

Abstract
We previously reported in atrial myocytes that inhibition of cAMP-dependent protein kinase (PKA) by laminin (LMN)-integrin signaling activates β2-adrenergic receptor (β2-AR) stimulation of cytosolic phospholipase A2 (cPLA2). The present study sought to determine the signaling mechanisms by which inhibition of PKA activates β2-AR stimulation of cPLA2. We therefore determined the effects of zinterol (0.1 μM; zint-β2-AR) to stimulate ICa,L in atrial myocytes in the absence (+PKA) and presence (-PKA) of the PKA inhibitor (1 μM) KT5720 and compared these results with atrial myocytes attached to laminin (+LMN). Inhibition of Raf-1 (10 μM GW5074), phospholipase C (PLC; 0.5 μM edelfosine), PKC (4 μM chelerythrine) or IP3 receptor (IP3R) signaling (2 μM 2-APB) significantly inhibited zint-β2-AR stimulation of ICa,L in–PKA but not +PKA myocytes. Western blots showed that zint-β2-AR stimulation increased ERK1/2 phosphorylation in–PKA compared to +PKA myocytes. Adenoviral (Adv) expression of dominant negative (dn) -PKCα, dn-Raf-1 or an IP3 affinity trap, each inhibited zint-β2-AR stimulation of ICa,L in + LMN myocytes compared to control +LMN myocytes infected with Adv-βgal. In +LMN myocytes, zint-β2-AR stimulation of ICa,L was enhanced by adenoviral overexpression of wild-type cPLA2 and inhibited by double dn-cPLA2S505A/S515A mutant compared to control +LMN myocytes infected with Adv-βgal. In–PKA myocytes depletion of intracellular Ca2+ stores by 5 μM thapsigargin failed to inhibit zint-β2-AR stimulation of ICa,L via cPLA2. However, disruption of caveolae formation by 10 mM methyl-β-cyclodextrin inhibited zint-β2-AR stimulation of ICa,L in–PKA myocytes significantly more than in +PKA myocytes. We conclude that inhibition of PKA removes inhibition of Raf-1 and thereby allows β2-AR stimulation to act via PKCα/Raf-1/MEK/ERK1/2 and IP3-mediated Ca2+ signaling to stimulate cPLA2 signaling within caveolae. These findings may be relevant to the remodeling of β-AR signaling in failing and/or aging heart, both of which exhibit decreases in adenylate cyclase activity.
Introduction
We previously reported that attachment of atrial myocytes to the extracellular matrix protein laminin (LMN) acts via β1 integrin receptors to decrease β1-adrenergic receptor (AR) and increase β2-AR stimulation of L-type Ca2+ current (ICa,L) [1]. Cell attachment to LMN decreases β1-AR signaling by inhibiting adenylate cyclase activity and diminishing cAMP levels via integrin-dependent activation of focal adhesion kinase (FAK)/phosphatidyinositol-3’ kinase (PI-3K)/protein kinase B (Akt) signaling [2]. We also reported that atrial cell attachment to LMN enhances β2-AR signaling by activating Gi/ERK/cytosolic phospholipase A2 (cPLA2)/arachidonic acid (AA) stimulation of ICa,L [3]. β2-AR activation of cPLA2 signaling is dependent on concomitant LMN-mediated inhibition of adenylate cyclase/cAMP-dependent kinase (PKA) [3]. In other words, cell attachment to LMN acts via inhibition of adenylate cyclase/PKA to both inhibit β1-AR signaling and enhance β2-AR signaling through activation of cPLA2. In embryonic chick ventricular myocytes [4] and rat ventricular myocytes [5] β2-AR stimulation also activates cPLA2/AA signaling. Moreover, these authors proposed that activation of β2-AR/cPLA2 signaling may compensate for depressed cAMP signaling [4]. Interestingly, in both of these studies by Pavoine et al. (1999) and Ait-Mamar et al., (2005) cardiomyocytes were cultured on LMN, supporting our findings that cell attachment to LMN may be responsible for inhibition of PKA and activation of β2-AR/cPLA2 signaling. However, the mechanism by which PKA inhibition activates β2-AR/cPLA2 signaling is not clear.
Our initial experiments indicated that in atrial myocytes β2-AR activation of cPLA2 is Ca2+-dependent and mediated via ERK1/2 signaling [3]. This is consistent with studies in embryonic chick ventricular myocytes (cultured on LMN) in which β2-AR stimulation acts via ERK1/2 signaling to activate cPLA2 [6]. Moreover, in a variety of cell systems Raf-1 activates downstream ERK1/2 and PKA inhibits Raf-1 [7, 8]. Therefore, inhibition of PKA should remove inhibition of Raf-1, thereby allowing β2-AR stimulation to act via Raf-1/MEK/ERK1/2 signaling. Moreover, protein kinase C (PKC) activates Raf-1 [9, 10]. In other words, PKA inhibits and PKC activates Raf-1/MEK/ERK1/2 signaling. Based on these considerations we sought to determine whether inhibition of PKA facilitates β2-AR stimulation to act via PKC/Raf-1/MEK/ERK1/2 to activate cPLA2. These findings may be relevant to the remodeling of β2-AR signaling in the failing and/or aging heart, both of which exhibit decreases in adenylate cyclase activity.
Materials and Methods
Ethics Statement
The animal and experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of Loyola University Medical Center, Maywood, IL. IACUC prescribed the rules for the animal care and supervised their enforcement. Animals were obtained from a licensed vendor (R & R Research, Howard City, MI., USA), and housed and fed in our AAALAC approved Comparative Medicine Department. Adult cats of either sex (n = 32 cats) were anesthetized with sodium pentobarbital (50 mg/kg, IP).
Isolation of atrial myocytes
Once fully anesthetized, a bilateral thoracotomy was performed, and the heart was rapidly excised and mounted on a Langendorff perfusion apparatus. After enzyme (collagenase; type II, Worthington Biochemical) digestion, atrial myocytes were isolated as previously reported [11].
Perforated patch clamp experiments
Electrophysiological recordings from atrial myocytes were performed in the perforated (nystatin) patch whole-cell configuration at room temperature, as previously described [11]. L-type Ca2+ current (ICa,L) was activated by depolarizing pulses from a holding potential of -40 mV to 0 mV for 200 ms every 5 s and measured in relation to steady-state current. β2-AR stimulation was achieved by 0.1 μM zinterol (zint-β2-AR), a specific β2-AR agonist [12]. Agonist was applied for approximately 4 min and the effects on peak ICa,L amplitude were recorded at the steady-state response.
Plating of atrial myocytes on LMN coated glass coverslips
Generally, we compared freshly isolated atrial myocytes obtained from the same hearts, as previously described [3]; atrial myocytes on uncoated glass cover-slips in the absence of PKA inhibitor (+PKA) and atrial myocytes on uncoated glass coverslips exposed to the specific PKA inhibitor (1 μM) KT5720 (-PKA). Because pharmacological inhibition of PKA elicits signaling mechanisms that are similar to those elicited by LMN-integrin signaling, we performed some experiments on atrial myocytes attached to glass cover-slips coated with laminin (+LMN; 40 ug/ml) for at least 2 hrs, as previously described [11]. Inhibition of PKA by KT5720 typically decreases basal ICa,L amplitude by 15–20% [3], consistent with the relatively high endogenous PKA activity in cat atrial myocytes [11]. In addition, a variety of experimental results indicate that atrial cell attachment to LMN is not restoring LMN-mediated signaling somehow lost during the cell isolation procedure. For example, control experiments have shown that atrial myocytes plated on poly-L-lysine, a non-specific substrate for cell attachment, fail to exhibit changes in β-AR signaling similar to cells attached to LMN [1]. Moreover, freshly isolated cardiomyocytes not plated on LMN exhibit responses to β-AR stimulation which are similar to multicellular cardiac preparations, i.e. exhibit predominantly β1-AR over β2-AR signaling [1]. However, cell attachment to LMN decreases the β1-/β2-AR signaling ratio resulting in predominantly β2-AR over β1-AR signaling [1]. Moreover, pharmacological inhibition of PKA in myocytes not attached to LMN mimics the effects of cell attachment to LMN [3].
