 965 and 1260 sites (p < 0.001), which were three of the four sites predicted to have high accessibility (One-way ANOVA F=9.4, p=2.1×10−9) (Fig. 5A). SEAP 800 hhRz showed 16.0 ±1.9 % knockdown (p=1.02×10−7, n=40), SEAP 965 hhRz showed 14.3 ± 2.7% knockdown (p=8.4×10−6, n=40), and SEAP 1260 hhRz showed 14.0 ± 3.0% knockdown (p=6.7×10−5, n=30). HhRzs targeting sites 800 and 965 promoted the greatest knockdown, and these were further evaluated. These modest levels of knockdown are most likely related to the very restricted annealing platforms expected for the SEAP mRNA in the regions of maximum predicted accessibility and due to preformed target mRNA and protein in the stable cell line. Nevertheless, this approach has the capacity to reliably measure small differences in mean SEAP activity values with statistical significance due to the low coefficient of variation (CV) (CV = [(standard deviation/mean) × 100]. The mean CV (±SEM) for the screen test of all SEAP hhRz samples (Fig. 5A) is 15.56 ± 1.23. The 96-well format for the SEAP assay offers the potential to test many PTGS candidates in a short period of time and could be subject to robotic approaches to handle much larger numbers of test agents. To determine whether the observed knockdown was related to catalytic function of the hhRzs in the VA1 chimeras, as opposed to simpler antisense effects, single nucleotide mutations were made in the catalytic core (G5C, G8C, and G12C mutations) (Hertel et al., 1992) and the catalytically inactivated VAI-hhRz expression constructs were tested vs. enzymatically active VAI-hhRz constructs. Mutations at these nucleotides in the conserved catalytic core abolish hhRz catalytic activity (Perriman et al., 1992; Ruffner et al., 1990). Catalytic inactive hhRzs targeting SEAP sites 800 and 965 showed no significant reversal of SEAP reporter protein knockdown compared to active hhRzs targeting these same sites (Fig. 5B). Tests of the null hypothesis (no difference of samples relative to controls) was refuted (One Way ANOVA, F=6.54, p=8.98×10−8) indicating that not all samples occur from an overall population with a single mean. All hhRz samples were tested relative to the control for significance of SEAP suppression. Most hhRzs (asterisks), including both active hhRzs, show significant knockdown (p < 0.05) relative to control VAI vector transfection except for the G5C and G8C inactive agents targeting the 800 site (p=0.07 (G5C), p=0.08 (G8C)). ANOVAs comparing the active and inactive subpopulations for hhRz 800 and 965 showed no significance (hhRz 800: F= 0.260, p=0.854; hhRz965: F= 0.896, p=0.446). None of the catalytic mutations showed a significant reversal of knockdown compared to their active hhRz versions (p>0.05), with 800 G5C mutation showing 11.9% knockdown (n=8), 800 G8C showing 12.4% knockdown (n=8), 800 G12C showing 16.6% knockdown (n=15), 965 G5C showing 14.7% knockdown (n=16), 965 G8C showing 20.3% knockdown (n=24), and 965 G12C showing 21.9% knockdown (n=24). These outcomes suggest that the knockdown of SEAP protein by the active hhRzs is due to a pure antisense or a catalytic antisense effect (single round of cleavage without Michaelis-Menten turnover which requires product dissociation). A pure catalytic effect (with turnover) would show full reversal of knockdown with mutation of the hhRz core. The mean CV (±SEM) for the screen of specific SEAP hhRz catalytic mutants (Fig. 5B) is 22.91 ± 2.34. Antisense type knockdown outcomes were reported with extended stem II RzA6 design hhRzs within an earlier adenoviral VAI scaffold (pgVaL-Ad) that targeted human RHO in a previous study from this lab (Abdelmaksoud et al., 2009). We also tested two shRNAs against the SEAP mRNA at sites 246 and 965 (Fig. 5C). The 246 site was predicted to be inaccessible and showed no hhRz mediated knockdown and the 965 was predicted to be accessible and showed significant hhRz-mediated knockdown. shRNA against site 246 failed to significantly suppress SEAP expression, whereas shRNA 965 significantly suppressed SEAP expression (37.1% knockdown) relative to control (scrambled shRNA). The 965 shRNA promoted a 39.1% SEAP knockdown while the 246 shRNA did not promote SEAP suppression (One-Way ANOVA: F= 25.42, p = 9.8×10−8; post-hoc Bonferroni t-tests: 965 shRNA: t-test= −5.76, p= 3.62×10−6; 246 shRNA: t-test= 0.60, p = 1; 246 vs 965: t-test= 6.48, p = 3.67×10−7). These results are in concert with computational predictions of SEAP mRNA accessibility and with outcomes from hhRz trials. Three out of four predicted accessible sites in SEAP lead to significant knockdown and both sites predicted to be inaccessible showed no statistically significant knockdown. This outcome demonstrates that both hhRz and shRNA efficacy is dependent upon accessibility in the target mRNA, that SEAP is a difficult target mRNA because of its tight folded structure, and that mppRNA is a relatively reliable predictor of possible knockdown.
Figure 5. Screening Intracellular Catalytic Efficacy of various hhRz-VA1 and shRNA Constructs Targeting SEAP mRNA.
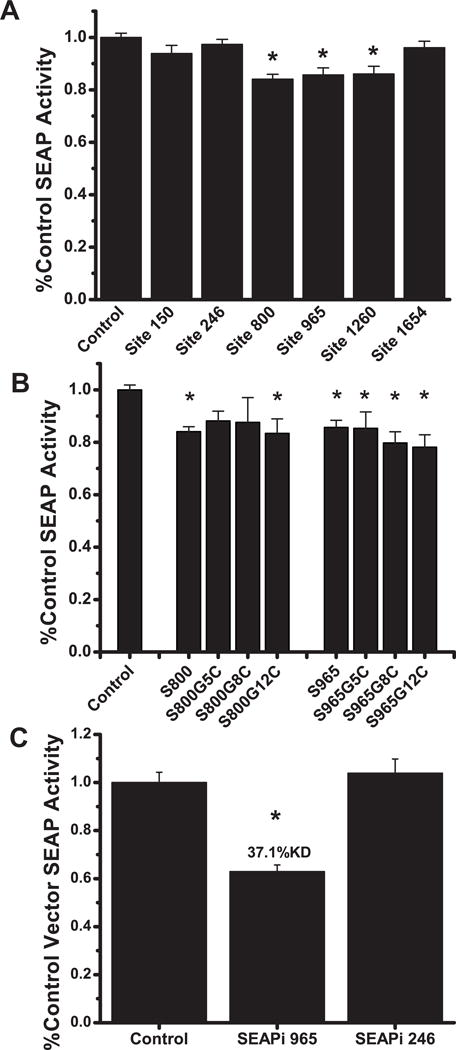
(A) HEK293S-SEAP cells were transiently transfected in 96-well plates with VAI-hhRz-1 constructs targeted to 6 different sites in SEAP mRNA. Total secreted SEAP protein activity was assayed 72 hours post-transfection and the mean fractions of control VAI-hhRz vector SEAP activity are shown ± SEM. Extended stem II hhRzs targeting sites 800, 965, and 1260 showed statistically significant knockdown (asterisks indicate p < 0.05 compared to control). Transfection efficiency was assessed by co-transfection with EGFP expression plasmid and measuring EGFP fluorescence using the Ascent Fluoroskan plate reader. No significant difference in EGFP fluorescence was observed between the different treatment wells (One-Way ANOVA p>0.1). (B) Inactivating mutations of the two lead candidate VAI-hhRz constructs targeting SEAP sites 800 and 965 were generated by single G→C mutations of the G5, G8, or G12 positions in the consensus catalytic core (Hertel et al., 1992). Mean fractions of control vector SEAP protein activity are shown ± SEM. The outcomes indicate that the suppression by the active hhRz is predominantly related to an antisense effect. (C) shRNA cDNAs were designed against the 246 and 965 regions of SEAP mRNA and ligated into the pSuper shRNA expression plasmid prior to transient transfections in to stable HEK293S-SEAP cells and SEAP protein was assayed after 72 hrs.
