

Abstract
Background:
The term renal tubular acidosis (RTA) describes a group of uncommon kidney disorders characterized by defective acid-base regulation. Reaching the diagnosis of RTA is complex and often delayed, resulting in suboptimal treatment.
Methods:
This article provides an overview of the clinical features of RTA and diagnostic approaches in a format accessible to physicians for everyday use.
Results:
The 3 major forms of disease are classified by their respective tubular transport defects, each of which produces persistent hyperchloremic metabolic acidosis. Distal RTA is characterized by limited urinary acid secretion, proximal RTA by restricted urinary bicarbonate reabsorption, and hyperkalemic RTA by absolute or relative hypoaldosteronism. RTA is often detected incidentally as a biochemical diagnosis in asymptomatic individuals. When present, clinical features may range from mild nonspecific complaints to life-threatening physiologic disturbances.
Conclusion:
RTA is a complex condition that requires thoughtful investigation. Physicians should be aware of the presentation of RTA and the investigative options available to confirm the diagnosis.
Keywords: Acid-base equilibrium, acidosis, acidosis–renal tubular
INTRODUCTION
Renal tubular acidosis (RTA) refers to a group of disorders characterized by defective renal acid-base regulation. The capacity for normal urinary acidification is impaired, resulting in net acid retention and hyperchloremic metabolic acidosis. Acid-base disequilibrium occurs despite a normal or only mildly reduced glomerular filtration rate (GFR). RTA is a poorly appreciated entity among physicians. A basic understanding of the mechanisms of disease is important for management, although a detailed discussion of renal physiology and RTA etiopathogenesis is beyond the scope of this article.
RTA is classified into 3 major forms: distal, proximal, and hyperkalemic RTA. Distal RTA is associated with reduced urinary acid secretion, proximal RTA is characterized by impaired bicarbonate (HCO3−) reabsorption, and hyperkalemic RTA is an acid-base disturbance generated by aldosterone deficiency or resistance. Electrolyte and acid-base disturbances are key components of each disorder. RTA is often detected incidentally through an abnormal blood workup, but some patients present with clinical features such as poor growth, dehydration, or altered mental state.
RTA can be triggered by many causes, from primary renal lesions to secondary disease processes. While knowledge of the entire range of etiologies implicated in RTA is not feasible, clinicians should maintain some appreciation of the common causes (Tables 1-3). RTA remains an infrequent disorder, although its detection and recognition as a distinct entity are widening.1 RTA is a troublesome condition to quantify, and exact data on prevalence and incidence are scant. Prognosis varies and often depends on the underlying cause. Some patients lead normal lives with minimal treatment, while others progress to end-stage renal failure with reduced survival.
Table 1.
Common Causes of Distal Renal Tubular Acidosis

Table 3.
Common Causes of Hyperkalemic Renal Tubular Acidosis

Table 2.
Common Causes of Proximal Renal Tubular Acidosis

Confirming the diagnosis of RTA is difficult and often delayed, resulting in suboptimal treatment. Most recent literature concerning RTA explores disease pathophysiology, and concise descriptions of its diagnostic evaluation are not easily obtained. This article provides a timely review of the clinical manifestations and diagnostic workup of RTA with the goal of improving clinician awareness and simplifying assessment of this condition. This information is particularly useful for those not routinely exposed to patients with nephrologic conditions. The mechanisms and etiology of RTA are not addressed in depth in the present paper.
CLINICAL FEATURES
Distal RTA
Distal RTA, also known as type 1 RTA, is most commonly observed in children as a primary genetic defect.1 Depending on the gene mutation, children can present with mild or severe disease. Severe forms of heritable distal RTA manifest as vomiting and profound dehydration, obtundation, restricted skeletal growth, and rickets. Such cases are usually detected in infancy. Milder disease presents later in childhood, and complaints may include weakness or polyuria, although many patients remain asymptomatic. A relatively frequent first manifestation—typically during adolescence—of mild forms of distal RTA is nephrolithiasis owing to altered renal calcium handling.2
Acquired deficits usually manifest in adults and are most typically associated with autoimmune conditions or nephrotoxins. However, secondary distal RTA may evolve at any age depending on the cause of kidney disease.1 In addition to the signs specific to RTA, patients may present with features of their underlying disorder, such as discoid rash in patients with systemic lupus erythematosus.