Adenoviral infection of atrial myocytes
In some experiments, atrial myocytes were attached to laminin (2h) and then infected (100 moi, 24h) with replication-defective adenovirus (Adv) prior to electrophysiological recording. PKCα was inhibited by infection with an Adv expressing kinase-inactive mouse PKCα [13], kindly provided by Dr. Trevor Biden, Garvan Institute of Medical Research, St. Vincent’s Hospital, Sydney Australia. An Adv expressing a dominant-negative (dn) mutant of rabbit PKCε [14] was kindly provided by Dr. Peipei Ping, University of California-Los Angeles. Adv expressing wild type, and a non-phosphorylatable mutant of human cPLA2 (S515A/S505A double mutant) [15] were generously provided by Dr. K.U. Malik, University of Tennessee Health Science Center, Memphis, TN. An Adv expressing a dn mutant of human Raf-1 [9] was kindly provided by Dr. Dan Kuppuswamy, Medical University of South Carolina, Charleston, SC. A control Adv expressing nuclear-encoded β-galactosidase (Adv-βgal) was used to control for nonspecific effects of adenoviral infection [16]. Adenoviruses were amplified and purified using HEK293 cells, and the multiplicity of infection (moi) for each virus was determined by dilution assay in HEK293 cells grown in 96 well clusters, as previously described [16]. Preliminary experiments using 5-bromo-4-chloro-3-indolyl-beta- D-galactopyranoside (X-gal) staining of Adv-βgal infected cells determined that a concentration of 100 moi infected 93±3% (n = 3 expts, 400–700 cells/expt) of cultured myocytes. The IP3 affinity trap consists of the ligand binding domain of the rat type 1 IP3R. The construction of this vector and subsequent production of adenovirus was previously described in detail [17]. Freshly isolated atrial myocytes were infected with Adv-IP3 affinity trap or the Adv-βgal (control) for 1 h, followed by 18 h short-term culture at 37°C.
Western blot
Atrial myocytes were plated on poly-L-lysine, a biologically inactive substrate and therefore represent +PKA myocytes. Atrial myocytes were either untreated or treated with Zinterol, KT5720 alone (PKA inhibitor) and KT combined with Zinterol. A positive control showing ERK1/2 and p38MAPK phosphorylation in A7r5 cells is used after stimulation with 1 μM angiotensin II (5 min). Briefly atrial homogenates were centrifuged at 10,000 g for 2 min and the supernatant was collected. The protein concentrations of the samples were determined using the Bradford protein assay (Bio-Rad). Aliquots of the samples (40 μg) were dissolved in a Laemmli Sample buffer containing: 60 mM Tris-HCl, 2% SDS, 20% Glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue (pH 6.8) and the proteins were separated on a 4–15% Mini-PROTEAN TGX Gel (Bio-Rad). After transfer, the membrane was blocked with TBS-T Buffer containing 20 mM Tris pH 7.5, 150 mM NaCl, 0.05% Tween, and a 5% blocking powder (Bio Rad) at 4°C for 1 hr. The blot was probed with primary and secondary antibodies for phorpho-ERK1/2 and phosphor-p38MAPK that were purchased from cell signaling technologies (Danvers, MA). The blot was subsequently probed with GAPDH primary antibody for 2 hrs, followed by HRP-conjugated goat anti-mouse secondary antibody (1:10,000, Santa Cruz Biotechnology) at 4°C for 1 hr. Specific binding was visualized by chemiluminescence (Immun-Star Western C Kit, Bio-Rad) using a ChemiDoc XRS imager (Bio-Rad Life Science Research, Hercules, CA). The intensities of the bands corresponding to each protein were quantified using ImageLab software (Bio-Rad). The relative intensity for each band was normalized to the intensity of the GAPDH staining.
Chemicals
Zinterol, AACOCF3, GW5074, edelfosine (ET-18-OCH3), chelerythrine, 2-aminoethyl diphenyl borate (2-APB), KT5720, methyl-β-cyclodextrin, ryanodine, thapsigargin (Sigma Chemical).
Statistics
Data are mean ± standard error (SE) of the mean. Measurements were analyzed using either paired or unpaired Student’s t test for significance at P<0.05. Multiple comparisons were performed by ANOVA followed by a Student–Newman–Keuls test with significance at P<0.05.
Results
Role of Raf-1 in +PKA and –PKA atrial myocytes
As shown in Fig 1we determined the effects of 10 μM GW5074, a potent Raf-1 inhibitor [8] on zint-β2-AR stimulation of ICa,L in +PKA and–PKA atrial myocytes. In +PKA myocytes (A) 0.1 μM zint-β2-AR stimulation elicited a typical increase in ICa,L (124±12%, N = 3). In another group of +PKA myocytes from the same hearts (A), prior exposure to GW5074 for 30 min had no significant effects on basal ICa,L amplitude (open bar) or zint-β2-AR stimulation of ICa,L (126±11%, N = 3). In contrast, as shown in panel B, control zint-β2-AR stimulation of ICa,L in–PKA myocytes (B; 207±23%, N = 3) was enhanced compared to +PKA myocytes (A; 124±12%, N = 3) due to activation of cPLA2 signaling, as previously reported [4]. In–PKA myocytes, GW5074 had no effects on basal ICa,L amplitudes (open bars) but in contrast to +PKA myocytes, GW5074 significantly inhibited zint-β2-AR stimulation of ICa,L (B; 40±5%, N = 3). Additional experiments showed that zint-β2-AR stimulation of ICa,L in +LMN myocytes produced results similar to those found in–PKA myocytes (control; 192±13%, N = 3) and (GW5074; 33±9%, N = 3) (see S1 Fig for data).
Fig 1.

Effects of Raf-1 inhibition (10 μM GW5074) on zint-β2-AR stimulation of ICa,L in +PKA (A), -PKA (B) atrial myocytes. A; In +PKA myocytes, GW5074 (30 min) had no significant effect on zint-β2-AR stimulation of ICa,L. B; In -PKA myocytes, zint-β2-AR stimulation of ICa,L was enhanced compared to control responses (A) and GW5074 significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.05.
Comparison of zint-β2-AR induced ICa,L currents in the Adv-βgal and dn-Raf-1 mutant infected atrial myocytes
To further establish the role of Raf-1, we infected atrial myocytes with an Adv that expresses a dn-Raf-1 mutant (generously provided by Dr. Kuppuswamy [9, 18]. Control cells were infected with Adv-βgal. Infected cells were cultured on LMN overnight and therefore represent +LMN myocytes. As shown in Fig 2A, in control +LMN myocytes expressing βgal, zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L (202±19%). In +LMN myocytes expressing the dn-Raf-1 mutant, zint-β2-AR stimulation of ICa,L was significantly inhibited (105±14%; P<0.02) compared to control. Together, these findings indicate that Raf-1 signaling plays no role in β2-AR stimulation of ICa,L in freshly isolated atrial myocytes not attached to LMN (Fig 1A). However, when PKA is inhibited by either PKA inhibitor or cell attachment to LMN, β2-AR stimulation acts via Raf-1 to activate cPLA2 and stimulate ICa,L.
Fig 2. Effects of dn-Raf-1-Adv on zint-β2-AR stimulation of ICa,L in +LMN myocytes.

In control +LMN myocytes (βgal) zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L. In +LMN myocytes infected with dn-Raf-1-Adv zint-β2-AR stimulation of ICa,L was significantly inhibited compared with controls. Numbers in parentheses indicate that number of cells studied. * = P<0.05.