3.73. Physical Coupling of SEAP Reporter into Disease Target mRNA for PTGS Lead Screening
Mutations in the human rod opsin (RHO) gene are responsible for many cases (~30%) of autosomal dominant retinitis pigmentosa, an inherited photoreceptor degeneration. Genetic disease identifies and validates RHO mRNA as a disease target mRNA for PTGS gene therapy (Dryja et al., 1990; Gal et al., 1997). Mutations in at least 25 other genes also promote autosomal dominant retinitis pigmentosa, and there are many other forms of dominant retinal degenerations (e.g. cone dystrophy) so the strategies developed here for PTGS development should be readily and rapidly transposed to other validated disease target mRNAs. Moreover, suppression of normal WT targets may also be a strategy for certain disease states (e.g. age-related macular degeneration). To achieve this goal one must first identify the most accessible region(s) of the target mRNA and then generally test a multitude of PTGS agents to identify a lead agent with greatest potency or efficacy (Sullivan et al., 2011). With the SEAP reporter model we invested extensive effort to develop a method to screen large numbers of hhRzs, shRNAs, or other PTGS agents (e.g. miRNAs) against disease targets, with approaches amenable to technological extension to the realm of HTS. Two generalized strategies emerged, reported here in proof-of-principle, for adapting the SEAP HTS cell-reporter assay platform to screen candidate PTGS agents targeting human RHO mRNA. These strategies can be extended to any disease target and both involve what is known as RNA fusion technology (Husken et al., 2003). The first strategy involves inserting a limited mRNA target region(s) (here RHO) into the early 3′UTR of the SEAP reporter mRNA (Fig. 6A). A stop codon insures that the inserted cDNA element is within the 3′UTR of SEAP. As the 3′UTR modulates mRNA lifetime, cleavage within the target RHO region is expected to reduce the half-life of the SEAP mRNA. The pSEAP-STOP-L57RHO vector was generated to test this strategy. The proven-accessible 250 region of RHO (region around the Leu57 codon) contains a secondary structure with an expected large loop (33 nt) capping a stable stem structure (Fig. 6C) (Abdelmaksoud et al., 2009). A small cDNA, encoding 62 nts of the entire predicted secondary structure (stem and loop) of region 250 in human RHO mRNA, was inserted into the early 3′-UTR of the SEAP expression vector between a set of unique engineered restriction sites (see Methods and Supplementary Methods). HhRzs (RzA6 design with 6 bp stem II) targeting RHO at site 266 and SEAP at site 965 were cloned into VAI-hhRz expression constructs and transiently co-transfected with the pSEAP-STOP-L57RHO into HEK293S cells. Both the RHO and SEAP targeting hhRz constructs reduced levels of SEAP activity by over 35% (p < 0.05) relative to control vector transfections that expressed the scaffold RNA without the hhRz, as measured by the HTS SEAP assay (Fig. 6B). Mean fractions of control vector (pSEAP-control) transfection SEAP activity are shown ± SEM (One-Way ANOVA F=7.34, p=4.32×10−4). Significant knockdown was observed by both hhRz constructs, with 36.5% knockdown for the RHO 266 construct (Bonferroni t=−4.01, p=0.0014, n=16) and 35.5% knockdown for the SEAP 965 construct (t= −3.18, p=0.016, n=8). The knockdown by the RHO hhRz is not statistically different from the knockdown by the SEAP hhRz (t=0.092, p=1). Also, catalytic core mutation reversed knockdown at the 266 RHO site, demonstrating cellular catalytic activity in this context with an extended Stem II hhRz. The catalytic core mutation in the 266 RHO hhRz completely obviated SEAP suppression relative to the catalytically active agent (t= 2.89, p=0.036) and brought the level of suppression to the level of control (t=−0.388, p=1, n=8). Using this strategy, any predicted or experimentally proven structured element(s) that embraces an accessible regions of a target mRNA, or control inaccessible regions, could be screened for intracellular sensitivity to PTGS agents. The most likely reason for higher levels of knockdown of SEAP activity by the SEAP 965 hhRz relative to tests of the same hhRz in stable SEAP secreting cell lines (Fig. 5A) is that in this experiment both target and enzyme plasmids were transfected into naïve cells that had zero levels of target mRNA already in the translation processing stream at the time of transfection. This is one of the limitations of conducting PTGS experiments in cell lines stably expressing the target, as there will always be a fraction of the target which can never be suppressed. This is not a concern for HTS as long as one can reliably determine the construct(s) that lead to the greatest knockdown for subsequent optimization to identify a lead candidate, which is the goal of the current development. This RNA fusion strategy requires ligating multiple target mRNA regions for analysis of the full range of predicted accessible regions, which adds complexity for HTS to achieve a lead PTGS agent. The low CV allows us to discriminate between candidates with a broad range of target suppression potential.
Figure 6. hhRz Screening Based on RHO structured Element in SEAP 3′UTR.
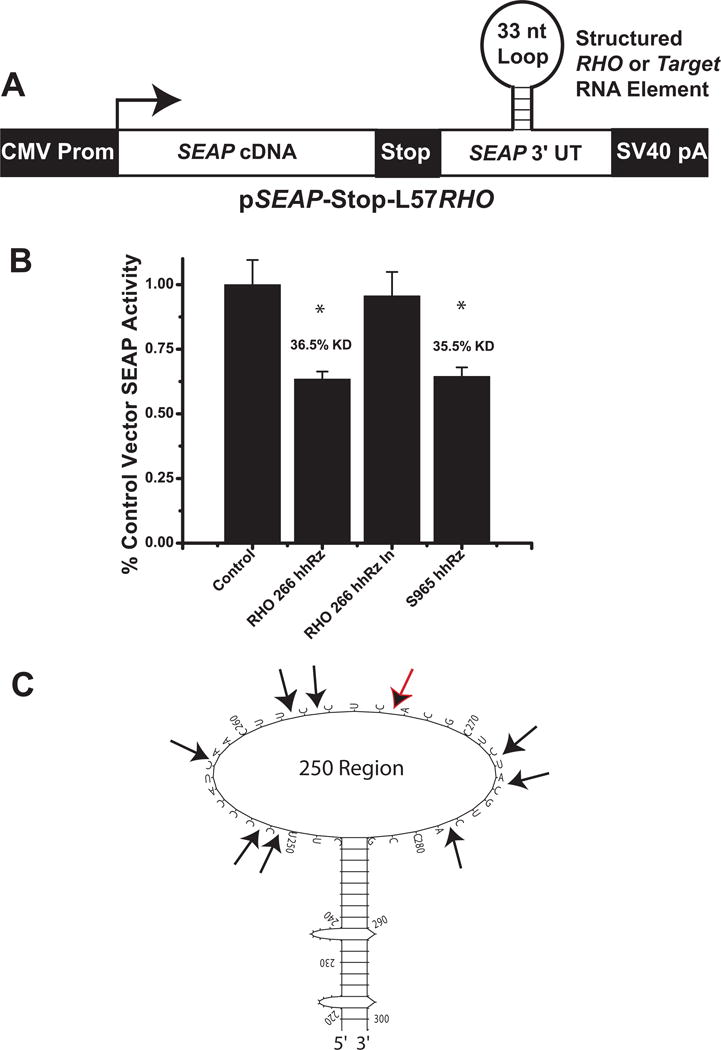
(A) The computationally predicted stable stem loop structure in the 250 region of the human RHO mRNA was inserted into the 3′UTR of SEAP mRNA (genetic construct shown schematically). (B) pSEAP-STOP-L57RHO construct was co-transfected with active or inactive VAI-hhRz constructs targeting the RHO structured element (at site 266) or an active construct targeting SEAP (at site 965) (RzA6 design hhRzs). (C) Predicted secondary structural motif of the region around the L57 codon. This regional element was embedded into the 3′UT region of the SEAP mRNA. HhRz cleavage sites are indicated with black arrows and the black arrow with the red outline is the 266 CUC↓ cleavage site.
A second more flexible RNA fusion strategy, utilizing an engineered bicistronic pRHO-IRES-SEAP vector, was developed to adapt the SEAP HTS cell-reporter assay platform to screen PTGS target sites in full-length human RHO target mRNA (Fig. 7A). This vector allows translation of both RHO and SEAP proteins from a single bicistronic mRNA transcript. Here, ribosome translation of RHO is 5′ cap-dependent while translation of SEAP is cap-independent and mediated by ribosome recognition of the tertiary-structured IRES element. The cDNA encoding the full length human RHO mRNA transcript (from transcription start to just preceding the first (dominant) polyadenylation signal (so that mRNA termination does not occur) was placed upstream of the encephalomyocarditis IRES sequence and the SEAP cDNA was placed downstream. Stable local RNA secondary structures emerge during transcription while the nascent RNA transcript is still associated with the RNA polymerase (Tinoco and Bustamante, 1999; Singh and Padgett, 2009). Hence, we placed the target mRNA upstream of the IRES and SEAP reporter sequences in order to favor proper folding of the target mRNA component of the bicistronic RNA independent from both IRES and SEAP elements, which are transcribed later in time. Our rationale is that this placement would lead to a more native structural folding of the upstream target RNA component, which is the focus of therapeutics development. The pRHO-IRES-SEAP bicistronic reporter strategy provides a sensitive and efficient measure to test independent hhRz or RNAi mediated cleavage within RHO (or any disease target) in the bicistronic mRNA. Cleavage anywhere within the 5′cap-RHO-IRES-SEAP-polyA-3′ bicistronic mRNA would decrease its half-life and yield a reduced steady-state level of intact bicistronic mRNA in the cell. The expected result is a new steady-state level of SEAP enzyme expression (and RHO expression) and secretion because these outcomes are directly proportional to the steady state level of its mRNA, with at least 98.5% of the protein being secreted into the medium (Berger et al., 1988). To conduct an initial proof-of-principle test of this strategy, hhRz and shRNA agents were designed against predicted and experimentally validated accessible regions in SEAP and RHO mRNA (regions around the SEAP 965 site and RHO 725 site) (Fig. 7). Strong accessibility around the 725 region has been determined (see Fig. 4, Abdelmaksoud et al., 2009; unpublished work Sullivan et al.). A 725 GUC↓ RHO-targeting hhRz and a 965 AUA↓ SEAP-targeting hhRz were embedded in the pUC-VaL scaffold, both with 4 bp Stem II regions. shRNA agents targeting these sites were cloned into the pSUPER RNAi expression system under control of the extrinsic H1 promoter (RHOi-725 and SEAPi-965). HEK293S cells were transiently co-transfected with both pRHO-IRES-SEAP and pUC-VaL-hhRz or shRNA agents. Media was removed to assess SEAP enzyme activity, and total RNA was extracted from the same cells. RHO-IRES-SEAP mRNA levels in total cellular RNA extracts were analyzed by qRT-PCR analysis with RHO cDNA component specific primers. Catalytically active hhRzs targeting RHO 725 or SEAP 965 significantly suppressed SEAP protein expression (p<0.05), while catalytic core mutants reversed suppression to control levels indicating catalytic function of the hhRzs (Fig. 7B); mRNA levels were not tested. One way ANOVA showed significant differences among samples (p=8.54×10−10). Active 725 RHO hhRz significantly suppress SEAP protein secretion (p=5.4×10−6), and active 965 SEAP hhRz significantly suppressed SEAP protein secretion (p= 3.4×10−6), while catalytically inactive hhRzs did not suppress in both cases, which suggests a pure catalytic effect. shRNA agents promoted SEAP protein knockdown as well as RHO-IRES-SEAP mRNA knockdown (p < 0.001) (Fig. 7C). SEAPi-965 showed significant knockdown of bicistronic mRNA and SEAP protein activity compared to a scrambled control shRNA. RHOi-725 significantly suppressed bicistronic mRNA and SEAP protein activity. Both SEAPi-965 and RHOi-725 showed significant SEAP protein knockdown relative to scrambled control (One-way ANOVA F=1035.8, p<0.001), with a 62.2% knockdown in SEAP activity for SEAPi-965 (p=4.19×10−24, n=9) and a 19.6% knockdown in SEAP activity for RHOi-725 (p=1.35×10−12, n=9). Knockdown of SEAP enzyme secretion was greater with the SEAP-targeting shRNA vs. the RHO-component targeting shRNA (p = 3.21×10−20, n=9). Corresponding RHO component bicistronic mRNA levels were also significantly decreased relative to scramble control (One-way ANOVA F=50.3, p=9.8×10−9) with a 68.6% knockdown for SEAPi-965 (p=5.6×10−9, n=8) and a 37.5% knockdown for RHOi-725 (p=5.99×10−5, n=8) relative to control. Knockdown of bicistronic mRNA levels was greater with the SEAP-targeting shRNA vs. the RHO-targeting shRNA (p=5.28×10−4, n=8). Non-equivalent knockdown of SEAP activity by the RHO and SEAP shRNA targeting agents was likely due to differences in RNAi-mediated targeting efficacy at the two sites in the bicistronic mRNA. RHO is upstream of the IRES element in the bicistronic mRNA, and cleavage within the RHO component may permit some persistent translation of SEAP protein from the cap-independent IRES element in the 3′ cleavage fragments during their cellular lifetime prior to degradation. The extent to which possible differential stability of downstream IRES-containing mRNA fragments plays a role with other target sites in RHO or in other upstream target mRNAs was not determined but stable 3′ mRNA cleavage fragments active in translation were observed in studies of antisense oligodeoxynucleotide-directed RNase H mediated cleavage of mRNA (Thoma et al., 2001; Hasselblatt et al., 2005) even though the fragments contained neither a 5′ cap nor IRES structures.