Proximal RTA
Proximal RTA is also known as type 2 RTA. Its demography is similar to the patient demographics of distal RTA in that most cases are discovered in children with heritable mutations.1 As with distal RTA, proximal RTA has a spectrum of disease severity. The clinical manifestations of mild disease are usually limited to short stature and lethargy. In severe cases, patients present with respiratory distress, vomiting, and feeding difficulties.
Most cases of proximal RTA occur as a component of generalized tubular dysfunction, referred to as Fanconi syndrome.3 Fanconi syndrome is a malabsorptive state of the proximal convoluted tubules, while proximal RTA refers only to the deficiency in HCO3− retention. As with RTA, Fanconi syndrome may be hereditary or acquired. Patients with Fanconi syndrome experience added nutritional deficiencies, and clinical sequelae are more common in patients with Fanconi syndrome than in patients with isolated proximal RTA. Typical signs include failure to thrive, volume depletion, rickets, and constipation.3 Several rare genetic disorders are associated with Fanconi syndrome and may be suggested by malformations such as ocular abnormalities, tooth deformities, and intellectual impairment.4
Both isolated proximal RTA and Fanconi syndrome can also arise as acquired lesions in children or adults. Extrarenal manifestations may reveal underlying conditions, such as Wilson disease or obstructive uropathy.
The physical and biochemical changes of hereditary isolated proximal RTA are often transient. Normal tubular function is generally restored after 3-5 years, although any delays experienced in physical development may be irreversible. The relatively infrequent progression of hereditary proximal RTA contrasts with the natural history of distal RTA, which is a lifelong disease. Patients with Fanconi syndrome have a variable duration of illness. When a secondary cause is present, morbidity is often dictated more by the underlying illness than by RTA or Fanconi syndrome per se.
Hyperkalemic RTA
The hyperkalemic RTA subtype is also commonly known as type 4 RTA. The manifestations of hyperkalemic RTA are a consequence of the effects of aldosterone deficiency or insensitivity. Biochemical findings are routinely the only detectable change in these patients.1 Potential symptoms include postural hypotension and lethargy. In many patients, the clinical manifestations are directed by the secondary disease rather than by RTA.
Hyperkalemic RTA occurs more frequently in adults than children and is usually an acquired rather than a heritable defect.5 The potential causes are broad because virtually any renal insult can produce this condition. Patients should therefore be examined for signs of secondary renal disease, including stigmata of diabetes mellitus or reports of nephrotoxic medication consumption.
DIAGNOSIS
Serum Biochemistry and Acid-Base Balance
The defining feature of most cases of RTA is impaired renal acid-base buffering and the development of non–anion gap metabolic acidosis. Many cases are detected inadvertently upon the discovery of this unexplained acid-base disturbance during routine investigations for other purposes. The first step in diagnosing RTA of any type is confirmation of a persisting hyperchloremic metabolic acidosis. Unreported chronic diarrhea must be excluded in this context because it is the most common reason for hyperchloremic acidosis.
RTA is a process limited to malfunction of pertinent acid-base mechanisms; renal performance is otherwise satisfactory. RTA is one of many conditions included within the ambit of chronic kidney disease (CKD) and may be simultaneously accompanied by other renal pathology. The GFR in isolated true RTA should be normal or only mildly reduced.