Inhibition of PKA, increases ERK1/2 phosphorylation in atrial myocytes
It is well established that Raf-1 activates downstream MEK/ERK1/2 signaling [7, 8]. Moreover, our previous results indicated that inhibition of MEK/ERK1/2 signaling by U0126 inhibited zint-β2-AR stimulation cPLA2 signaling [3]. Therefore, inhibition of PKA should activate β2-AR stimulation of ERK1/2 phosphorylation. As shown in Fig 3, we performed Western blots and probed for zint-β2-AR-mediated ERK1/2 and p38MAPK phosphorylation in the absence (+PKA) and presence of PKA inhibitor (–PKA). Atrial myocytes were plated on poly-L-lysine, a biologically inactive substrate and therefore represent +PKA myocytes. As shown in Fig 3in control +PKA myocytes zint-β2-AR stimulation modestly increased ERK1/2 phosphorylation (solid bars). Exposure of +PKA myocytes to the PKA inhibitor (1 μM) KT5720, (–PKA myocytes) further increased ERK/1/2 phosphorylation, suggesting that inhibition of basal PKA activity removes PKA-induced inhibition of basal ERK1/2 phosphorylation. In–PKA myocytes, zint-β2-AR stimulation prominently stimulated ERK1/2 phosphorylation. Moreover, zint-β2-AR stimulation only modestly stimulated p38MAPK phosphorylation (open bars), and was not significantly affected by KT5720. The last lane on the Western blot is a positive control showing ERK1/2 and p38MAPK phosphorylation in A7r5 cells stimulated by 1μM angiotensin II (5 min). In conjunction with our previous findings [3], the present results indicate that inhibition of PKA preferentially activates β2-AR stimulation of ERK1/2 signaling.
Fig 3. Effect of PKA inhibition on β2-AR-medited phosphorylation of ERK1/2.

Western blots of atrial cell homogenates show that in control -PKA myocytes zinterol modestly increased ERK1/2 phosphorylation (solid bars). Inhibition of PKA by 1 μM KT5720 (KT) increased basal ERK/1/2 phosphorylation. In the presence of KT5720, zinterol prominently stimulates ERK1/2 phosphorylation. Zinterol only modestly stimulated p38MAPK phosphorylation (open bars), and without significant effect by KT5720. The last lane on the Western blot is a positive control showing ERK1/2 and p38MAPK phosphorylation in A7r5 cells stimulated by 1 μM angiotensin II (5 min). Similar results were obtained in a total of 3 experiments. * = P<0.05.
Role of PLC in the enhanced zint-β2-AR induced ICa,L currents in –PKA atrial myocytes
It is well established that Raf-1 activates downstream MEK/ERK1/2 signaling [7, 8]. Moreover, our previous results indicated that inhibition of MEK/ERK1/2 signaling by U0126 inhibited zint-β2-AR stimulation of cPLA2 signaling [3]. This raises the question of how β2-AR stimulation activates Raf-1. In a variety of cell systems [10], including cardiac muscle [9], protein kinase C (PKC) activates Raf-1/MEK/ERK1/2. Receptor-mediated activation of phospholipase C (PLC) and the hydrolysis of phospholipids results in the production of diacylglycerol and stimulation of PKC isoenzymes. Therefore, as shown in Fig 4we determined the effects of 0.5 μM edelfosine, a specific inhibitor of phospholipase C [19], on zint-β2-AR stimulation of ICa,L in +PKA and–PKA myocytes. In control cells (A), edelfosine had little effect on basal ICa,L amplitude (open bars) or zint-β2-AR stimulation of ICa,L (zint, 125±3% vs edelfosine, 114±7%). In–PKA myocytes (B) zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L (215±5%) compared to control responses (A) due to activation of cPLA2 [3]. In contrast to +PKA myocytes, edelfosine now significantly inhibited zint-β2-AR stimulation of ICa,L (78±11%; P<0.0005). Additional experiments in +LMN myocytes showed results similar to those found in–PKA myocytes, i.e. zint, 204±2% vs edelfosine, 84±8%; P<0.03 (see S2 Fig for data). In other words, inhibition of PKA allows β2-AR stimulation to act via PLC to stimulate cPLA2 signaling.
Fig 4.

Effects of 0.5 μM edelfosine on zint-β2-AR stimulation of ICa,L in +PKA (A), -PKA(B) and atrial myocytes. A; in control +PKA myocytes edelfosine had no significant effects on zint-β2-AR stimulation of ICa,L. B; in -PKA myocytes zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L compared to controls (A) and edelfosine significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.05.
Role of PKC in the enhanced zint-β2-AR induced ICa,L currents in –PKA atrial myocytes
Because activation of PLC produces diacylglycerol and activation of PKC we next determined the effects of 4 μM chelerythrine, an inhibitor of Ca2+-dependent and Ca2+ -independent PKCs [20] on zint-β2-AR stimulation of ICa,L in +PKA and–PKA atrial myocytes. As shown in Fig 5, in +PKA myocytes (panel A), chelerythrine had no significant effects on basal ICa,L amplitude (open bars) or zint-β2-AR stimulation of ICa,L (control,106±8% vs cheler, 116±3%). In–PKA myocytes (panel B) zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L (278±13%) compared to control (panel A) due to activation of cPLA2 [3]. In contrast to +PKA myocytes, chelerythrine now significantly inhibited zint-β2-AR stimulation of ICa,L (71±13%; P<0.002). These findings indicate PKC signaling plays no role in β2-AR stimulation of ICa,L in freshly isolated atrial myocytes. However when PKA is inhibited β2-AR stimulation acts via PKC to stimulate cPLA2, consistent with β2-AR stimulation of PLC.
Fig 5.

Effects of 4 μM chelerythrine (cheler) on zint-β2-AR stimulation of ICa,L in +PKA(A) and -PKA (B) myocytes and effects of dn-PKCα-Adv (C) on zint-β2-AR stimulation of ICa,L in +LMN myocytes. A; in control +PKA myocytes, chelerythrine had no significant effects on zint-β2-AR stimulation of ICa,L. B; in—PKA myocytes, zint-β2-AR stimulation elicited a typically enhanced in increase in ICa,L compared to controls (A) and chelerythrine significantly inhibited zint-β2-AR stimulation of ICa,L. C; in control +LMN myocytes (βgal) zint-β2-AR stimulation of ICa,L was typically enhanced compared to controls (A). In +LMN myocytes infected with dn-PKCα-Adv, zint-β2-AR stimulation of ICa,L was significantly inhibited. Numbers in parentheses indicate that number of cells studied. * = P<0.05.
In another approach we infected atrial myocyte with an Adv expressing a dominant-negative mutant of Ca2+-dependent PKCα (dn-PKCα). Control cells were infected with Adv-βgal. Infected cells were cultured overnight on LMN and therefore represent +LMN myocytes. As shown in Fig 5C, in control +LMN myocytes (Adv-βgal) zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L (208±8%) compared to +PKA myocytes (panel A). In +LMN myocytes expressing the dn-PKCα mutant, zint-β2-AR stimulation of ICa,L was significantly inhibited (control, 208±8% vs dn-PKCα, 57±5%; P<0.002). Similar experiments performed on +LMN myocytes expressing a dn-PKCε mutant showed no differences with control +LMN (β-gal) myocytes (control, 158±21% vs dn-PKCε, 165±31%, N = 4). These findings provide additional support for the idea that β2-AR stimulation acts via Ca2+-dependent PKCα to activate cPLA2.
Role of cPLA2 in the enhanced zint-β2-AR induced ICa,L currents in LMN plated atrial myocytes
Activation of cPLA2 requires phosphorylation at serine sites S505 and S515 [21–23]. We therefore determined the effect of β2-AR stimulation in +LMN myocytes infected with Adv that either expressed a non-phosphorylatable dominant-negative cPLA2 (dn-cPLA2S505A/S515A) mutant, wild-type cPLA2 (wt-cPLA2) or βgal as control. Infected atrial myocytes were cultured on LMN overnight and therefore represent +LMN myocytes. Fig 6shows that in control +LMN myocytes expressing βgal, β2-AR stimulation elicited a typical increase in ICa,L (166±11%). In +LMN myocytes expressing the double dn-cPLA2S505A/S515A mutant β2-AR stimulation of ICa,L was significantly attenuated (84±8%) compared to control. In +LMN myocytes overexpressing wt-cPLA2, β2-AR stimulation of ICa,L was significantly enhanced (307±8%) compared to either control or dn-cPLA2S505A/S515A. These results confirm that cell attachment to LMN activates β2-AR/cPLA2 signaling and further indicates that β2-AR stimulation of cPLA2 requires serine phosphorylation at one or both sites.