Figure 7. Evaluation of Ribozyme and shRNA Cleavage of RHO-IRES-SEAP Components.
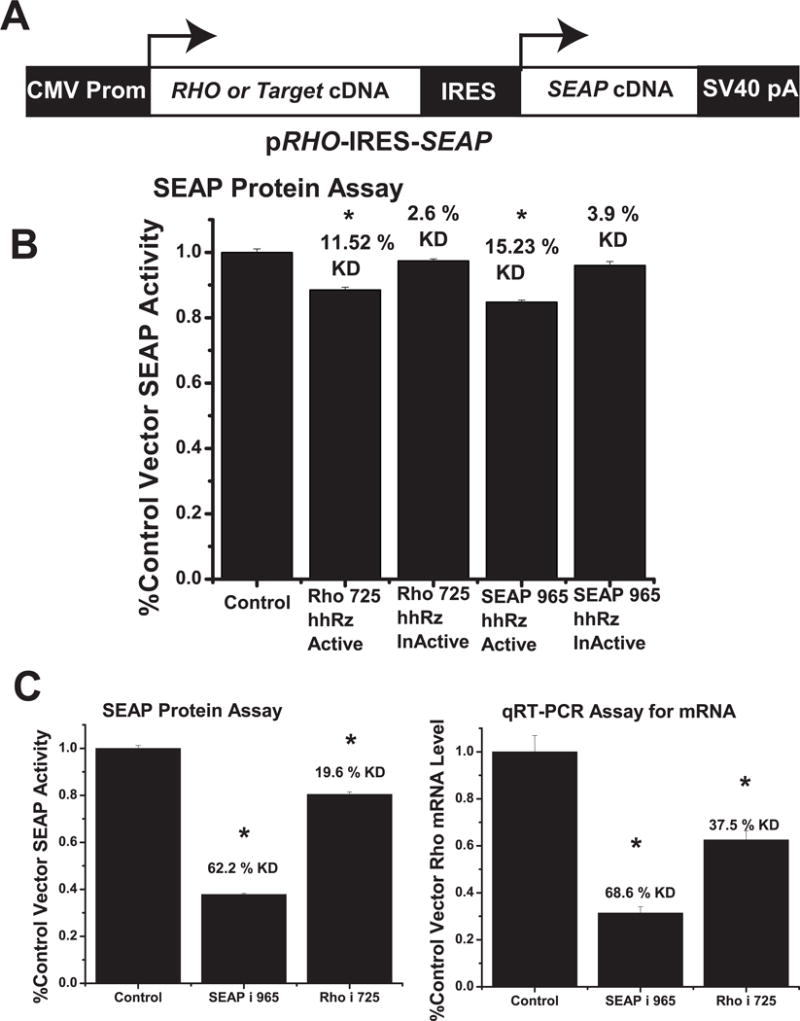
(A) Schematic of pRHO-IRES-SEAP construct. The leftmost arrow shows the position of transcription start for the bicistronic mRNA and the position of cap-dependent translation initiation. The rightmost arrow shows the position of cap-independent translation mediated by the folded IRES RNA element. (B) Test of hhRzs (active and inactive) against 725 GUC cleavage site in RHO and the 965 cleavage site in SEAP components of the RHO-IRES-SEAP dicistronic mRNA. HhRzs with 4 bp stem II regions targeting the two sites were ligated into the pUC-VaL scaffold. (C) pSUPER shRNA constructs were designed to target the SEAP 965 region (SEAPi-965) and the RHO 725 region (RHOi-725), which was also known to be accessible. The shRNA constructs were transiently co-transfected with pRHO-IRES-SEAP plasmid into HEK293S cells. Media was removed 48 hours post-transfection and assayed for SEAP enzyme activity (left panel). Total RNA was then extracted and RHO component bicistronic mRNA levels quantified by qRT-PCR (right panel). Mean fractions of control vector transfection SEAP activity or relative RHO mRNA level are shown ± SEM. Asterisks indicate significant (p<0.05) knockdown relative to control transfection. While SEAP protein and RHO component bicistronic mRNA knockdown levels were relatively similar for SEAP- targeting shRNA agents, SEAP protein level knockdown is proportionally smaller than the knockdown of RHO component bicistronic mRNA levels by the RHO targeting shRNA agent.
3.74. IRES as a Structured RNA Insulator Element
Use of the bicistronic Target-IRES-SEAP mRNA represents a suitable model for development of PTGS agents against the “natural” target mRNA provided that the IRES and SEAP sequences do not influence the folding of the upstream target mRNA component. Using a bioinformatics RNA folding approach we investigated the extent to which the IRES element, intervening between the upstream full-length cDNA of the target and the downstream SEAP reporter cDNA, isolates the upstream (disease target) region of the bicistronic mRNA for independent folding. This issue is critical to this strategy because identification of lead PTGS candidates by an initial HTS type screen should occur under conditions that most closely simulate the true in cellulo native structure(s) and accessibility of the full length target mRNA. In tests of three bicistronic mRNAs engaging three human retinal mRNA targets of relevance to human disease, the IRES element appears to strongly insulate the folding of the upstream target element from downstream IRES and SEAP components (Supp. Fig. 2).
3.75. Investigating the kinetics of PTGS target knockdown in live cells
SEAP is secreted in bulk from live human cells into the culture medium, where it has an exceptionally long half-life (~500hrs). Given that secreted SEAP reflects the steady-state levels of the mRNA from which it is translated (Berger et al., 1988), a live cell SEAP reporter assay can be exploited to measure the in vivo kinetics of target mRNA suppression by a given hhRz, shRNA, or other PTGS agent. We transiently transfected expression plasmids for hhRzs known to successfully target the RHO (hhRz CUC↓ 266) (Abdulmaksoud et al, 2009) or SEAP components (hhRz CUA↓ 800, hhRz AUA↓ 965) of the bicistronic mRNA into stable expression HEK293S-RHO-IRES-SEAP cells. The time course of SEAP expression was measured over 72 hours post completion of transfection (t =0) (Fig. 8A). By the 72 hr time point the three hhRzs had exerted statistically significant knockdown of SEAP activity normalized to control (SEAP 800 hhRz: 36.2%; SEAP 965 hhRz: 38.6%; RHO 266 hhRz: 41.2%). Control samples which express the VAI scaffold without an hhRz showed increased expression and SEAP secretion over the same time period. Control sample showed steady increase in SEAP activity over the time course assessed (slope 174.3 ± 20.8, R2 = 0.996, p = 0.06). The SEAP 965 hhRz (slope −51.8 ± 14.9, R2 = 0.927, p = 0.122), the SEAP 800 hhRz (slope −41.0 ± 0.13, R2 = 0.963, p= 0.09), and the RHO 266 hhRz (slope = −42.2 ± 0.135, R2 = 0.78, p = 0.21) showed suppression of SEAP expression. HhRz-mediated suppression or knockdown emerged over time which was examined in bar graph analysis at different time points (Fig. 8B). At the 24 hr time point there is no significant difference of any hhRz agent with respect to control at criterion level (p ≤0.05) (ANOVA, F=2.567, p = 0.074). Statistically significant suppression of SEAP emerges by 48 hrs for all three hhRzs (ANOVA, F=38.96, p=3.98×10−10; Bonferroni t-tests: SEAP 800: t= −7.44, p=2.55×10−7; SEAP 965: t=−7.80, p=1.02×10−7; RHO 266: t= −10.20, p=3.71×10−10). There were no significant differences between the levels of suppression by the three hhRzs at 48 hrs. There was no significant difference between the SEAP 965 hhRz samples relative to RHO 266 (t = −2.4, p = 0.14). There was no significant difference between the SEAP 800 hhRz samples relative to RHO 266 (t = −2.8, p = 0.06). Levels of suppression are slightly greater and apparently stabilizing by 72 hrs for all three hhRzs (ANOVA, F=28.19, p= 1.32×10−8; Bonferroni t-tests: SEAP 800: t=−6.99, p= 7.98×10−7; SEAP 965: t= −7.46, p= 2.40×10−7; RHO 266: t= −7.95, p = 6.98×10−8). At the 72 hr time point there were no significant differences between the levels of suppression by the three active hhRzs. At 72 hours the relative knockdown by SEAP 800 hhRz is 36.2%, SEAP 965 hhRz is 38.6%, and by RHO 266 hhRz is 41.2%. The essentially constant SEAP secretion in cells transfected with these active hhRzs suggests that abundant and rapid transcription of VAI-hhRz RNA by RNA polymerase III occurs in human cells and is followed by target mRNA recognition and suppression to yield a new target mRNA steady state within 24–48 hrs. We investigated the upregulation of the pUC-VaL-hhRz RNA expression in HEK293S cells by RNA Polymerase III after transient transfection by endpoint RT/PCR and found that the candidate therapeutic RNA is expressed by 24 hrs and appears to increase in abundance by 72 hrs (Fig. 8C).
Figure 8. Evaluation of Kinetics of Target mRNA Knockdown in Live Human Cells.
