

INTRODUCTION
Neurofibromatosis type 1 (NF1) or von Recklinghausen disease is an inherited autosomal dominant disorder and the most common of the phakomatoses (neurocutaneous syndromes), affecting 1 in 3,000 individuals.1 Pathogenesis involves mutations in the NF1 gene that encodes the protein neurofibromin. Neurofibromin belongs to the family of GTPase-activating proteins and is found in many organs such as the brain, kidney, spleen, and thymus.2
The disease demonstrates 100% penetrance but has high variability of expression.3 Patients with the same NF1 gene mutation often display different clinical manifestations of NF1.4 Some common clinical manifestations include café au lait macules, axillary or inguinal freckling, Lisch nodules (iris hamartomas), and neurofibromas.2,5 Café au lait spots—flat hyperpigmented macules—are the earliest clinical finding in NF1. They typically present at birth or appear during early childhood.
Peripheral neurofibromas are benign peripheral nerve sheath tumors composed of Schwann cells, fibroblasts, perineurial cells, and mast cells. The 3 general types of neurofibroma are cutaneous, nodular, and plexiform. Plexiform lesions are less common than the cutaneous and nodular forms. Plexiform neurofibromas can involve the orbit and are associated with increased mortality in patients with NF1 because of malignant transformation or compression of the airway or spinal cord. They can also result in significant disfigurement and morbidity.6
We report the case of a 16-year-old male with NF1 who demonstrated a plexiform neurofibroma and associated orbital findings of sphenoid wing dysplasia and buphthalmos. We discuss the computed tomography (CT) and magnetic resonance imaging (MRI) findings of these NF1 manifestations as well as common central nervous system (CNS) manifestations of NF1, including focal areas of signal intensity (FASIs), optic nerve gliomas, parenchymal gliomas, and dural ectasia of the optic nerve sheath and spinal canal.
HISTORY
A 16-year-old male with a history of NF1 presented for an annual follow-up visit in November 2012, with the craniofacial team at Ochsner Clinic Foundation for evaluation and treatment of a large plexiform neurofibroma in the right orbit. The patient demonstrated multiple clinical features consistent with NF1 such as café au lait spots and axillary freckling during physical examination. He also had notable facial asymmetry, right blepharoptosis, right lower lid retraction, ectropion, and right epiphora with insufficient tear drainage through the nasolacrimal duct.
RADIOGRAPHIC APPEARANCE
MRI demonstrated a typical right-sided periorbital neurofibroma consisting of an area of diffuse edema with associated heterogeneous enhancement within the soft tissues overlying the anterior frontal calvarium that extended into the periorbital/preseptal soft tissues and extraconal space of the right orbit (Figures 1 and 2). In addition, MRI showed dysplasia of the greater wing of the right sphenoid bone and abnormal enlargement of the right globe (Figure 3).
Figure 1.
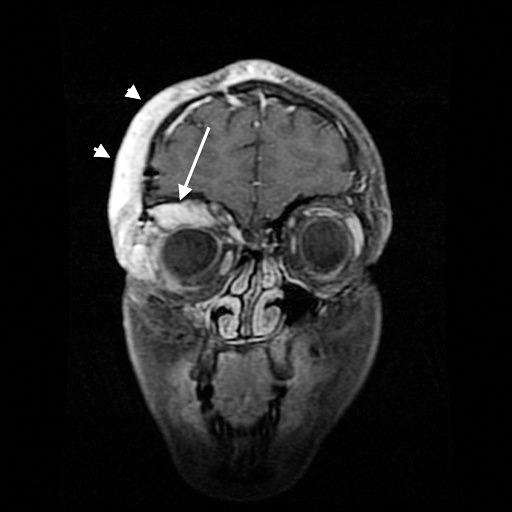
Coronal T1 fat-saturated contrast-enhanced magnetic resonance imaging shows the right plexiform neurofibroma. The lesion heterogeneously enhances and extends along the anterior frontal calvarium into the periorbital/preseptal soft tissues (arrowheads) and superolateral extraconal space of the right orbit (long arrow).
Figure 2.