Fig 6. Effects of dn-cPLA2S515A/S505A and wt-cPLA2 on zint-β2-AR stimulation of ICa,L in +LMN myocytes.
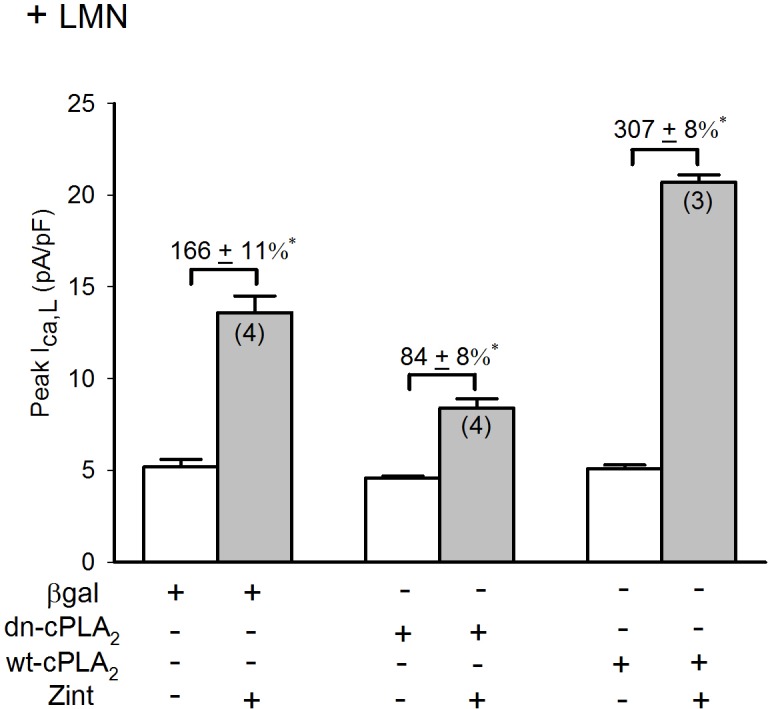
Compared to control +LMN myocytes (βgal), zint-β2-AR stimulation of ICa,L was significantly inhibited and enhanced in myocytes overexpressing dn-cPLA2S515A/S505A and expressing wt-cPLA2, respectively. Numbers in parentheses indicate that number of cells studied. * = P<0.05.
Role of IP3 receptor in the enhanced zint-β2-AR induced ICa,L currents in -PKA atrial myocytes
Our previous work showed that β2-AR stimulation of cPLA2 signaling is dependent on intracellular Ca2+ [3]. The fact that β2-AR stimulation acts via PLC to activate cPLA2 (Fig 3) suggests the potential involvement of IP3-mediated Ca2+ signaling. We therefore determined the effects of 2 μM 2-APB, a putative IP3 receptor (IP3R) blocking agent [24], on β2-AR stimulation of ICa,L in +PKA and–PKA myocytes. As shown in Fig 7A, in +PKA myocytes 2-APB had no significant effect on basal ICa,L (open bars) or zint-β2-AR stimulation of ICa,L (control, 121±4% vs 2-APB, 113±5%). In–PKA myocytes (B) zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L compared to control responses (panel A). In contrast to +PKA myocytes, 2-APB significantly inhibited zint-β2-AR stimulation of ICa,L (control, 245±25% vs 2-APB, 89±19%; P<0.02). Similar results were obtained in +LMN myocytes where zint-β2-AR stimulation of ICa,L also was significantly blocked by 2-APB (control, 186±26%; N = 4 vs 87±7%; N = 4, P<0.01) (see S3 Fig online data).
Fig 7.

Effects of 2 μM 2-APB on zint-β2-AR stimulation of ICa,L in +PKA (A), -PKA atrial myocytes. A; in control +PKA myocytes, 2-APB had no significant effects on zint-β2-AR stimulation of ICa,L. B; in -PKA myocytes, zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L compared to controls (A) and 2-APB significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.05.
In another approach, we infected atrial myocytes with an adenovirus that expresses an IP3 affinity trap which binds to IP3 in the cytosol and thereby inhibits IP3-mediated Ca2+ signaling by preventing IP3 from reaching and activating the IP3 receptor (IP3R) [17]. Cells were cultured overnight on LMN and therefore represent +LMN myocytes. As shown in Fig 8, in control +LMN myocytes expressing βgal, zint-β2-AR stimulation elicited a typical increase in ICa,L (182±4%). However, in +LMN myocytes expressing the IP3 trap zint-β2-AR stimulation of ICa,L (111±17%) was significantly inhibited compared to controls (P<0.002). Together, these findings suggest that IP3R signaling plays no role in zint-β2-AR stimulation of ICa,L in freshly isolated atrial myocytes. However, when PKA is inhibited by either PKA inhibitor (Fig 7) or cell attachment to LMN (Fig 8), β2-AR stimulation of cPLA2 is dependent on IP3-mediated Ca2+ signaling.
Fig 8. Effect of adenovirus IP3 affinity trap on zint-β2-AR stimulation of ICa,L in +LMN myocytes.

Compared with control +LMN myocytes (βgal), zint-β2-AR stimulation of ICa,L was significantly inhibited in +LMN myocytes infected with adenovirus expressing the IP3 affinity trap. Numbers in parentheses indicate the number of myocytes studied. * = P<0.05.
Role of IP3 receptors located on SR or nuclear envelope in the enhanced zint-β2-AR induced ICa,L currents in -PKA atrial myocytes
IP3Rs are thought to be located primarily on the sarcoplasmic reticulum (SR) and nuclear envelope membranes. Because the SR and nuclear envelope membranes are highly interconnected, inhibition of SR Ca2+ uptake by thapsigargin depletes intracellular Ca2+ stores from both sites [25]. Therefore, in Fig 9we determined whether depletion of SR and nuclear Ca2+ by 5 μM thapsigargin (10 min; Thaps) inhibits β2-AR stimulation of ICa,L in +PKA (A) and–PKA (B) myocytes. In +PKA myocytes (A) thapsigargin had no effect on basal ICa,L amplitude (open bars). Compared to control responses (107±8%), thapsigargin slightly enhanced zint-β2-AR stimulation of ICa,L (143±4%), although the change was not statistically significant. This modest increase in β2-AR stimulation of ICa,L is consistent with the effects of thapsigargin to inhibit SR Ca2+ release and thereby inhibit Ca2+-mediated inactivation of ICa,L. In fact, separate experiments showed that thapsigargin abolished SR Ca2+ transients (data not shown). In–PKA myocytes (B), zint-β2-AR stimulation of ICa,L (207±17%) was typically enhanced compared to control responses (107±8%) obtained in +PKA myocytes (A). Interestingly, in–PKA myocytes treated with thapsigargin β2-AR stimulation of ICa,L (238±12%) was still enhanced. In other words, depletion of Ca2+ from SR and nuclear membranes failed to prevent PKA inhibition from enhancing β2-AR signaling. Moreover, the last column in Fig 9B shows that in–PKA myocytes treated with thapsigargin, AACOCF3 (cPLA2 inhibitor) significantly inhibited zint-β2-AR stimulation of ICa,L (62±12%), indicating that the enhanced response to zint-β2-AR stimulation was in fact due to activation of cPLA2 signaling and that depletion of SR and nuclear Ca2+ stores failed to prevent β2-AR/cPLA2 signaling. Similar results were obtained when Ca2+ stores were depleted by treatment (10 min) with 10 μM ryanodine (data not shown). These results indicate that IP3-dependent Ca2+ signaling is not mediated via IP3Rs located on SR or nuclear envelope membranes.
Fig 9.