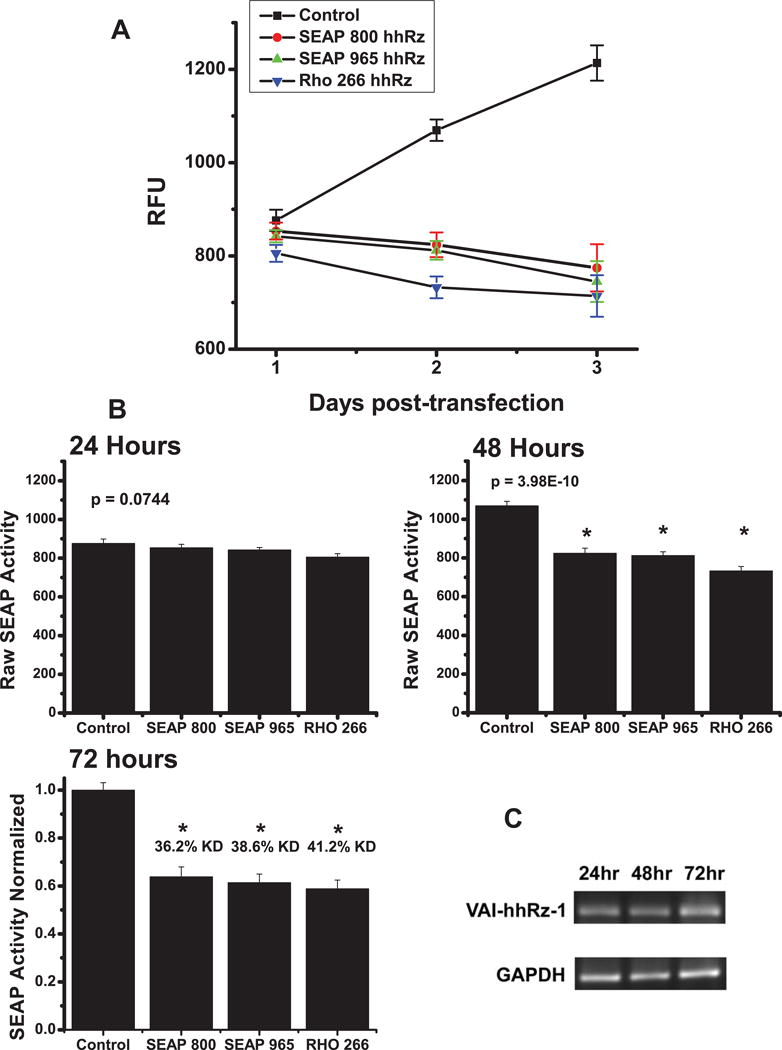
HhRzs in pNEB-VAI-hhRz targeting RHO (CUC↓ 266) and SEAP (CUA↓ 800, AUA↓ 965) mRNAs and the control plasmid expressing the chimeric VAI RNA without a hhRz (pNEB-VAI) were transiently transfected into HEK293S cells stably expressing the pRHO-IRES-SEAP construct. (A) Graphs for the control, RHO CUC↓ 266, SEAP AUA↓ 965, and SEAP CUA↓ 800 time courses are displayed. The initial time point (t = 0) reflects the completion of the transfection protocol. Media was removed for SEAP protein assay and replaced with fresh media at 24 hours, 48 hours, and 72 hours post-transfection. Cells transfected with active hhRz constructs targeting either the RHO or SEAP components of the bicistronic mRNA showed no increase in SEAP protein levels while cells transfected with the control plasmid (pNEB-VAI with no hhRz ligated) showed a constant increase in SEAP protein levels over time. (B) At 24 hours there is no difference between the control and hhRz samples. At 48 hrs there are significant differences between samples. At 72 hrs there are significant differences between samples and control, but no significant difference between the SEAP or RHO hhRz samples (p > 0.05). At 72 hours the relative knockdown by SEAP 800 hhRz is 36.2%, SEAP 965 hhRz is 38.6%, and by RHO 266 hhRz is 41.2%. (C) Cytoplasmic expression of VAI-hhRz-1 in transiently transfected cells. RT-PCR results from cytoplasmic RNA extracted from HEK293S cells transiently transfected with pUC-VaL-hhRz plasmid. At 24, 48, and 72 hours post-transfection, cells were harvested and cytoplasmic RNA was extracted. 1st strand cDNA was synthesized using reverse transcriptase with gene specific primers. VAI and GAPDH sequences were amplified using PCR and the expected amplified products of 170 bp for VAI and 453 bp for GAPDH are shown after 2% agarose gel electrophoresis and ethidium bromide staining. Expression of VAI-hhRz-1 RNA was observed at 24 hours post-transfection and up to 72 hours post-transfection. Corresponding GAPDH RNA levels from the same cellular cytoplasmic extracts are shown below.
3.76. RNA Fusion Expression Plasmids
We constructed a generic plasmid for conducting HTS PTGS screens (Fig. 9A). The pTarget-IRES-SEAP plasmid allows ligation of a full length cDNA for the target of interest upstream of the IRES element. This cDNA should fully represent the entire mature mRNA from transcription start to just before the first polyadenylation signal. The plasmid demonstrating the ligation of the full length human RHO cDNA is shown (Fig. 9B). The proximal promoter and multiple cloning site in the pTarget-IRES-SEAP plasmid is engineered to initiate CMV expression immediately upstream of the multiple cloning site to minimize addition of vector sequences in the transcript (Fig. 9C). The region around the 3′ UTR site for structured Target element insertions is also shown (Fig. 9D). The multiple cloning site and adapter for ligation allows positive selection for desired clones.
Figure 9. Fusion RNA Expression Plasmids.
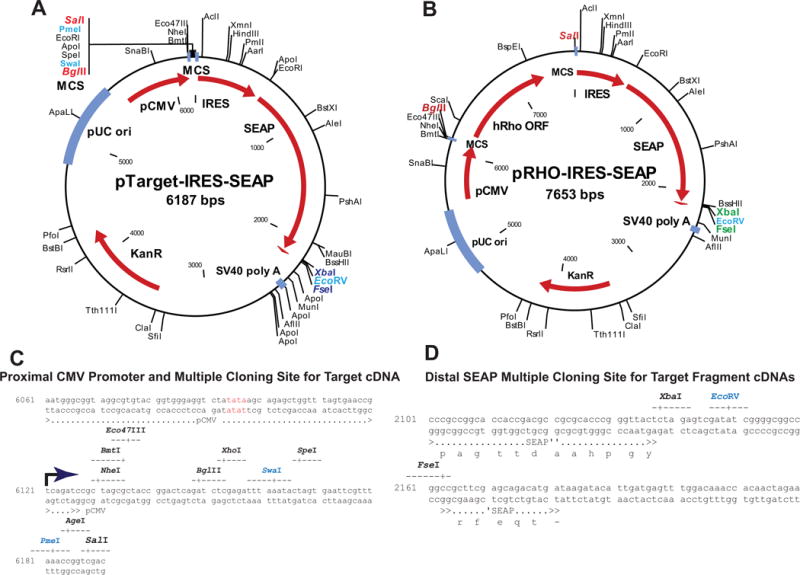
(A) In pTarget-IRES-SEAP there is an adapter cloned between the Bgl II and Sal I sites which includes the following unique restriction sites (Bgl II, Xho I, Swa I, Spe I, Eco RI, Pme I, Age I, Sal I). Depending upon the target cDNA inserted one of these rare cutting restriction enzymes can be used after ligation to linearize unligated parental plasmid and minimize its transfection potential and enhance for positive clones. (B) In pRHO-IRES-SEAP (parent plasmid for pTarget-IRES-SEAP) the target cDNA is amplified from another plasmid source with a BglII site at transcription start and a Sal I site just prior to the first (assumed dominant) polyA signal. The PCR product is digested with Bgl II and Sal I (both are unique in the plasmid) and ligated directionally into the pTarget-IRES-SEAP plasmid cut with the same restriction enzymes. (C) These plasmids are engineered such that the Blg II (AGATCT) site is located at the transcriptional start site for the CMV promoter. This secures that the mRNA of the Target element transcribed in cells, as the upstream component of the bicistronic mRNA, faithfully represents the 5′UTR of the folded mRNA with minimal vector sequence tags. (D) Similarly, at the terminus of the SEAP coding sequence there is an Xba I, Eco RV, Fse I adapter that allows ligation of structured elements into the SEAP mRNA and selection post-ligation with EcoRV. We expect that the pTarget-IRES-SEAP plasmid can be used directly for structured element insertions into the 3′ region of SEAP when there is no upstream Target cDNA inserted.
4. Potential Pitfalls and Trouble shooting
4.1. Proof-of-Principle with SEAP mRNA as Target
Successful development of therapeutic PTGS agents requires that one rigorously address both the structural and functional biocomplexity of the target mRNA and the structure-based function of the PTGS agents themselves. Toward this overall goal, we developed a HTS screening platform to identify lead candidate trans-acting hhRz and shRNA constructs, as a first step prior to optimizing PTGS agents for cellular efficacy as therapeutic agents. While the HTS approach was developed with hhRz expression constructs, and further validated with shRNA constructs, an equivalent approach can be used for expressed antisense RNA or miRNA constructs. This HTS system permits rapid confirmation of predicted or experimentally-demonstrated accessible regions in a given disease target mRNA in human cells, it allows identification of a lead candidate to optimize for therapeutics development, and it could also be used in the optimization process of lead agents (e.g. hhRz or shRNA). An additional and powerful novelty of this platform is that it allows kinetic assessment of lead PTGS agent function within the context of the live human cellular milieu. This approach was first validated against the reporter element, SEAP, which is then utilized in RNA fusion technology for disease target PTGS screening. We present the initial identification of novel lead candidate therapeutics for human RHO mRNA, which with further development, have potential as PTGS agents for mutation-independent therapy of adRP, and possibly for other retinal degenerative and potentially vascular conditions. The HTS platform described here represents a strong, effective, and easily utilized tool for RNA drug discovery in academic and corporate pharmaceutic labs.
Inspired by RNA fusion concepts we hypothesized that SEAP would be useful as a reporter construct for PTGS development. We first tested hhRzs against SEAP mRNA target and were able to demonstrate a moderate knockdown of SEAP expression by two hhRzs embedded within a chimeric engineered adenoviral VA1 RNA (Lieber and Strauss, 1995). Potential accessible sites in the SEAP mRNA were computationally predicted using algorithms based upon different physical and chemical principles. Three out of four predicted accessible sites allowed significant hhRz or shRNA suppression and two predicted inaccessible sites did not allow knockdown, further establishing the viability of our mppRNA bioinformatics computational approach for predicting accessible target regions. The 1654 site near the terminal 3′ end of the target mRNA was predicted to be accessible but proved insensitive to knockdown. This result emphasizes that while in silico predictions of accessibility are largely effective with our multi-parameter prediction approach (mppRNA), experimental validation of predicted results is also needed, and can be achieved rapidly with this HTS system. A potential pitfall is that the structure of the target mRNA can never be fully determined with contemporary technology. We applied robust bioinformatics approaches to rationally estimate target mRNA structure. And we have shown that the IRES element in the bicistronic Target-IRES-SEAP mRNA likely insulates the upstream target from the downstream reporter. The pitfall is addressed by this approach in that one can rapidly screen many different constructs to identify a lead candidate. As we develop greater knowledge the bioinformatics mppRNA model can be refined to appropriately accommodate experimental findings so that the predicted sites of accessibility are likely to lead to the most potent PTGS agents.