Other forms of renal insufficiency are known to induce both anion gap and non–anion gap acidosis through various separate processes. In CKD, acid retention generally occurs largely because of impaired ammonium (NH4+) excretion facilitated by inadequate generation of ammonia buffer.6 This process is in contradistinction to distal RTA in which the fault lies with NH4+ transport. In CKD, acidosis worsens and anion gap widens as the number of functional nephrons declines, as measured by GFR.7,8 Late in the disease course, refractory RTA may be gradually accompanied by declining GFR.1
RTA is associated with abnormal potassium (K+) distribution in the kidney. K+ derangement is also a frequent finding in RTA and can guide evaluation and treatment. Hypokalemia is characteristic of distal RTA and proximal RTA, while hyperkalemia is indicative of hyperkalemic RTA.
Some patients with proximal RTA may also have findings indicative of Fanconi syndrome. Nutritional deficiencies typical of Fanconi syndrome include hypophosphatemia, hyponatremia, and rarely, hypoglycemia or hypoproteinemia.
Urine Biochemistry
Urine composition is important for the diagnosis of RTA. Patients with normal renal function and intact urinary acidification mechanisms who are exposed to metabolic acidosis will usually produce a urine pH ≤5.3 in attempt to restore the plasma pH. Under normal conditions in individuals with a normal acid-base status, the average urinary pH is 5-6.9
In patients with hyperchloremic metabolic acidosis and alkaline urine, considered in this context as urine pH >5.5, RTA of some type is strongly suggested. However, urinary pH alone is a poor diagnostic tool. In several circumstances, pH can lead to false diagnosis. Both intravascular volume depletion and urinary tract infection by urea-splitting organisms can elevate urinary pH while simultaneously causing non–anion gap metabolic acidosis, mimicking the appearance of RTA. Additionally, urinary pH is often <5.5 in cases of distal and hyperkalemic RTA.
Elevated urinary calcium is prevalent in distal RTA. This elevation explains the proclivity for nephrocalcinosis and stone formation in patients with distal RTA.
Confirmatory Testing
Definitive testing to confirm the diagnosis of RTA is complex and primarily involves urinary measurement of indices of acid and HCO3− secretion. Before explaining such tests, revisiting the basic mechanisms of acid-base imbalance in the different forms of RTA will be helpful. Knowledge of the expected physiologic disturbance will guide the clinician in selecting an appropriate provocation test.
Patients with distal RTA have a distal tubular defect characterized by the inability to excrete hydrogen (H+). Patients with proximal RTA have a problem with proximal tubular HCO3− reabsorption that results in a decline of serum HCO3− and pH. Finally, hyperkalemic RTA is typified by hypoaldosteronism. Hypoaldosteronism in this context refers to the effects of reduced aldosterone activity that may be a consequence of low aldosterone secretion from primary or secondary adrenal insufficiency or a result of renal insensitivity to aldosterone. Hyperkalemic RTA is said to be present once this condition upsets the acid-base balance and is accompanied by a normal anion gap metabolic acidosis. The acidosis is thought to be induced by hyperkalemia that retards urinary NH4+ excretion.5,10,11
The distinction between hyperkalemic RTA and distal and proximal RTA is relatively straightforward. Hyperkalemic RTA is readily diagnosed by hyperkalemia and serum aldosterone measurements in the presence of near-normal GFR. Serum aldosterone may be high or low depending on the reason for hypoaldosteronism. Distinguishing between proximal and distal RTA requires different methods.
An HCO3− loading test (Sidebar) is used to confirm proximal RTA.12,13 The principle is that in patients with acidemia, low plasma HCO3− concentration, and intact proximal tubule function, administering HCO3− should not alter urine HCO3− levels because the HCO3− is avidly reabsorbed from the urine. In patients with proximal RTA, urinary HCO3− concentration will rise following HCO3− infusion because of impaired reabsorption.