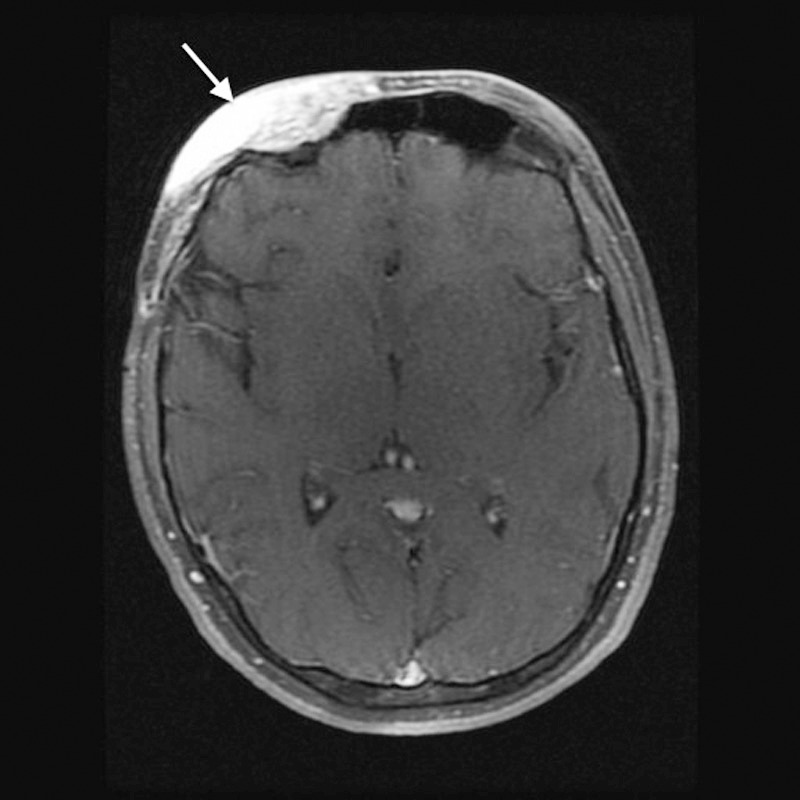
Axial T1 fat-saturated contrast-enhanced magnetic resonance imaging shows enhancing soft tissue overlying the right anterior frontal calvarium consistent with plexiform neurofibroma (arrow).
Figure 3.
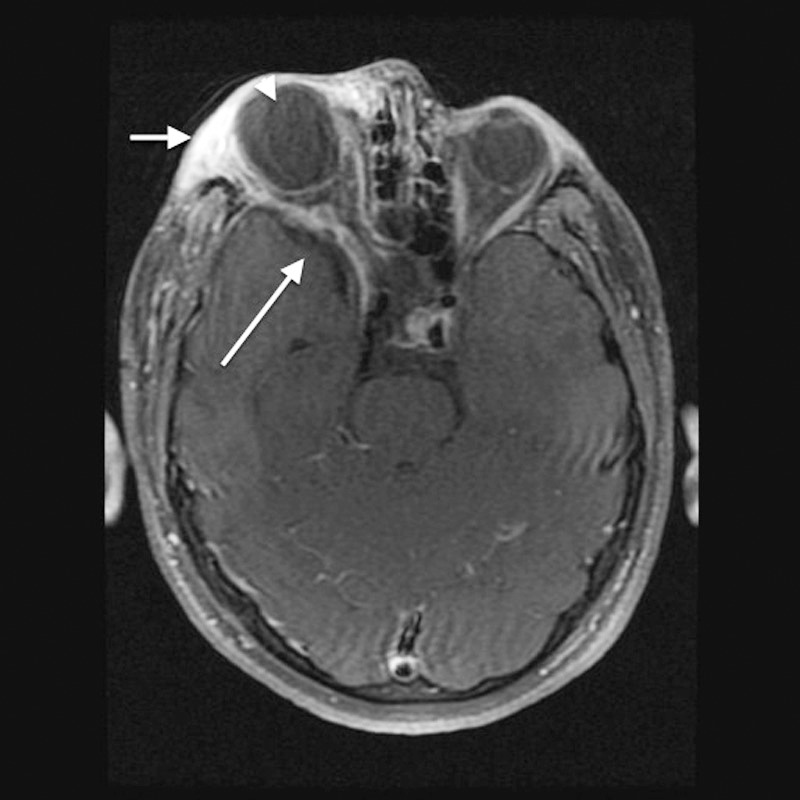
Axial T1 fat-saturated contrast-enhanced magnetic resonance imaging shows diffuse edema with heterogeneous enhancement of the preseptal soft tissue and superolateral extraconal right orbit consistent with plexiform neurofibroma (short arrow). Enlargement of the right middle cranial fossa as a result of greater sphenoid wing dysplasia is visible (long arrow). The globes are asymmetric in size, with marked buphthalmos on the right (arrowhead).