The ability of this HTS platform to statistically discriminate between small differences in secreted SEAP expression among many possible PTGS agents is an important advantage of this technology. The SEM bars are typically small and, for example, the CVs for the data in Fig. 5A (15.6 ± 1.2%) and Fig. 5B (14.4 ± 6.4%) are noted. When we compare our CV to other studies on HTS siRNA/shRNA screens our CVs (~15%) approach that of small molecule HTS screens (13.4%) compared to siRNA assays (~26.5%) (Birmingham et al. 2009). Our CV values represent nearly a 300% reduction in CV compared to outcomes in a previous PTGS study from this lab that used western analysis as a primary measure of target knockdown (41.7 ± 6.0%) (Abdelmaksoud et al., 2009). This level of variance is common in western analysis, which is typically used to screen efficacy of PTGS agents. In the prior study we used western analysis to screen for hhRz knockdown after co-transfection and were able to evaluate only 8 potentially active agents over a 2 year period, given the higher levels of variance in western analysis and the much larger number of experiments needed for power to achieve solid statistical inference (Abdelmaksoud et al., 2009; Zar, 1984). The current HTS platform, designed to identify lead candidates that could be further optimized for therapeutic RNA Drug Discovery, was used to screen 34 potentially active hhRzs against human RHO in a matter of two to three months without any robotic tools. The SEAP-based HTS platform established here has improved throughput between one and two orders of magnitude and substantially reduced error of measurement. We expect that these attributes will aid investigators in screening small or large sets of PTGS agents in order to identify lead candidates, a critical initial component in the process of therapeutic RNA drug discovery for arbitrary mRNA disease targets. We demonstrated the use of the platform to identify a lead candidate hhRz against the human RHO mRNA. While not demonstrated here, this HTS platform can also be used to optimize a single lead candidate by testing many different variations, or combinatorial libraries, of a single PTGS agent that targets a single site in the target mRNA. With robotic tools for pipetting and transfection we estimate that there is potential to expand the number of agents tested by an additional 1–2 log-fold.
Another potential pitfall is that stable cell lines with preformed SEAP mRNA and protein, or any target RNA and protein, prior to transfection of a PTGS agent, have a minimum (floor) level of expression beyond which knockdown cannot occur (Supp. Fig. 1C). This result occurs because of the delay in expression of the PTGS agent in the target stable cell line after transfection, while the cell continues to constitutively express the target mRNA and its protein. Comparing Supp. Figs 1B and 1C a substantial fraction (~50%) of the dynamic range of the fluorescence assay is lost in cell lines already stably expressing SEAP protein because of preformed mRNA and protein already in the processing streams. Therefore, we estimate that the maximum level of suppression measurable in stable cell lines is only approximately 50%, but that the absolute level of knockdown of freshly synthesized target and protein is likely to be higher. Hence, while the dynamic range of knockdown is reduced with stable cell lines, the low CV of the data and the HTS capacity of the approach are advantages for screening in RNA Drug Discovery that far outweigh any potential disadvantages. To resolve this potential pitfall one can use naïve cells in transient transfection experiments where there is no loss of dynamic range. However, one expects greater variability of measures due to the additional variables of transfection efficiency and expression from both target and hhRz expression plasmids. Or, one could generate a stable cell line with the target placed under control of a doxycycline-inducible promoter (e.g. TetON). HEK293 cells were used because they offer very high transfection efficiencies (>90%) to minimize the impact of this variable on evaluating PTGS agents in transient transfection experiments in stable target expressing cell lines or in naïve cells. In RNA Drug Discovery the identification of a lead candidate requires the reliable measure of relative target knockdown of by the set of agents, and their rank ordering of efficacy, and is initially more important than determining absolute levels of knockdown. We can reliably measure significant relative differences in knockdown among different PTGS agents to within a few percent of control. The high reliability of this approach to discriminate among differences in efficacy is a substantial advantage for RNA Drug Discovery. Other HTS approaches have been developed in this lab to further refine the lead PTGS agents once identified by this approach, and to evaluate PTGS agents for cellular toxicity (Kolniak and Sullivan, 2011).
Another potential pitfall is that measured fraction of SEAP enzyme activity is not a direct measure of expressed SEAP protein. The assays depend upon the established work that expressed SEAP protein is secreted in bulk into the medium (Berger et al., 1988). To resolve this we determined that our assay for SEAP activity is linear with respect to PLAP protein over the time frame of the assay (Supp. Fig. 1). Hence, we expect that the reduction in SEAP activity is proportional to the levels of SEAP protein knockdown but this was not explicitly measured as our goal was a system capable of HTS. Another potential pitfall is that since the activity is measured relative to an appropriate control (e.g. expression vector without embedded PTGS agent) it is a measure of relative knockdown, not a measure of absolute protein knockdown. One would generally expect levels of target protein knockdown in proportion to the percentage of target mRNA suppression. This is especially true for RHO as a target mRNA and protein, which are highly stable molecular entities. Given the linearity of the assay, activity measures could be scaled and referenced to absolute PLAP protein values.
4.2. HTS SEAP Platform to Identify Leads and Optimize PTGS Agents against Disease Targets
A major contribution of this work is a tool that exploits the SEAP HTS platform to screen sets of hhRz or RNAi agents designed against arbitrary mRNA targets. Our HTS approach was initially focused on a full-length disease target mRNA (RHO) expressed in live human cells where protein: RNA interactions and mRNA trafficking play important roles in RNA accessibility. Demonstrated success of PTGS functionality within a human cell testing environment is a strong indicator of functional performance (efficacy) in animal disease models in vivo because PTGS agents operate within the common housekeeping level of cellular metabolism. Through this HTS approach we are able to identify the best RNA drug (lead) among many potential candidates at regions of the target mRNA that are expected to be accessible. Leads may then be further optimized to maximize efficacy, and optimization can be managed, at least in part, on the same HTS platform. Such optimizations could include testing the impact of variation in the length of the antisense flanks on efficacy and specificity, testing alterations of Stem II and its loop on efficacy and specificity, or trials of upstream tertiary accessory elements that enhance catalytic activity under intracellular environmental conditions (De la Pena et al., 2003; Khvorova et al., 2003; Penedo et al., 2004; Martick and Scott, 2006). Evaluation of such tertiary accessory elements and their influence on efficacy of our lead 725 candidate is a topic of ongoing RNA structure-activity investigation.
We demonstrated proof-of-principle of this approach toward therapeutic RNA Drug Discovery by integrating the SEAP cDNA into constructs that express all or part of a model disease target mRNA (human RHO mRNA). Expression of the target RNA is directly and physically linked to SEAP protein expression, allowing HTS testing of hhRz, shRNA, or other PTGS agents against the model target fusion RNA. Two formats were explored, first a SEAP mRNA in which structured elements of the target mRNA are embedded in the 3′-UTR region, and second a bicistronic mRNA containing Target-IRES-SEAP. In both strategies cleavage of the fusion RNA in the local structural target mRNA domain or in the full-length target sequence is expected to promote intracellular mRNA degradation (decreased half-life) and suppression of SEAP protein expression. A critical feature of both approaches is that they are immediately extendable to any arbitrary RNA target. The first strategy embeds a local RNA secondary structural element for targeting. Once a region of the target mRNA is assessed as truly accessible by in silico and/or experimental means, one can embed a piece of the target cDNA into the 3′UTR of SEAP with sufficient primary sequence to generate a range of possible local secondary and tertiary structures that otherwise would be likely to occur in the native target in vivo. As secondary RNA structural folding is driven by strong local neighborhood base pairing (Liphardt et al., 2001), one expects that the presence of a sufficiently large piece of the cDNA will allow such independent disease target structural folding elements to emerge embedded within the 3′UT of the SEAP mRNA. An accessible region often contains many potential cleavage sites for an hhRz as the probability of finding an NUH motif occurs approximately once in every twelve nt. Many different PTGS agents could then be used to probe knockdown within this disease target region to identify the most efficacious site-specific PTGS agent, or to optimize a single lead agent. The second strategy places the full-length target cDNA immediately downstream of the transcription start site of the promoter (e.g. CMV) such that the target mRNA component can begin folding while the emerging bicistronic mRNA is still coupled with the RNA Pol-II polymerase. As strong RNA secondary structural folding is coincident with transcription (hierarchical RNA folding) (Tinoco and Bustamante, 1999; Singh and Padgett, 2009), this approach is expected to generate a reliable fold for the target component of the bicistronic mRNA that simulates in vivo folding of the native full-length target mRNA. We tested this hypothesis with an RNA folding bioinformatics approach which suggests that, at least for three retina-expressed mRNA targets, that the folding of the upstream target is not grossly impacted by the remainder of the dicistronic mRNA and that the IRES structure acts as an insulator element to isolate the folding of the upstream target mRNA component (Supp. Fig. 2). Our Target-IRES-SEAP approach offers potential to test every site in the target mRNA in order to identify the highest ranking lead candidate for further optimization among many possible identified accessible regions. Here we showed how to vastly reduce the size of the potential target site ensemble by a bioinformatics accessibility mapping (mppRNA) of the target mRNA.
With this approach we showed that the prior known 266 CUC↓ RHO cleavage site is accessible for PTGS suppression and we showed that a site not previously tested at 725 GUC↓ was also accessible for suppression. The 725 cleavage site is found in a region shown computationally to be accessible in our prior study (Fig. 4, Abdelmaksoud et al., 2009) but was not previously tested. While the methodology is presented here, a full description of the lead identification (725 GUC↓) and its initial evaluation will be provided in a subsequent manuscript. A screen for PTGS agents against arbitrary target mRNAs can be conducted simply by cloning the full length target cDNA (from transcription start to just before the first polyA signal) into the pTarget-IRES-SEAP plasmid and forming a stable HEK293S cell line. A series of PTGS expression plasmids are made and transfected into the Target-IRES-SEAP cell line and the SEAP activity assay conducted and statistically evaluated to find the agent that exerts the greatest knockdown. This constitutes a lead therapeutic candidate to which optimization protocols can be applied to further enhance efficacy.