Effects of 5 μM thapsigargin (10 min; Thaps) on zint-β2-AR stimulation of ICa,L in +PKA(A) and -PKA (B) myocytes. A; in +PKA myocytes, compared to control responses, thapsigargin slightly enhanced zint-β2-AR stimulation of ICa,L. B; in -PKA myocytes, zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L compared to control (A) that was not prevented by treatment with thapsigargin. The addition of 10 μM AACOCF3 (+ thaps) significantly inhibited zint-β2-AR stimulation of ICa,L indicating that thapsigargin did not prevent zint-β2-AR stimulation of cPLA2. Numbers in parentheses indicate the number of myocytes studied. * = P<0.05.
Role of IP3 receptors located in caveolae in the enhanced zint-β2-AR induced ICa,L currents in -PKA atrial myocytes
Alternatively, IP3R protein is present in caveolae [26, 27], which are abundant in atrial myocytes [28]. We therefore determined the effects of 10 mM methyl-β-cyclodextrin (MCD; 30 min), an agent that disrupts caveolae formation [29], on zint-β2-AR stimulation of ICa,L in +PKA and–PKA myocytes. As shown in Fig 10A, in +PKA myocytes MCD caused a modest but significant inhibition of zint-β2-AR stimulation of ICa,L (control,122±5% vs MCD, 93±7%; P<0.05), representing a 24% decrease. This is consistent with the idea that β2-ARs are normally localized to caveolae [30]. However, in–PKA myocytes (B) MCD elicited a significantly larger inhibition of zint-β2-AR stimulation of ICa,L (control, 235±7% vs MCD, 54±12%; P<0.008), representing a 77% decrease. The fact that MCD elicited a significantly larger inhibition of β2-AR signaling in–PKA compared to +PKA myocytes supports the idea that the IP3Rs that are essential for β2-AR/cPLA2 signaling are localized to the caveolae.
Fig 10.

Effect of 2 mM methyl-β-cyclodextrin (MCD; 30 min) on zint-β2-AR stimulation of ICa,L in +PKA (A) and -PKA (B) myocytes. A; in +PKA myocytes, MCD significantly decreased zint-β2-AR stimulation of ICa,L compared to control responses (-24%). B; in—PKA myocytes, zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L compared to +PKA myocytes (A) and the effect of MCD to decrease zint-β2-AR stimulation of ICa,L was enhanced (-77%) compared to +PKA myocytes (A). Numbers in parentheses indicate the number of myocytes studied. * = P<0.05.
Discussion
We previously reported that in atrial myocytes inhibition of adenylate cyclase/PKA by either cell attachment to LMN or inhibition of PKA in cells not attached to LMN activates β2-AR/cPLA2 signaling [2, 3]. The present study extends those findings by showing that when PKA is inhibited, β2-AR stimulation acts via PKCα/Raf-1/MEK/ERK1/2 and IP3-dependent Ca2+ signaling to activate cPLA2.
In the present experiments, we showed that inhibition of PKA prominently stimulates β2-AR-mediated phosphorylation of ERK1/2, consistent with β2-AR stimulation of ICa,L via MEK/ERK1/2 signaling [3]. Moreover, inhibition of Raf-1 by either GW5074 or adenoviral expression of a dn-Raf-1 mutant significantly inhibited β2-AR stimulation of ICa,L in–PKA or +LMN myocytes but failed to affect β2-AR signaling in +PKA myocytes. These findings can be explained by the fact that PKA inhibits Raf-1 and Raf-1 activates MEK/ERK1/2 signaling [7, 31, 32]. Because atrial myocytes normally exhibit relatively high endogenous levels of adenylate cyclase/PKA activity [11], Raf-1 is normally inhibited and therefore β2-AR stimulation is unable to activate Raf-1/MEK/ERK1/2 signaling in +PKA myocytes. However, when PKA is inhibited (–PKA or +LMN myocytes) β2-AR stimulation acts via Raf-1/MEK/ERK1/2 to activate cPLA2. The present experiments also show that adenoviral overexpression of wt-cPLA2 or expression of a dn-cPLA2S505A/S515A mutant in +LMN myocytes significantly enhanced or inhibited, respectively, β2-AR stimulation of ICa,L compared to control +LMN myocytes expressing βgal. In various cell systems including vascular smooth muscle and fibroblasts ERK1/2 phosphorylates cPLA2 at S505 and S515 [15, 21–23]. In cardiac ventricular myocytes (cultured on LMN) β2-AR stimulation acts via ERK1/2 and p38MAPK to phosphorylate cPLA2 at S505 [6].
In atrial myocytes, β2-ARs are coupled to both Gs- and Gi-mediated signaling pathways [12]. We [3] and others [4] have reported that β2-ARs act via pertussis toxin-sensitive Gi to activate cPLA2. In various cell systems, including rat ventricular myocytes [33] activation of PKA phosphorylates β2-ARs and thereby switches β2-AR coupling from Gs-mediated adenylate cyclase to Gi-mediated ERK1/2 signaling [34, 35]. Moreover, inhibition of PKA by H-89 inhibited β2-AR/Gi-mediated ERK1/2 signaling. These findings are not consistent with the present results, which indicate that in adult atrial myocytes inhibition of PKA by H-89 or KT5720 activates (rather than inhibits) β2-AR/Gi-mediated ERK1/2 signaling [3]. However, our findings are consistent with reports that PKA inhibits Raf-1 and its downstream MEK/ERK1/2 signaling pathway [7, 31, 32]. Therefore, in atrial myocytes inhibition of PKA removes Raf-1 inhibition and thereby activates β2-AR/Gi stimulation of Raf-1/MEK/ERK1/2 signaling.
The present results also indicate that inhibition of PLC (edelfosine) or PKC (chelerythrine or dn-PKCα) significantly inhibited β2-AR stimulation of ICa,L in–PKA or +LMN myocytes but not in +PKA myocytes. Moreover, in +LMN myocytes expression of dn-PKCα significantly inhibited β2-AR signaling while expression of dn-PKCε had no effect, indicating that β2-ARs act via Ca2+-dependent PKCα to activate cPLA2. These findings are consistent with reports that PKC activates Raf-1/MEK/ERK1/2 signaling in adult cardiac myocytes [9, 18] and that PKC activates cPLA2 signaling [10].
Our previous findings showed that strong chelation of intracellular Ca2+ by BAPTA prevented β2-AR stimulation via cPLA2. This is consistent with the fact that several of the signaling molecules involved in the proposed β2-AR/cPLA2 signaling cascade are Ca2+-dependent, including PLC, PKCα and cPLA2. In the present study, we investigated more specifically which source of intracellular Ca2+ is required for β2-AR/cPLA2 signaling. We found that 2-APB, an agent that inhibits IP3-mediated Ca2+ release in cat atrial myocytes [24] significantly inhibited β2-AR stimulation of ICa,L in both–PKA and +LMN myocytes but not in +PKA myocytes. Moreover, adenoviral expression of an IP3 affinity trap which inhibits IP3-mediated Ca2+ signaling [17] resulted in a similar inhibition of β2-AR signaling in +LMN myocytes. The fact that 2-APB had no effect on β2-AR signaling in control +PKA myocytes and that it exerted effects similar to those of the Adv-IP3 affinity trap suggests that 2-APB acted specifically to inhibit IP3-mediated Ca2+ signaling. Together, these results indicate that β2-AR stimulation of cPLA2 is dependent on IP3-mediated Ca2+ signaling. Although IP3Rs are typically located on SR and nuclear membranes, the present results showed that thapsigargin, an agent that depletes both SR and nuclear envelope Ca2+ stores [25] failed to prevent β2-AR stimulation of ICa,L via cPLA2. However, disruption of caveolae formation by methyl-β-cyclodextrin elicited a significantly larger inhibition of β2-AR stimulation in–PKA myocytes than control +PKA myocytes. We therefore conclude that IP3Rs on the SR and nuclear envelope membranes are not involved in the Ca2+ signaling required for β2-AR stimulation of cPLA2. Alternatively, β2-AR stimulation of ICa,L via cPLA2 is dependent on IP3-mediated Ca2+ signaling in caveolae. In endothelial and smooth muscle cells plasmalemmal caveolae contain IP3R protein that is speculated to mediate Ca2+ influx through the caveolar membrane [26, 27]. Moreover, in rat ventricular myocytes (cultured on laminin) β2-ARs stimulate translocation of cPLA2 to the low density caveolin-3 enriched membrane fraction suggesting that cPLA2 translocates to caveolae [5]. In fact, lipid rafts i.e. caveolae contain a wide variety of signaling components including β2-ARs, Gi, PKC, Raf-1, ERK1/2, IP3Rs [36], involved in β2-AR/cPLA2 signaling.