CT of the head showed right-sided induration of the periorbital and perizygomatic soft tissues corresponding to the area of heterogeneous enhancement noted on the MRI. The sphenoid wing dysplasia was again noted and better characterized on CT. The right bony orbit was enlarged compared to the left. The right maxillary antrum was hypoplastic and partially opacified by fat and soft tissue. CT also showed dehiscence of the right posterior lamina papyracea. Finally, the right globe was enlarged, consistent with buphthalmos (Figure 4).
Figure 4.
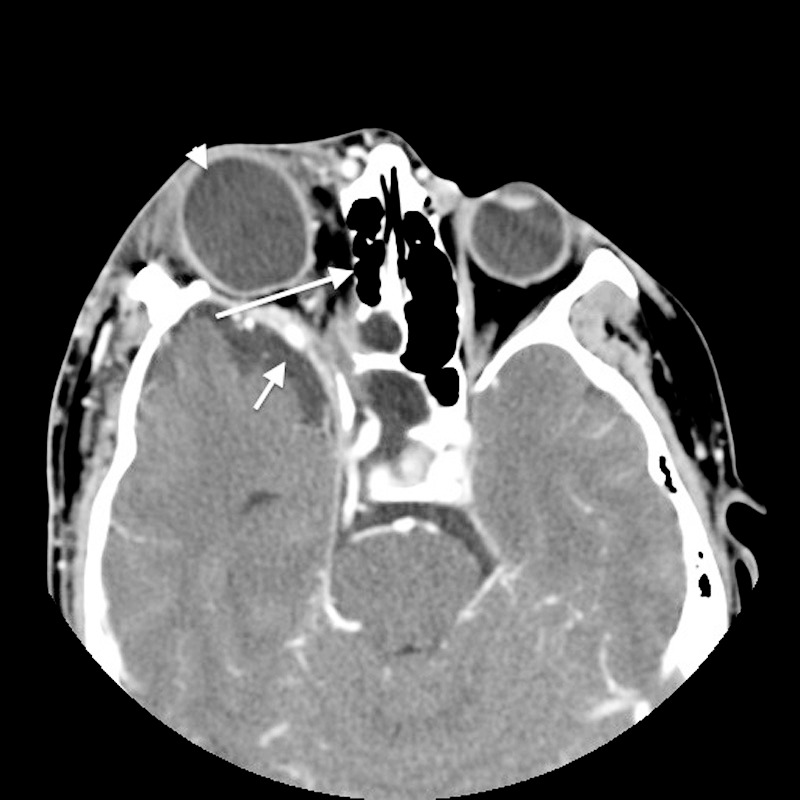
Axial postcontrast computed tomography shows deformity of the right greater wing of the sphenoid bone with dehiscence of the right posterior lamina papyracea (long arrow). Enlargement of the right middle cranial fossa is visible (short arrow) with resulting right-sided proptosis with right buphthalmos (arrowhead).
DISCUSSION
Diagnosis of NF1 requires the presence of at least 2 of the following clinical features: at least 6 café au lait spots (>5 mm in diameter if the patient is prepubertal or >15 mm diameter postpuberty), at least 2 neurofibromas (1 if plexiform), freckling in the axillary or inguinal region, optic glioma, at least 2 Lisch nodules, distinctive body lesions (sphenoid dysplasia or pseudarthrosis), or a known first-degree relative with NF1.7 Our patient met the criteria with café au lait spots, plexiform neurofibroma, and sphenoid dysplasia. We observed the latter 2 features on imaging.