4.3. Improvement over Prior Approaches
Our fusion mRNA and SEAP-mediated reporter approach to HTS screening for PTGS agents improves upon prior methods. One approach fused the N-terminal portion of the p300 protein in-frame with the luciferase cDNA to generate a fusion mRNA and protein and then screened ribozymes against the target portion (Kawasaki et al., 1996). Similarly, another group fused an EGFP in frame with an oncogene protein (c-erbB-2) (Wichen et al., 1998). In these approaches the full length target could not be screened, and in generic form a fusion protein strategy would obviate screening of all potential target sites within the 3′UT, which are lost or displaced from the target coding sequences by interceding reporter sequences. Potential cleavage sites in the 5′UT and coding region are the only motifs that can be tested, but there may be accessible regions for potent knockdown in the 3′UT of any mRNA. Also, there is no isolation of target from reporter RNA sequences with this strategy, which could impact true target accessibility relative to what would occur in a native mRNA target. A luciferase reporter assay, albeit potentially useful as a HTS screen, requires cellular extraction, and cannot be used to follow PTGS kinetics in real time in live cells as can occur with a SEAP reporter HTS screen, and is complicated as typically two luciferase reporters are required to reference and normalize outcomes. Another approach sought to develop a HTS for antisense agents by fusing a full-length cDNA for a target into the 3′UT of either the luciferase or enhanced yellow fluorescent protein reporters (Husken et al., 2003). This strategy also fails to isolate the target and reporter RNA sequences, which can complicate equivalent accessibility of the true target mRNA component. While EYFP does not require cellular extraction for quantitative measurement, fluorescent proteins commonly have long cellular half-lives (>24 hrs) and any protein that is made from mRNA that is not destroyed by the PTGS agent simply accumulates over time intracellularly to compress the dynamic range of measure of knockdown. We initially explored short half life forms of EGFP for this HTS approach (not shown) and decided on the SEAP reporter because of the reliable and efficient measure of the secreted reporter enzyme. Our approach with the bicistronic Target-IRES-SEAP mRNA allows a full-length screen of all potential sites in a native target mRNA. The IRES element and the intrinsic biology of RNA folding, occurring concurrent with transcription, both act to isolate the upstream target RNA sequences from intrusion by downstream IRES isolator and reporter elements. Discrete secondary elements in such a dicistronic mRNA are expected to remain intrinsically stable (Liphardt et al., 2001). Computational analysis indicated that the placement of an IRES element between any of three arbitrary upstream target mRNAs (that play roles in hereditary retinal degenerations) (i.e. RHO, RPE65, RDH5) and the downstream SEAP element appears to function as an insulator to help to preserve native folding of the target mRNA. We expect that this Target-IRES-SEAP construction strategy will act to preserve native type folding and accessibility in true mRNA target sequences and further validate the identification of novel leads against the upstream target mRNAs.
A major issue in RNA Drug Discovery for PTGS agents is to be able to anticipate functional cellular performance before expensive and time consuming preclinical testing in animals. Our approach can identify lead candidates, could rationally assess optimizing variations on a single lead, and critically has potential to evaluate the kinetic landscape of PTGS in cellulo. As the reporter, SEAP, is secreted the cellular reaction mechanism for target knockdown can be sampled over time without cell perturbation or harvesting. Finally, and critically, this approach reduced the time needed to develop and optimize a PTGS agent between one and two orders of magnitude compared to the classical approach used previously in this lab that involved slow, variable, and cumbersome tools (e.g. Western analysis). Incorporating robotic tools onto this platform would greatly extend current HTS capacity by enhancing speed in sample processing and further minimizing variation due to manual pipetting. Robotic tools are available to conduct transfections, fluid transfers, and to integrate with fluorescent plate readers. We anticipate that this SEAP-based HTS approach can set a new standard for large scale HTS screening of antisense, ribozyme, or RNAi agents. HTS and high content screening are necessary to relieve the substantial bottlenecks in RNA drug discovery and allow refined and optimized strongly efficacious PTGS agents to emerge for therapeutic preclinical studies (Sullivan et al., 2008). We further expect that such HTS tool development will strongly aid in wrestling RNA biocomplexity that impacts both target mRNA accessibility and PTGS ligand conformational landscapes that both influence functional knockdown performance.
4.4. Advantages, Potential Disadvantages, and Possible Improvements of the PTGS Screen Design
4.4.1. Advantages
Relative to our prior study for lead identification that used Western analysis the current SEAP-based screen is on the order of 60-fold faster, with lower variance and increased sensitivity. The approach is readily adapted to HTS through robotics for fluid handling operations. The bicistronic Target-IRES-SEAP reporter fusion RNA approach allows a broad based screen of many hhRz or shRNA plasmid constructs to identify a lead candidate. The bicistronic approach can also be used for lead optimization, although final outcomes should always be tested against full length target mRNA without the reporter and insulator elements. The target fragment approach, cloned into the boundary of the coding/3′UTR of SEAP cDNA, allows a more focused approach to develop or optimize PTGS agents targeting local structural regions of the target mRNA, but is more tedious because of the required construction of multiple expression constructs.
Another advantage of our HTS SEAP approach is that approximately 99% of SEAP protein is secreted from the cells into the media such that the extracellular levels of SEAP protein reflect the steady-state levels of the mRNA encoding the reporter protein (Berger et al., 1988). There is little intracellular SEAP protein accumulation that would otherwise preclude reliable and prolonged measures of proportional levels of the steady-state mRNA. And, since the extracellular accumulated SEAP protein has an extremely long half-life at 37°C (>500 hrs) (Weber et al., 2007), its measure in sampled extracellular media directly reflects the temporally integrated effect of the PTGS agent to knockdown target mRNA in live cells. We showed that the SEAP HTS expression assay can be reliably used to examine the long-term kinetics of PTGS suppression of steady-state bicistronic target mRNA concentration inside live human cells. This property, while not exploited here, will allow assessment of the impact of varying molar template ratios of hhRz/target expression plasmids, to achieve varying enzyme/target RNA ratios in cells, on the level of target suppression or knockdown. In cellulo hhRz kinetics under varying enzyme/substrate RNA ratio is expected to provide a strong assessment of relative potency of different lead agents in cells. For example, one could evaluate PTGS kinetics in live human cells over longer periods of time under conditions of varying levels of PTGS plasmid. In vivo kinetic measures of PTGS in live cells are feasible with this approach and are relevant to understanding real time intracellular performance of candidate RNA drugs that can help to estimate behavior prior to expensive and slow testing in animal disease models.
4.4.2. Potential Disadvantages
The current approach to lead identification is entirely a rational one that is driven by initial computational or experimental approaches to assess target mRNA accessibility. Any errors in the accessibility mapping could severely limit identification of efficacious lead agents. While the mppRNA approach to target mRNA has clearly lead us to catalytic agents functional in human cells for the target RHO mRNA, we cannot prove that there are not other regions in the target mRNA that are more amenable to hhRz or shRNA attack. This strategy needs to be extended to other target mRNAs to further establish and validate its utility. Another vision science lab, aware of our initial prior results, successfully utilized this SEAP-based reporter approach to identify ribozyme and siRNA agents to target retinitis pigmentosa GTPase regulator (RPGR), a disease target implicated in X-linked retinitis pigmentosa (Chen B, O’Donoghue MB, Lewin AS, Gorbatyuk MS. 2008. Testing RPGR specific ribozyme and siRNA in vitro: tools for the treatment of dominant X-linked retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 49: E-Abstract 5345). Concerns over the intrusion of IRES or SEAP components of the fusion RNA into true upstream target folding in the bicistronic mRNA appear low given the RNA folding outcomes and the cellular expression of SEAP from the bicistronic RNA. Indeed, the IRES element appears to function as an insulator element.
4.4.3. Potential Improvements
SEAP is a simple reporter enzyme phosphatase that is secreted. The signals could be normalized to transfected cells by added pEGFP-N1 to the transfection mix. In some cases it may be of interest to use an alternative secreted enzyme such as Luciferase and to use that to conduct an alternative luminescence quantitative assay. The secreted Luciferase could be normalized to an alternative cell based form of luciferase. There could be some inference to try to reduce the size of the IRES insulator element (e.g. to a P2A element); however, shorter elements, while potentially effective, are unlikely to serve equivalent insulator functions relative to the full length IRES element from ECMV because the larger structures offer more folding stability (insulation) between upstream target and downstream SEAP components. It may be feasible to convert the Target-IRES-SEAP plasmid into one in which one can positively select for effective knockdown sequences (e.g. replacing SEAP with HSV thymidine kinase for selection of cells in ganciclovir-cells that survive GCV must have substantially reduced bicistronic mRNA and thymidine kinase protein). Finally, it may be feasible to model the gene expression paradigm established here in order to better relate the suppression of the fusion mRNA to the variation in secretion of the SEAP protein (e.g. Hoops et al., 2006)
4.5. Transcription of Target mRNAs from cDNAs as Opposed to Genomic Constructs
In the context of HTS for lead agents in RNA Drug Discovery does the transcription of the target mRNA from a cDNA impact identification of the most potent lead agent relative to testing against a target transcribed from a genomic construct as would occur in vivo? Pre-mRNAs are folding into stable secondary structures concurrent with ongoing transcription, splicing and further processing in the nucleus to a mature mRNA on the timescale of only tens of minutes for most average sized mRNAs. The slow kinetic timescale of PTGS agents (ribozymes, shRNAs), at approximately 1/min, indicates that attempting to cleave the target mRNA while it is a pre-mRNA during its brief sojourn in the nucleus is fraught with difficulty. The best location for targeting is in the cytoplasm where most mature mRNAs encoding proteins relevant to retinal or ocular diseases have their greatest lifetime and where the probability of substantial target knockdown is greatly improved. But does the history of intron splicing events possibly impact the structure of a mature mRNA which may not be represented in drug discovery when the target is transcribed by a cDNA? We conducted mppRNA on both the full length human RHO and on the full length human RHO pre-mRNA (with introns and exons). We investigated Exon 3 which contains the lead 725 hhRz/shRNA cleavage site. There was similar or precise accessibility maps over the Exon 3 region comparing the RHO mature and pre-mRNAs (data not shown). While this singular result must be tested against multiple potential cleavage sites in other exons and in other disease targets it makes sense that there would be similarities. RNA folding occurs co-transcriptionally into stable local secondary structures that are likely to be present before and after the introns are removed, or to emerge in the local neighborhood between exonic boundaries. The slow settling of mature mRNAs into native states after intron processing are likely to reflect stable local secondary structures that appear to be well-sampled by our mppRNA approach. Nevertheless, further bioinformatics and empirical work is needed to more rigorously assess this hypothesis.