We therefore propose (see Fig 11) that under normal conditions (A; +PKA myocytes), β2-AR/Gs stimulation acts via adenylate cyclase (AC)/cAMP/PKA to stimulate ICa,L. The relatively high basal levels of endogenous PKA activity and β2-AR-stimulated PKA activity both inhibit Raf-1 signaling, thereby preventing β2-AR stimulation from normally activating cPLA2. However, inhibition of PKA by either PKA inhibitor (B;–PKA myocytes) or by cell attachment to LMN (C; +LMN myocytes), removes inhibition of Raf-1. Under these conditions, β2-AR/Gi stimulation of PLC activates PKCα and IP3R-mediated Ca2+ signaling in caveolae. With PKA inhibited, PKCα is now able to stimulate Raf-1/MEK/ERK1/2 signaling. Therefore, β2-AR/Gi-mediated activation of both ERK1/2 and IP3-dependent Ca2+ signaling stimulates cPLA2/AA. We previously reported that arachidonic acid (AA) stimulates ICa,L [3]. This scenario is consistent with the fact that cPLA2 activation requires both elevation of submicromolar intracellular [Ca2+] and phosphorylation by various kinases (see also [21–23]).
Fig 11.

Schematic summary showing the proposed signaling mechanisms underlying zint-β2-AR stimulation of ICa,L in +PKA (A), -PKA (B) atrial myocytes. A; in +PKA myocytes, zint-β2-AR stimulation acts via Gs to activate adenylate cyclase (AC)/cAMP-dependent kinase (PKA) which in turn stimulates ICa,L. Both basal and stimulated PKA activity inhibits Raf-1 signaling, thereby preventing zint-β2-AR stimulation of ICa,L via cPLA2. B; in cells not attached to LMN, inhibition of PKA by KT5720 removes inhibition of Raf-1. C; cell attachment to LMN acts via β1 integrins and FAK/PI-(3)K/Akt signaling to inhibit adenylate cyclase (AC)/cAMP-dependent kinase (PKA) activity, thereby removing inhibition of Raf-1. β2-AR stimulation acts via Gi to stimulate PLC leading to activation of PKCα and IP3R-mediated Ca2+ signaling within caveolae. With PKA inhibited, PKCα stimulates Raf-1/MEK/ERK1/2 signaling. Together, ERK1/2 and IP3-mediated Ca2+ signaling activate cPLA/AA, resulting in stimulation of ICa,L.
Conclusions
These findings may have important implications with respect to the aging and/or failing heart, both of which exhibit decreases in adenylate cyclase activity. In both animal models [37] and in the human right atrium [38], increasing age is associated with a decrease in β1-AR function that results from a decrease in adenylate cyclase activity. Likewise, adenylate cyclase activity is depressed in the failing human heart [39] and canine pacing-induced heart failure [40, 41] and yet β2-AR signaling is preserved. Previous studies by Nalli et al suggest that PKA and PKG phosphorylates PLCβ3 in gastric smooth muscle cells and decreases PI hydrolysis [42]. The decrease in PI hydrolysis has been suggested to diminish IP3-mediated Ca2+ release decreasing muscle contraction. Along these lines, the present studies also suggest that feline atrial cardiomyocytes exhibit a similar regulation as that seen in gastric muscle. Our research also suggests that increases in extracellular matrix proteins, i.e. fibrosis, may contribute to decreases in adenylate cyclase/PKA activity in the aging and/or failing heart, which in turn switches β2-AR signaling from cAMP/PKA to cPLA2/AA. This mechanism may be responsible for the preservation of β2-AR signaling while β1-AR signaling is depressed. Thus, the diminished PKA activity in cardiac fibrosis may decrease phosphorylation of PLC resulting in an increase in IP hydrolysis, Ca2+ release and greater contraction. Indeed, our present results indicate that inhibition of PLC significantly inhibited β2-AR stimulation of ICa,L in–PKA or +LMN myocytes but not in +PKA myocytes. In addition, our results suggest that 2-APB, an agent that inhibits IP3-mediated Ca2+ release in cat atrial myocytes [24] significantly inhibited β2-AR stimulation of ICa,L in both–PKA and +LMN myocytes but not in +PKA myocytes. Together, these results indicate that β2-AR stimulation of cPLA2 in both–PKA and +LMN myocytes is dependent on PLC/IP3-mediated Ca2+ signaling. Given that cPLA2/AA signaling is a potentially pro-inflammatory mediator, the present findings suggest that fibrosis in the aging and/or failing heart may predispose inflammation and atrial dysfunction. In fact, AA is reported to slow atrial conduction and has been implicated in the development of postoperative atrial fibrillation [43]. On the other hand, activation of the Raf/MEK/ERK pathway may be cardioprotective [44]. In fact, inhibition of adenylate cyclase/PKA and subsequent activation of Raf-1/MEK/ERK1/2 signaling enhances cardiac resistance to oxidative stress, increases cell survival, and extends lifespan [45]. Therefore, inhibition of adenylate cyclase/PKA activity and the remodeling of β2-AR signaling to activate Raf/MEK/ERK1/2 may help ameliorate the deterioration of function that occurs in the aging and/or failing atrium. Previous studies have indicated that, beside inhibition of COX, aspirin has COX-independent mechanisms that plays a role in tumor suppression [46, 47]. Aspirin reportedly inhibits PI3K/Akt kinase activity in epithelial ovarian cancer cells [48]. Aspirin down regulates the expression of PI3K, Akt and ERK in rodent model of acute pulmonary embolism [49] and in turn inhibit the release of inflammatory cytokines [50]. On the other hand, our studies indicate that laminin acts via FAK/PI-(3)K/Akt signaling to inhibit adenylate cyclase-mediated stimulation of ICa,L. Although, there is no direct evidence suggesting that aspirin or NSAIDs play a role in cardiomyocyte aging, we hypothesize that aspirin or NSAIDs may influence cPLA2/AA pathway and this requires further investigation.
Supporting Information
A; In +LMN myocytes, zint-β2-AR stimulation of ICa,L was enhanced and GW5074 significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.004.
(TIF)
A; in +LMN myocytes zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L and edelfosine significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.03.
(TIF)
A; +LMN myocytes, zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L and 2-APB significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.01.
(TIF)
Acknowledgments
We thank Ms. Rekha Iyenger for her expert technical assistance with the adenoviral experiments.
Abbreviations
- LMN
laminin
- cPLA2
cytosolic phospholipase A2
- AA
arachidonic acid
- ERK
extracellular-signal regulated kinase
- MEK
mitogen-activated protein kinase kinase
- Raf-1 (also known as c-Raf)
MAP kinase kinase kinase
- PLC
phospholipase C
- FAK
focal adhesion kinase
- PI-(3)K
phosphatidyinositol-3’ kinase
- Akt
protein kinase B
- zint-β2-AR
zinterol β2-adrenergic receptor
- ICa,L
L-type Ca2+ current
- Adv
adenovirus
- dn
dominant negative
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by NIH grants HL079038 (S.L.L.), HL62426-Project 4 and AHA Scientist Development Grant (13SDG14000006) (M.R.P.) and a grant from the Dr. Ralph and Marian Falk Medical Research Trust (A.M.S.).