As previously stated, plexiform neurofibroma is a type of peripheral nerve sheath tumor, and it is essentially pathognomonic of NF1. While the most common nerve sheath tumors seen in NF1 are the cutaneous and subcutaneous neurofibromas, plexiform neurofibromas are much rarer and have greater potential for morbidity. These tumors develop from angiogenic and invasive Schwann cells that create either a superficial overgrowth of skin and soft tissues or a deep palpable cord of thickened nerves. They are important to recognize and monitor because a painful expanding lesion may indicate malignant transformation. These plexiform lesions develop from cranial and large peripheral nerve sheaths and occur in approximately half of all NF1 cases.8 They can cause significant disfigurement, disability, and morbidity if cranial nerves or peripheral nerve roots are compressed. They transform into malignant peripheral nerve sheath tumors in approximately 10% of NF1 patients.9
The cross-sectional imaging of plexiform neurofibroma has many distinctive characteristics. Imaging appearance is often compared to a “bag of worms” and may involve large portions of the face in the form of a serpentine soft tissue mass. T1-weighted MRI should display a heterogeneous signal, and T2-weighted MRI should display a hyperintense signal. The lesion should show heterogeneous enhancement with contrast.6,10
Sphenoid wing dysplasia, another common manifestation of NF1 seen in our patient's imaging, is a bony defect, decalcification, or remodeling of the greater wing of the sphenoid bone. Sphenoid wing dysplasia can occur with enlargement of the middle fossa, resulting in herniation of the intracranial contents into the orbit. In addition to enlargement, the middle cranial fossa often displays distinctive arachnoid cysts. On CT, this osseous defect with herniation of the middle cranial fossa is readily seen with associated proptosis.11
Buphthalmos is diffuse enlargement of the eye secondary to increased intraocular pressure. Our patient's buphthalmos, proptosis, and middle cranial fossa herniation contributed to the striking right-sided prominence seen on MRI and CT imaging. Pathogenesis of buphthalmos in NF1 involves increased intraocular pressure resulting from obstruction of the canal of Schlemm. This anatomic structure is located between the cornea and iris and is responsible for resorption of the aqueous humor in the anterior chamber. In NF1, abnormal mesodermal tissue results in canal obstruction. In contrast, congenital or infantile glaucoma is typically the cause of buphthalmos in patients who do not have NF1. Because the sclera is soft and deformable at an early age, this manifestation is almost uniquely seen during childhood.12
Common CNS manifestations of NF1 that were not seen in our patient include FASIs, optic nerve gliomas, parenchymal gliomas, and dural ectasia of the optic nerve sheath and spinal canal.
FASIs—the most common neuroimaging finding in patients with NF1—are hyperintense foci detected on T2-weighted MRI that result from abnormal myelination with waxing and waning dysplastic white matter lesions. FASIs occur in the brain of patients with NF1 when the tissue microstructure is disrupted, allowing compartmentalization and accumulation of fluid in myelin vacuoles. FASIs are commonly identified in the cerebellum, thalami, internal capsule, brainstem, basal ganglia, and subcortical hemispheric white matter. The globus pallidus of the basal ganglia is the most frequently involved site.13
FASIs in the setting of NF1 are usually transient and asymptomatic, occurring in patients aged 4-12 years and regressing by adulthood.14 Approximately 80% of children with NF1 experience moderate to severe cognitive impairment.13 Studies have shown a relationship between FASIs and cognitive dysfunction in patients with NF1.15-18 Hyman and colleagues found that children with NF1 and discrete FASI lesions in the thalamus specifically, a relatively rare location for T2 hyperintensity (approximately 8% of cases), score 18 intellectual quotient points lower than children without discrete thalamic lesions.19
Optic nerve glioma is the most common primary tumor of the optic nerve. The term optic nerve glioma is used to describe tumors confined to the precortical visual pathway. They may occur anywhere along the optic tract and occasionally may involve the hypothalamus.20 Most optic nerve gliomas are classified as World Health Organization Grade 1 juvenile pilocytic astrocytoma. While malignant tumors may occur in adults, these cases are extremely rare. Optic nerve gliomas are an important radiologic finding because they occur in approximately 20% of patients with NF1.21 Often these optic tumors are bilateral in cases of NF1.10
On cross-sectional imaging, findings of optic nerve gliomas include an isointense signal on T1-weighted MRI, isointense to hyperintense signal on T2-weighted MRI, variable enhancement on postcontrast T1-weighted MRI, and rare calcification on CT. In addition, on T2 images, a rim of hyperintensity can sometimes be seen along the periphery of the lesion, indicating leptomeningeal infiltration and proliferation rather than expanded subarachnoid space. The appearance of optic nerve gliomas is different in patients with and without NF1. Patients without NF1 commonly show fusiform lesions and isolated chiasmal gliomas. In contrast, patients with NF1 often show a diffusely enlarged optic nerve that can be torturous, kinked, or buckled. The optic nerve glioma may involve the optic nerve, optic radiation, optic tracts, and/or optic chiasm.10 Children with chiasm tumors may present with premature or delayed puberty caused by hypothalamic involvement.