Conclusions
We designed and tested strategies to conduct efficient, low variability HTS screens of PTGS agents in the cultured cell environment. The goal was to develop an effective strategy to identify, among many possibilities, a lead candidate PTGS agent that can be subsequently optimized in further studies. The strategy can be rapidly applied to an arbitrary disease target mRNA and is relatively simple such that it can be conducted in a routine molecular biological laboratory or in academic or corporate RNA drug discovery laboratories.
Supplementary Material
Highlights.
Method to rapidly screen for effective and potent Post-transcriptional gene silencing agents under conditions amenable to high throughput screening.
Exploits RNA fusion technology to evaluate entire target mRNAs or explicit regions known to be accessible to annealing.
Has been used to identify strong lead ribozyme and shRNA agents against human RHO mRNA, a retinal degeneration disease target mRNA.
Can be applied to an arbitrary disease target mRNA.
Acknowledgments
We acknowledge the contribution of R. Thomas Taggart, Ph.D. to the construction of a plasmid (to be described in full elsewhere) in which hRHO transcription is initiated at true transcription start by the CMV promoter. We acknowledge the effort of the lab of Caroline E. Bass, Ph.D. (Department of Pharmacology, University at Buffalo) who removed the RHO gene from the pRHO-IRES-SEAP plasmid and ligated in a multiple cloning site adapter to form the pTarget-IRES-SEAP plasmid for their own use. We appreciate the critical read of the manuscript by Steven J. Fliesler, Ph.D. (Director of Vision Research, Dept. of Ophthalmology, University at Buffalo; and Research Service, VA Western New York Healthcare System). The Department of Ophthalmology at the University at Buffalo is a member of the SUNY Eye Institute. This work was conducted at, and supported in part by, facilities and resources provided by the Veterans Administration Western New York Healthcare System. The contents do not represent the views of the Department of Veterans Affairs or the US Government. We appreciate the constructive comments of the reviewers in particular the insightful concern about use of cDNAs vs genomic expression constructs to evaluate target suppression by PTGS agents.
Funding
National Eye Institute (R01 EY13433-09, PI: JMS); VA Merit Award (1I01-BX000669-05, PI: JMS); NEI R24 UB Vision Infrastructure grant (NEI/NIH R24 grant EY016662 (UB Vision Infrastructure Center, PI: M Slaughter, Director-Biophotonics Module: JMS)); SUNY Health Now Award (Coordinating PI: JMS); Research to Prevent Blindness-Challenge and Unrestricted Grants (to the Department of Ophthalmology at the University at Buffalo); grant from the Oishei Foundation (Buffalo, NY) to the Department of Ophthalmology at University at Buffalo.
ABBREVIATIONS
- ANOVA
analysis of variance
- bp
base pair
- CV
coefficient of variation
- EGFP
enhanced green fluorescent protein
- FWHM
full width half maximum
- HEK293S
human embryonic kidney cells-suspension adapted
- HEK293S-RHO-IRES-SEAP
HEK293S cells stably expressing RHO-IRES-SEAP
- HEK293S-SEAP
HEK293S cells stably expressing SEAP
- hhRz
hammerhead ribozyme
- HTS
high-throughput screening
- IRES
internal ribosome entry site
- MFE
minimum folding energy
- mppRNA
multi-parameter prediction of RNA accessibility
- 4-MUP
4-methylumbelliferyl-phosphate
- nt
nucleotide
- NUH↓
ribozyme cleavage motif where N is any nucleotide, U is uridine, and H is any nucleotide excluding guanosine
- PLAP
placental alkaline phosphatase
- PTGS
post transcriptional gene silencing
- RHO
rod opsin
- RISC
RNA-induced silencing complex
- RNAi
RNA interference
- SEAP
secreted alkaline phosphatase
- SEM
standard error of mean
- shRNA
short-hairpin RNA
- siRNA
short interfering RNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceived and designed the experiments: JMS, EHY. Performed the experiments: EHY, MCB, JMS. Analyzed the data: EHY, JMS. Wrote the paper: EHY, JMS.
References
- Abdelmaksoud HEB, Yau EH, Zuker M, Sullivan JM. Development of lead hammerhead ribozyme candidates against human rod opsin for retinal degeneration therapy. Exp Eye Res. 2009;88(5):859–879. doi: 10.1016/j.exer.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarzguioui M, Prydz H. Hammerhead ribozyme design and application. Cell Mol Life Sci. 1998;54(11):1175–1202. doi: 10.1007/s000180050247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarzguioui M, Brede G, Babale E, Grotli M, Sproat B, Prydz H. Secondary structure prediction and in vitro accessibility of mRNA as tools in the selection of target sites for ribozymes. Nucleic Acids Res. 2000;28:4113–4124. doi: 10.1093/nar/28.21.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KP, Fox MC, Brown-Driver V, Martin MJ, Azad RF. Inhibition of cytomegalovirus immediate-early gene expression by an antisense oligonucleotide complementary to immediate-early RNA. Antimicrob Agents Chemother. 1996;40(9):2004–2011. doi: 10.1128/aac.40.9.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Hauber J, Hauber R, Geiger R, Cullen B. Secreted placental alkaline phosphatase: a powerful new quantitative indicator of gene expression in eukaryotic cells. Gene. 1988;66(1):1–10. doi: 10.1016/0378-1119(88)90219-3. [DOI] [PubMed] [Google Scholar]
- Bertrand E, Pictet R, Grange T. Can hammerhead ribozymes be efficient tools to inactivate gene function? Nucleic Acids Res. 1994;22(3):293–300. doi: 10.1093/nar/22.3.293. Corrigendum 22(7): 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E, Castanotto D, Zhou C, Carbonnelle C, Lee NS, Good P, Chatterjee S, Grange T, Pictet R, Kohn D, Engelke D, Rossi JJ. The expression cassette determines the functional activities of ribozymes in mammalian cells by controlling their intracellular localization. RNA. 1997;3:75–88. [PMC free article] [PubMed] [Google Scholar]
- Birikh KR, Heaton PA, Eckstein F. The structure, function and application of the hammerhead ribozyme. Eur J Biochem. 1997;245(1):1–16. doi: 10.1111/j.1432-1033.1997.t01-3-00001.x. [DOI] [PubMed] [Google Scholar]
- Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, Smith Q, Beijersbergen RL, Ghazal P, Shamu CE. Statistical methods for analysis of high throughput RNA interference screens. Nature Methods. 2009;6(8):569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Chu CY, Rana TM. Target accessibility dictates the potency of human RISC. Nat Struct Mol Biol. 2005;12:469–470. doi: 10.1038/nsmb931. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296(5567):550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Cashman SM, Binkley EA, Kumar-Singh R. Towards mutation–independent silencing of genes involved in retinal degeneration by RNA interference. Gene Therapy. 2005;12:1223–1228. doi: 10.1038/sj.gt.3302512. [DOI] [PubMed] [Google Scholar]
- De la Pena M, Gago S, Flores R. Peripheral regions of natural hammerhead ribozymes greatly increase their self-cleavage activity. EMBO J. 2003;22(20):5561–5570. doi: 10.1093/emboj/cdg530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Chan C, Lawrence C. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004;32:W135–W141. doi: 10.1093/nar/gkh449. (Web Server Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja T, McGee T, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343(6256):364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Farrar GJ, Kenna PF, Humphries P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 2002;21:857–864. doi: 10.1093/emboj/21.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores R, Serra P, Minoia S, DiSerio F, Navarro B. Viroids: from genotype to phenotype just relying on RNA sequence and structural motifs. Front Microbiol. 2012;3:217. doi: 10.3389/fmicb.2012.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal A, Apfelstedt-Sylla E, Janecke A, Zrenner E. Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Prog Retin Eye Res. 1997;16(1):51–79. [Google Scholar]
- Gorbatyuk MS, Pang JJ, Thomas J, Jr, Hauswirth WW, Lewin AS. Knockdown of wild-type mouse rhodopsin using an AAV vectored ribozyme as part of an RNA replacement approach. Mol Vis. 2005;11:648–656. [PubMed] [Google Scholar]
- Gorbatyuk M, Justilien V, Hauswirth WW, Lewin AS. Preservation of photoreceptor morphology and function in P23H rats using an allele independent ribozyme. Exp Eye Res. 2007;84(1):44–52. doi: 10.1016/j.exer.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseloff J, Gerlach WL. Simple RNA enzymes with new and highly specific endoribonuclease activities. Nature. 1988;334(6183):585–91. doi: 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- Hasselblatt P, Hockenjos B, Thoma C, Blum HE, Offensperger WB. Translation of stable hepadnaviral mRNA cleavage fragments induced by the action of phosphorothioate-modified antisense oligodeoxynucleotides. Nucleic Acids Res. 2005;33(1):114–125. doi: 10.1093/nar/gki155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth WW, Lewin AS. Ribozyme uses in retinal gene therapy. Prog Retin Eye Res. 2000;19(6):689–710. doi: 10.1016/s1350-9462(00)00007-0. [DOI] [PubMed] [Google Scholar]