References
- 1.Wang YG, Samarel AM, Lipsius SL. Laminin binding to beta1-integrins selectively alters beta1- and beta2-adrenoceptor signalling in cat atrial myocytes. The Journal of physiology. 2000;527 Pt 1:3–9. Epub 2000/08/16. PubMed Central PMCID: PMCPMC2270063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang YG, Ji X, Pabbidi M, Samarel AM, Lipsius SL. Laminin acts via focal adhesion kinase/phosphatidylinositol-3' kinase/protein kinase B to down-regulate beta1-adrenergic receptor signalling in cat atrial myocytes. The Journal of physiology. 2009;587(3):541–50. Epub 2008/12/10. 19064616; PubMed Central PMCID: PMCPMC2670079. 10.1113/jphysiol.2008.163824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pabbidi MR, Ji X, Samarel AM, Lipsius SL. Laminin enhances beta(2)-adrenergic receptor stimulation of L-type Ca(2+) current via cytosolic phospholipase A(2) signalling in cat atrial myocytes. The Journal of physiology. 2009;587(Pt 20):4785–97. Epub 2009/08/26. PubMed Central PMCID: PMCPMC2770147. 10.1113/jphysiol.2009.179226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavoine C, Magne S, Sauvadet A, Pecker F. Evidence for a beta2-adrenergic/arachidonic acid pathway in ventricular cardiomyocytes. Regulation by the beta1-adrenergic/camp pathway. The Journal of biological chemistry. 1999;274(2):628–37. Epub 1999/01/05. [DOI] [PubMed] [Google Scholar]
- 5.Ait-Mamar B, Cailleret M, Rucker-Martin C, Bouabdallah A, Candiani G, Adamy C, et al. The cytosolic phospholipase A2 pathway, a safeguard of beta2-adrenergic cardiac effects in rat. The Journal of biological chemistry. 2005;280(19):18881–90. Epub 2005/02/25. 10.1074/jbc.M410305200 [DOI] [PubMed] [Google Scholar]
- 6.Magne S, Couchie D, Pecker F, Pavoine C. Beta(2)-adrenergic receptor agonists increase intracellular free Ca(2+) concentration cycling in ventricular cardiomyocytes through p38 and p42/44 MAPK-mediated cytosolic phospholipase A(2) activation. The Journal of biological chemistry. 2001;276(43):39539–48. Epub 2001/08/17. 10.1074/jbc.M100954200 [DOI] [PubMed] [Google Scholar]
- 7.Gerits N, Kostenko S, Shiryaev A, Johannessen M, Moens U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility. Cellular signalling. 2008;20(9):1592–607. Epub 2008/04/22. 10.1016/j.cellsig.2008.02.022 [DOI] [PubMed] [Google Scholar]
- 8.Kohno M, Pouyssegur J. Pharmacological inhibitors of the ERK signaling pathway: application as anticancer drugs. Progress in cell cycle research. 2003;5:219–24. Epub 2003/11/05. [PubMed] [Google Scholar]
- 9.Iijima Y, Laser M, Shiraishi H, Willey CD, Sundaravadivel B, Xu L, et al. c-Raf/MEK/ERK pathway controls protein kinase C-mediated p70S6K activation in adult cardiac muscle cells. The Journal of biological chemistry. 2002;277(25):23065–75. Epub 2002/04/10. 10.1074/jbc.M200328200 [DOI] [PubMed] [Google Scholar]
- 10.van Rossum DB, Patterson RL. PKC and PLA2: probing the complexities of the calcium network. Cell calcium. 2009;45(6):535–45. Epub 2009/04/07. 10.1016/j.ceca.2009.02.008 [DOI] [PubMed] [Google Scholar]
- 11.Wang YG, Samarel AM, Lipsius SL. Laminin acts via beta 1 integrin signalling to alter cholinergic regulation of L-type Ca(2+) current in cat atrial myocytes. The Journal of physiology. 2000;526 Pt 1:57–68. Epub 2000/07/06. PubMed Central PMCID: PMCPMC2269985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YG, Dedkova EN, Steinberg SF, Blatter LA, Lipsius SL. Beta 2-adrenergic receptor signaling acts via NO release to mediate ACh-induced activation of ATP-sensitive K+ current in cat atrial myocytes. The Journal of general physiology. 2002;119(1):69–82. Epub 2002/01/05. PubMed Central PMCID: PMCPMC2233856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carpenter L, Cordery D, Biden TJ. Protein kinase Cdelta activation by interleukin-1beta stabilizes inducible nitric-oxide synthase mRNA in pancreatic beta-cells. The Journal of biological chemistry. 2001;276(7):5368–74. Epub 2000/11/23. 10.1074/jbc.M010036200 [DOI] [PubMed] [Google Scholar]
- 14.Ping P, Zhang J, Cao X, Li RC, Kong D, Tang XL, et al. PKC-dependent activation of p44/p42 MAPKs during myocardial ischemia-reperfusion in conscious rabbits. The American journal of physiology. 1999;276(5 Pt 2):H1468–81. Epub 1999/05/18. [DOI] [PubMed] [Google Scholar]
- 15.Pavicevic Z, Leslie CC, Malik KU. cPLA2 phosphorylation at serine-515 and serine-505 is required for arachidonic acid release in vascular smooth muscle cells. Journal of lipid research. 2008;49(4):724–37. Epub 2008/01/12. 10.1194/jlr.M700419-JLR200 [DOI] [PubMed] [Google Scholar]
- 16.Heidkamp MC, Bayer AL, Martin JL, Samarel AM. Differential activation of mitogen-activated protein kinase cascades and apoptosis by protein kinase C epsilon and delta in neonatal rat ventricular myocytes. Circulation research. 2001;89(10):882–90. Epub 2001/11/10. [DOI] [PubMed] [Google Scholar]
- 17.Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. American journal of physiology Heart and circulatory physiology. 2008;294(2):H596–604. Epub 2007/12/07. 10.1152/ajpheart.01155.2007 [DOI] [PubMed] [Google Scholar]
- 18.Moschella PC, Rao VU, McDermott PJ, Kuppuswamy D. Regulation of mTOR and S6K1 activation by the nPKC isoforms, PKCepsilon and PKCdelta, in adult cardiac muscle cells. Journal of molecular and cellular cardiology. 2007;43(6):754–66. Epub 2007/11/03. PubMed Central PMCID: PMCPMC2170873. 10.1016/j.yjmcc.2007.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Powis G, Seewald MJ, Gratas C, Melder D, Riebow J, Modest EJ. Selective inhibition of phosphatidylinositol phospholipase C by cytotoxic ether lipid analogues. Cancer research. 1992;52(10):2835–40. Epub 1992/05/15. [PubMed] [Google Scholar]
- 20.Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochemical and biophysical research communications. 1990;172(3):993–9. Epub 1990/11/15. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh M, Tucker DE, Burchett SA, Leslie CC. Properties of the Group IV phospholipase A2 family. Progress in lipid research. 2006;45(6):487–510. Epub 2006/07/04. 10.1016/j.plipres.2006.05.003 [DOI] [PubMed] [Google Scholar]
- 22.Gijon MA, Leslie CC. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. Journal of leukocyte biology. 1999;65(3):330–6. Epub 1999/03/18. [DOI] [PubMed] [Google Scholar]
- 23.Hirabayashi T, Murayama T, Shimizu T. Regulatory mechanism and physiological role of cytosolic phospholipase A2. Biological & pharmaceutical bulletin. 2004;27(8):1168–73. Epub 2004/08/12. [DOI] [PubMed] [Google Scholar]
- 24.Zima AV, Blatter LA. Inositol-1,4,5-trisphosphate-dependent Ca(2+) signalling in cat atrial excitation-contraction coupling and arrhythmias. The Journal of physiology. 2004;555(Pt 3):607–15. Epub 2004/02/03. PubMed Central PMCID: PMCPMC1664857. 10.1113/jphysiol.2003.058529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X, Bers DM. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circulation research. 2006;99(3):283–91. Epub 2006/06/24. 10.1161/01.RES.0000233386.02708.72 [DOI] [PubMed] [Google Scholar]