Parenchymal gliomas are less prevalent than the optic nerve gliomas seen in NF1. They are usually low-grade astrocytomas that are diagnosed earlier in childhood compared to the population without NF1, at approximately 4-5 years of age. However, the risk of tumor development remains into adulthood. Patients with NF1 usually present with findings of increased intraocular pressure, or the tumor is discovered as an incidental finding on brain imaging. Gliomas can also develop in the brainstem, hypothalamus, and third ventricle in patients with NF1.22
Finally, dural ectasia of the optic nerve sheath often occurs in patients with NF1. Distinctive saccular dilatation of the meninges surrounding the orbital portion of the optic nerve is present. In cross-sectional imaging, increased perioptic fluid can be visualized.12 In the spinal canal, dural ectasia may be visualized as posterior vertebral scalloping on plain radiography and increased anteroposterior diameter of the lumbar dural sac on MRI. Dural ectasia is associated with nerve root herniation and radicular pain.
Neurofibromatosis type 2 (NF2) is a differential diagnosis to consider when NF1 is suspected. NF2 can be distinguished from NF1 by its high prevalence of bilateral acoustic schwannomas that rarely transform to malignant peripheral nerve sheath tumors. In addition to bilateral vestibular schwannomas, other CNS manifestations of NF2 are meningiomas and ependymomas. Additionally, café au lait macules occur infrequently, and Lisch nodules do not occur in NF2, further differentiating this diagnosis from NF1.23
The variability of NF1 manifestations makes imaging challenging; however, imaging can be invaluable to distinguish NF1 from differential diagnoses. Radiologists should be familiar with the key imaging findings associated with this disorder. In the case of our patient, the external disfigurement caused by the plexiform neurofibroma and the bony orbital changes caused by the greater sphenoid wing dysplasia required extensive surgical reconstructive procedures. The patient lost the functionality of his right eye and will require continued follow-up to monitor for growth of new plexiform lesions. Recognizing distinctive radiographic findings and planning treatment early in this disease can thus significantly benefit patients with NF1.
ACKNOWLEDGMENTS
The authors have no financial or proprietary interest in the subject matter of this article.