- Hertel KJ, Pardi A, Uhlenbeck OC, et al. Numbering system for the hammerhead. Nucleic Acids Res. 1992;20(12):3252. doi: 10.1093/nar/20.12.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann M, Tabler M, Tzortzakaki S, Sczakiel G. Extension of helix II of an HIV-1-directed hammerhead Rz with long antisense flanks does not alter kinetic parameters in vitro but causes loss of the inhibitory potential in living cells. Nucleic Acids Res. 1994;22:3951–3957. doi: 10.1093/nar/22.19.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. COPASI- a COmplex PAthway Simulator. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- Hormes R, Homann M, Oelze I, Marschall P, Tabler M, Eckstein F, Sczakiel G. The subcellular localization and length of hammerhead ribozymes determine efficacy in human cells. Nucleic Acids Res. 1997;25:769–775. doi: 10.1093/nar/25.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husken D, Asselbergs F, Kinzel B, et al. mRNA fusion constructs serve in a general cell-based assay to profile oligonucleotides activity. Nucleic Acids Res. 2003;31(17):e102. doi: 10.1093/nar/gng103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabs DA, Griffiths PD. Fomivirsen for the treatment of cytomegalovirus retinitis. Amer J Ophthalmol. 2002;133(4):552–556. doi: 10.1016/s0002-9394(02)01325-9. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Ohkawa J, Tanishige N, et al. Selection of the best target site for ribozyme-mediated cleavage within a fusion gene for adenovirus E1A-associated 300 kDa protein (p300) and luciferase. Nucleic Acids Res. 1996;24(15):3010–3016. doi: 10.1093/nar/24.15.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Lescoute A, Westhof E, Jayasena S. Sequence elements outside the hammerhead ribozyme catalytic core enable intracellular activity. Nat Struct Biol. 2003;10(9):708–712. doi: 10.1038/nsb959. [DOI] [PubMed] [Google Scholar]
- Koizumi M, Kamiya H, Ohtsuka E. Inhibition of c-Ha-ras gene expression by hammerhead ribozymes containing a stable C(UUCG)G hairpin loop. Biol Pharm Bull. 1993;16:879–883. doi: 10.1248/bpb.16.879. [DOI] [PubMed] [Google Scholar]
- Kolniak TA, Sullivan JM. Rapid, cell-based toxicity screen of potentially therapeutic post-transcriptional gene silencing agents. Exp Eye Res. 2011;92:328–337. doi: 10.1016/j.exer.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin AS, Hauswirth WW. Ribozyme gene therapy: applications for molecular medicine. Trends Mol Med. 2001;7(5):221–228. doi: 10.1016/s1471-4914(01)01965-7. [DOI] [PubMed] [Google Scholar]
- Lieber A, Strauss M. Selection of efficient cleavage sites in target RNAs by using a ribozyme expression library. Mol Cell Biol. 1995;15(1):540–51. doi: 10.1128/mcb.15.1.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima WF, Monia BP, Ecker DJ, Freier SM. Implication of RNA structure on antisense oligonucleotide hybridization kinetics. Biochemistry. 1992;31:12055–12061. doi: 10.1021/bi00163a013. [DOI] [PubMed] [Google Scholar]
- Liphardt J, Onoa B, Smith SB, Tinoco I, Jr, Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–737. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- Martick M, Scott W. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126(2):309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Disney MD, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH. Revolutions in RNA secondary structure prediction. J Mol Biol. 2006;359:526–532. doi: 10.1016/j.jmb.2006.01.067. [DOI] [PubMed] [Google Scholar]
- Merki E, Graham MJ, Mullick AE, Miller ER, Crooke RM, Pitas RE, Witztum JL, Tsimikas S. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118:743–753. doi: 10.1161/CIRCULATIONAHA.108.786822. [DOI] [PubMed] [Google Scholar]
- Millington-Ward S, O’Neill B, Tuohy G, Al-Jandel N, Kiang AS, Kenna PF, Palfi A, Hayden P, Mansergh F, Kennan A, Humphries P, Farrar GJ. Strategems in vitro for gene therapies directed to dominant mutations. Hum Mol Genet. 1997;6:1415–1426. doi: 10.1093/hmg/6.9.1415. [DOI] [PubMed] [Google Scholar]
- Montgomery RA, Dietz HC. Inhibition of fibrillin 1 expression using U1 snRNA as a vehicle for the presentation of antisense targeting sequence. Hum Mol Genet. 1997;6:519–525. doi: 10.1093/hmg/6.4.519. [DOI] [PubMed] [Google Scholar]
- Patzel V, Sczakiel G. Theoretical design of antisense RNA structures substantially improves annealing kinetics and efficacy in human cells. Nat Biotechnol. 1998;16(1):64–68. doi: 10.1038/nbt0198-64. [DOI] [PubMed] [Google Scholar]
- Patzel V, Steidl U, Kronenwett R, Haas R, Sczakiel G. A theoretical approach to select effective antisense oligodeoxyribonucleotides at high statistical probability. Nucleic Acids Res. 1999;27(22):4328–4334. doi: 10.1093/nar/27.22.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penedo J, Wilson T, Jayasena S, Khvorova A, Lilley D. Folding of the natural hammerhead ribozyme is enhanced by interaction of auxiliary elements. RNA. 2004;10(5):880–888. doi: 10.1261/rna.5268404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriman R, Delves A, Gerlach WL. Extended target-site specificity for a hammerhead ribozyme. Gene. 1992;113(2):157–163. doi: 10.1016/0378-1119(92)90391-2. [DOI] [PubMed] [Google Scholar]
- Rossi JJ. Expression strategies for short hairpin RNA interference triggers. Hum Gene Ther. 2008;19(4):313–317. doi: 10.1089/hum.2008.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner DE, Stormo GD, Uhlenbeck OC. Sequence requirements of the hammerhead RNA self-cleavage reaction. Biochemistry. 1990;29(47):10695–10702. doi: 10.1021/bi00499a018. [DOI] [PubMed] [Google Scholar]
- Scherr M, Rossi JJ. Rapid determination and quantitation of the accessibility to native RNAs by antisense oligodeoxynucleotides in murine cell extracts. Nucleic Acids Res. 1998;26:5079–5085. doi: 10.1093/nar/26.22.5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherr M, Rossi J, Sczakiel G, Patzel V. RNA accessibility prediction: a theoretical approach is consistent with experimental studies in cell extracts. Nucleic Acids Res. 2000;28(13):2455–2461. doi: 10.1093/nar/28.13.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Wu S, Chan CY, Klapper JR, Schneider E, Ding Y. A structural analysis of in vitro catalytic activities of hammerhead ribozymes. BMC Bioinformatics. 2007;8:469. doi: 10.1186/1471-2105-8-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimayama T, Nishikawa S, Taira K. Generality of the NUX rule: kinetic analysis of the results of systematic mutations in the trinucleotide at the cleavage site of hammerhead ribozymes. Biochemistry. 1995;34:3649–3654. doi: 10.1021/bi00011a020. [DOI] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16(11):1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stigbrand T. Present status and future trends of human alkaline phosphatases. Prog Clin Biol Res. 1984;166:3–14. [PubMed] [Google Scholar]
- Stillman B, Gluzman Y. Replication and supercoiling of Simian virus 40 DNA in cell extracts from human cells. Mol Cell Biol. 1985;5(8):2051–2060. doi: 10.1128/mcb.5.8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JM, Satchwell M. Development of stable cell lines expressing high levels of point mutants of human opsin for biochemical and biophysical studies. Methods Enzymol. 2000;315:30–58. doi: 10.1016/s0076-6879(00)15833-1. [DOI] [PubMed] [Google Scholar]
- Sullivan JM, Pietras K, Shin B, Misasi J. Hammerhead ribozymes designed to cleave all human rod opsin mRNAs which cause autosomal dominant retinitis pigmentosa. Mol Vis. 2002;8:102–113. [PubMed] [Google Scholar]
- Sullivan JM, Yau EH, Taggart RT, Butler MC, Kolniak TA. Bottlenecks in development of retinal therapeutic post-transcriptional gene silencing agents. Vision Res. 2008;48(3):453–469. doi: 10.1016/j.visres.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JM, Yau EH, Kolniak TA, Sheflin LG, Taggart RT, Abdelmaksoud HE. Variables and strategies in development of therapeutic post-transcriptional gene silencing agents. Journal of Ophthalmology. 2011;2011 doi: 10.1155/2011/531380). Article ID 531380, 31 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JM, Yau EH, Taggart RT, Butler MC, Kolniak TA. Relieving bottlenecks in RNA drug discovery for retinal diseases. Adv Exp Med Biol. 2012;2012; 723:145–153. doi: 10.1007/978-1-4614-0631-0_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Breaker R. Examination of the catalytic fitness of the hammerhead ribozyme by in vitro selection. RNA. 1997;3(8):914–925. [PMC free article] [PubMed] [Google Scholar]
- Thoma C, Hasselblatt P, Kock J, et al. Generation of stable mRNA fragments and translation of N-truncated proteins induced by antisense oligodeoxynucleotides. Mol Cell. 2001;8(4):865–872. doi: 10.1016/s1097-2765(01)00364-1. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Ayers DF, Malmstrom TA, McKenzie TL, Ganousis L, Chowrira BM, Couture L, Stinchcomb DT. Improved accumulation and activity of ribozymes expressed from a tRNA-based RNA polymerase III promoter. Nucleic Acids Res. 1995;23:2259–2268. doi: 10.1093/nar/23.12.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco I, Bustamante C. How RNA folds. J Mol Biol. 1999;293(2):271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gauss P, Thermes C, Groebe DR, Gayle M, Guild N, Stormo G, D’Aubenton-Carafa Y, Uhlenbeck OC, Tinoco I, Brody EN, Gold L. CUUCGG hairpins: extraordinarily stable RNA secondary structures associated with various biochemical processes. Proc Natl Acad Sci USA. 1988;85:1364–1368. doi: 10.1073/pnas.85.5.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenbeck OC. A small catalytic oligoribonucleotide. Nature. 1987;328(6131):596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- Vaish NK, Kore AR, Eckstein F. Recent developments in the hammerhead ribozyme field. Nucleic Acids Res. 1998;26(23):5237–5242. doi: 10.1093/nar/26.23.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W, Stelling J, Rimann M, Keller B, Daoud-El Baba M, Weber CC, Aubel D, Fussenegger M. A synthetic time-delay circuit in mammalian cells and mice. Proc Natl Acad Sci USA. 2007;104(8):2643–2648. doi: 10.1073/pnas.0606398104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechen K, Zimmer C, Dietel M. Selection of a high activity c-erbB-2 ribozyme using a fusion gene c-erbB-2 and the enhanced green fluorescent protein. Cancer Gene Ther. 1998;5(1):45–51. [PubMed] [Google Scholar]
- Zakharchuk A, Doronin K, Karpov V, Krougliak V, Naroditsky B. The fowl adenovirus type 1 (CELO) virus-associated RNA-encoding gene: a new ribozyme-expression vector. Gene. 1995;161(2):189–193. doi: 10.1016/0378-1119(95)00251-z. [DOI] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. Second. Prentice-Hall; Englewood Cliffs, NJ: 1984. p. 718. [Google Scholar]
- Zoumadakis M, Tabler M. Comparative analysis of cleavage rates after systematic permutation of the NUX consensus target motif for hammerhead ribozymes. Nucleic Acids Res. 1995;23(7):1192–1196. doi: 10.1093/nar/23.7.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.