- 26.Fujimoto T, Nakade S, Miyawaki A, Mikoshiba K, Ogawa K. Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. The Journal of cell biology. 1992;119(6):1507–13. Epub 1992/12/01. PubMed Central PMCID: PMCPMC2289753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnitzer JE, Oh P, Jacobson BS, Dvorak AM. Caveolae from luminal plasmalemma of rat lung endothelium: microdomains enriched in caveolin, Ca(2+)-ATPase, and inositol trisphosphate receptor. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(5):1759–63. Epub 1995/02/28. PubMed Central PMCID: PMCPMC42599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang YG, Dedkova EN, Ji X, Blatter LA, Lipsius SL. Phenylephrine acts via IP3-dependent intracellular NO release to stimulate L-type Ca2+ current in cat atrial myocytes. The Journal of physiology. 2005;567(Pt 1):143–57. Epub 2005/06/11. PubMed Central PMCID: PMCPMC1474159. 10.1113/jphysiol.2005.090035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smart EJ, Anderson RG. Alterations in membrane cholesterol that affect structure and function of caveolae. Methods in enzymology. 2002;353:131–9. Epub 2002/06/25. [DOI] [PubMed] [Google Scholar]
- 30.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of beta -adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. The Journal of biological chemistry. 2000;275(52):41447–57. Epub 2000/09/28. 10.1074/jbc.M006951200 [DOI] [PubMed] [Google Scholar]
- 31.Cook SJ, McCormick F. Inhibition by cAMP of Ras-dependent activation of Raf. Science (New York, NY). 1993;262(5136):1069–72. Epub 1993/11/12. [DOI] [PubMed] [Google Scholar]
- 32.Hafner S, Adler HS, Mischak H, Janosch P, Heidecker G, Wolfman A, et al. Mechanism of inhibition of Raf-1 by protein kinase A. Molecular and cellular biology. 1994;14(10):6696–703. Epub 1994/10/01. PubMed Central PMCID: PMCPMC359200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefkowitz RJ, Pierce KL, Luttrell LM. Dancing with different partners: protein kinase a phosphorylation of seven membrane-spanning receptors regulates their G protein-coupling specificity. Molecular pharmacology. 2002;62(5):971–4. Epub 2002/10/23. [DOI] [PubMed] [Google Scholar]
- 34.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390(6655):88–91. Epub 1997/11/18. 10.1038/36362 [DOI] [PubMed] [Google Scholar]
- 35.Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ. Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. The Journal of biological chemistry. 2002;277(34):31249–56. Epub 2002/06/14. 10.1074/jbc.M202753200 [DOI] [PubMed] [Google Scholar]
- 36.Anderson RG. The caveolae membrane system. Annual review of biochemistry. 1998;67:199–225. Epub 1998/10/06. 10.1146/annurev.biochem.67.1.199 [DOI] [PubMed] [Google Scholar]
- 37.Farrell SR, Howlett SE. The age-related decrease in catecholamine sensitivity is mediated by beta(1)-adrenergic receptors linked to a decrease in adenylate cyclase activity in ventricular myocytes from male Fischer 344 rats. Mechanisms of ageing and development. 2008;129(12):735–44. Epub 2008/11/01. 10.1016/j.mad.2008.09.017 [DOI] [PubMed] [Google Scholar]
- 38.Brodde OE, Zerkowski HR, Schranz D, Broede-Sitz A, Michel-Reher M, Schafer-Beisenbusch E, et al. Age-dependent changes in the beta-adrenoceptor-G-protein(s)-adenylyl cyclase system in human right atrium. Journal of cardiovascular pharmacology. 1995;26(1):20–6. Epub 1995/07/01. [DOI] [PubMed] [Google Scholar]
- 39.Bristow MR, Hershberger RE, Port JD, Minobe W, Rasmussen R. Beta 1- and beta 2-adrenergic receptor-mediated adenylate cyclase stimulation in nonfailing and failing human ventricular myocardium. Molecular pharmacology. 1989;35(3):295–303. Epub 1989/03/01. [PubMed] [Google Scholar]
- 40.Ishikawa Y, Sorota S, Kiuchi K, Shannon RP, Komamura K, Katsushika S, et al. Downregulation of adenylylcyclase types V and VI mRNA levels in pacing-induced heart failure in dogs. The Journal of clinical investigation. 1994;93(5):2224–9. Epub 1994/05/01. PubMed Central PMCID: PMCPMC294370. 10.1172/JCI117219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marzo KP, Frey MJ, Wilson JR, Liang BT, Manning DR, Lanoce V, et al. Beta-adrenergic receptor-G protein-adenylate cyclase complex in experimental canine congestive heart failure produced by rapid ventricular pacing. Circulation research. 1991;69(6):1546–56. Epub 1991/12/01. [DOI] [PubMed] [Google Scholar]
- 42.Nalli AD, Kumar DP, Al-Shboul O, Mahavadi S, Kuemmerle JF, Grider JR, et al. Regulation of Gbetagammai-dependent PLC-beta3 activity in smooth muscle: inhibitory phosphorylation of PLC-beta3 by PKA and PKG and stimulatory phosphorylation of Galphai-GTPase-activating protein RGS2 by PKG. Cell biochemistry and biophysics. 2014;70(2):867–80. Epub 2014/04/30. PubMed Central PMCID: PMCPMC4184950. 10.1007/s12013-014-9992-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tselentakis EV, Woodford E, Chandy J, Gaudette GR, Saltman AE. Inflammation effects on the electrical properties of atrial tissue and inducibility of postoperative atrial fibrillation. The Journal of surgical research. 2006;135(1):68–75. Epub 2006/05/03. 10.1016/j.jss.2006.03.024 [DOI] [PubMed] [Google Scholar]
- 44.Taylor RP, Starnes JW. Age, cell signalling and cardioprotection. Acta physiologica Scandinavica. 2003;178(2):107–16. Epub 2003/06/05. 10.1046/j.1365-201X.2003.01132.x [DOI] [PubMed] [Google Scholar]
- 45.Yan L, Vatner DE, O'Connor JP, Ivessa A, Ge H, Chen W, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130(2):247–58. Epub 2007/07/31. 10.1016/j.cell.2007.05.038 [DOI] [PubMed] [Google Scholar]
- 46.Goel A, Chang DK, Ricciardiello L, Gasche C, Boland CR. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9(1):383–90. Epub 2003/01/23. [PubMed] [Google Scholar]
- 47.Luciani MG, Campregher C, Gasche C. Aspirin blocks proliferation in colon cells by inducing a G1 arrest and apoptosis through activation of the checkpoint kinase ATM. Carcinogenesis. 2007;28(10):2207–17. Epub 2007/05/19. 10.1093/carcin/bgm101 [DOI] [PubMed] [Google Scholar]
- 48.Uddin S, Ahmed M, Hussain A, Assad L, Al-Dayel F, Bavi P, et al. Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian cancer. International journal of cancer. 2010;126(2):382–94. Epub 2009/07/22. 10.1002/ijc.24757 [DOI] [PubMed] [Google Scholar]
- 49.Wang L, Wu J, Zhang W, Zhi Y, Wu Y, Jiang R, et al. Effects of aspirin on the ERK and PI3K/Akt signaling pathways in rats with acute pulmonary embolism. Molecular medicine reports. 2013;8(5):1465–71. 10.3892/mmr.2013.1676 [DOI] [PubMed] [Google Scholar]
- 50.Liu YC, Kao SJ, Chuang IC, Chen HI. Nitric oxide modulates air embolism-induced lung injury in rats with normotension and hypertension. Clinical and experimental pharmacology & physiology. 2007;34(11):1173–80. Epub 2007/09/21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A; In +LMN myocytes, zint-β2-AR stimulation of ICa,L was enhanced and GW5074 significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.004.
(TIF)
A; in +LMN myocytes zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L and edelfosine significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.03.
(TIF)
A; +LMN myocytes, zint-β2-AR stimulation elicited a typically enhanced increase in ICa,L and 2-APB significantly inhibited zint-β2-AR stimulation of ICa,L. Numbers in parentheses indicate the number of myocytes studied. * = P<0.01.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.