REFERENCES
- 1. Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999. March 26; 89 1: 1- 6. [PubMed] [Google Scholar]
- 2. Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet. 1996. January; 33 1: 2- 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crow FW. A clinical, pathological, and genetic study of multiple neurofibromatosis. Springfield, IL: Thomas; 1956. [Google Scholar]
- 4. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000. January 1; 151 1: 33- 40. [DOI] [PubMed] [Google Scholar]
- 5. Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993. August; 53 2: 305- 313. [PMC free article] [PubMed] [Google Scholar]
- 6. DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000. March; 105 3 Pt 1: 608- 614. [DOI] [PubMed] [Google Scholar]
- 7. Prada CE, Rangwala FA, Martin LJ, et al. Pediatric plexiform neurofibromas: impact on morbidity and mortality in neurofibromatosis type 1. J Pediatr. 2012. March; 160 3: 461- 467. doi: 10.1016/j.jpeds.2011.08.051. [DOI] [PubMed] [Google Scholar]
- 8. Plotkin SR, Bredella MA, Cai W, et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS One. 2012; 7 4: e35711 doi: 10.1371/journal.pone.0035711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Annu Rev Pathol. 2012; 7: 469- 495. doi: 10.1146/annurev-pathol-011811-132441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tailor TD, Gupta D, Dalley RW, Keene CD, Anzai Y. Orbital neoplasms in adults: clinical, radiologic, and pathologic review. Radiographics. 2013. October; 33 6: 1739- 1758. doi: 10.1148/rg.336135502. [DOI] [PubMed] [Google Scholar]
- 11. Jacquemin C, Bosley TM, Liu D, Svedberg H, Buhaliqa A. Reassessment of sphenoid dysplasia associated with neurofibromatosis type 1. AJNR Am J Neuroradiol. 2002. April; 23 4: 644- 648. [PMC free article] [PubMed] [Google Scholar]
- 12. Smith M, Castillo M. Imaging and differential diagnosis of the large eye. Radiographics. 1994. July; 14 4: 721- 728. [DOI] [PubMed] [Google Scholar]
- 13. Billiet T, Mädler B, D'Arco F, et al. Characterizing the microstructural basis of “unidentified bright objects” in neurofibromatosis type 1: A combined in vivo multicomponent T2 relaxation and multi-shell diffusion MRI analysis. Neuroimage Clin. 2014. April 13; 4: 649- 658. doi: 10.1016/j.nicl.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hegde AN, Mohan S, Lath N, Lim CC. Differential diagnosis for bilateral abnormalities of the basal ganglia and thalamus. Radiographics. 2011. Jan-Feb; 31 1: 5- 30. doi: 10.1148/rg.311105041. [DOI] [PubMed] [Google Scholar]
- 15. North K, Joy P, Yuille D, et al. Specific learning disability in children with neurofibromatosis type 1: significance of MRI abnormalities. Neurology. 1994. May; 44 5: 878- 883. [DOI] [PubMed] [Google Scholar]
- 16. Denckla MB, Hofman K, Mazzocco MM, et al. Relationship between T2-weighted hyperintensities (unidentified bright objects) and lower IQs in children with neurofibromatosis-1. Am J Med Genet. 1996. February 16; 67 1: 98- 102. [DOI] [PubMed] [Google Scholar]
- 17. Goh WH, Khong PL, Leung CS, Wong VC. T2-weighted hyperintensities (unidentified bright objects) in children with neurofibromatosis 1: their impact on cognitive function. J Child Neurol. 2004. November; 19 11: 853- 858. [DOI] [PubMed] [Google Scholar]
- 18. Moore BD, Slopis JM, Schomer D, Jackson EF, Levy BM. Neuropsychological significance of areas of high signal intensity on brain MRIs of children with neurofibromatosis. Neurology. 1996. June; 46 6: 1660- 1668. [DOI] [PubMed] [Google Scholar]
- 19. Hyman SL, Gill DS, Shores EA, Steinberg A, North KN. T2 hyperintensities in children with neurofibromatosis type 1 and their relationship to cognitive functioning. J Neurol Neurosurg Psychiatry. 2007. October; 78 10: 1088- 1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Avery RA, Fisher MJ, Liu GT. Optic pathway gliomas. J Neuroophthalmol. 2011. September; 31 3: 269- 278. doi: 10.1097/WNO.0b013e31822aef82. [DOI] [PubMed] [Google Scholar]
- 21. Blazo MA, Lewis RA, Chintagumpala MM, Frazier M, McCluggage C, Plon SE. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004. June 15; 127A 3: 224- 229. [DOI] [PubMed] [Google Scholar]
- 22. Gutmann DH, Rasmussen SA, Wolkenstein P, et al. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1). Neurology. 2002. September 10; 59 5: 759- 761. [DOI] [PubMed] [Google Scholar]
- 23. Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009. March 17; 14 3: 102- 105. [DOI] [PMC free article] [PubMed] [Google Scholar]