In patients with clearly low serum HCO3− and metabolic acidosis, a single urinary measurement of the fractional excretion of HCO3− can sometimes confirm proximal RTA without the need for an HCO3− loading test. If serum HCO3− is low, under normal circumstances urinary reabsorption is expected to be vigorous and to be accompanied by an allowed fractional excretion <5%. A single inappropriately basic urine pH is also suggestive. In patients with equivocal readings, the HCO3− loading test is the gold standard for diagnosis.
On the other hand, definitive diagnosis of distal RTA is best reached with an NH4+ loading test (Sidebar).14,15 Administration of an acid load in the form of ammonium chloride (NH4Cl) should acidify urine in a normally functioning kidney in an effort to buffer blood pH. In patients with distal RTA and impaired urinary H+ and NH4+ secretion, urinary pH will not decrease as expected. In contrast to the HCO3− loading test used to diagnose proximal RTA, NH4+ concentration cannot be directly measured in urine. The urine anion gap (UAG) is used as a surrogate marker of NH4+ secretion.
The UAG is positive under normal circumstances, similar to the serum anion gap. It is derived by subtracting urine chloride (Cl−), which is an anion, from urinary cations, specifically sodium (Na+) and K+. The usual UAG ranges from 20-90 mEq/L. The kidneys acidify urine by secreting NH4+, which is usually secreted coupled to Cl−. Therefore, urinary Cl− provides an indirect measurement of NH4+ secretion. In a properly functioning nephron, the UAG becomes less positive and eventually becomes negative following administration of an NH4Cl load because increased NH4+ secretion is accompanied by increased urinary Cl−. Patients with impaired NH4+ excretion maintain an inappropriately positive UAG.
For diagnosis of distal RTA, the NH4+ loading test is the gold standard test, but it may occasionally be bypassed in patients with obvious hyperchloremic metabolic acidosis and inappropriately high urine pH. In these cases, a single UAG measurement may be sufficient to verify the diagnosis of distal RTA (Figure 1).16 Because the purpose of NH4+ loading is to generate acidosis, the test is unnecessary and possibly unsafe in patients who are already acidemic. The test must be undertaken cautiously because adverse effects, including nausea and vomiting or dysrhythmia, may occur.
Figure 1.

Suggested algorithm for suspected renal tubular acidosis (RTA) in patients with non–anion gap metabolic acidosis and hypokalemia. HCO3−, bicarbonate; UAG, urine anion gap.
The UAG also helps differentiate between diarrhea and distal RTA as the culprit for hyperchloremic metabolic acidosis. Patients with diarrhea and intact urinary acidification should be expected to produce a negative UAG. However, this result must be interpreted carefully in patients with volume depletion. Hypovolemia associated with a urine Na+ concentration <20 mEq/L alters Cl− reabsorption and may falsely raise the UAG.17
OTHER CONSIDERATIONS
Type 3 RTA
A fourth subtype, known as type 3 RTA, is rarely seen in clinical practice but deserves mentioning. This mixed syndrome has diagnostic findings characteristic of both distal and proximal RTA. Type 3 RTA manifests with debilitating congenital syndromes in children and is principally associated with carbonic anhydrase II deficiency.18
Type 3 RTA was formerly thought to be more widespread when it was first identified in 1972.19 Infants with distal RTA were routinely found to possess coexisting significant urinary HCO3− wasting.16 It is now acknowledged that most young children with distal RTA experience an initial transient phase of bicarbonaturia as part of the syndrome's natural history, and this reference to type 3 RTA is no longer in use.
Voltage-Dependent RTA
A small number of patients with distal RTA express hyperkalemia rather than hypokalemia. This variety of distal RTA is known as voltage-dependent RTA. Impaired acid secretion is a result of reduced distal tubular Na+ delivery or poor reabsorption, in contrast to the primary defect in H+ transport observed in classic distal RTA.20 A favorable transepithelial voltage gradient cannot be maintained, forcing retention of both K+ and H+. Voltage-dependent distal RTA may be confused with hyperkalemic RTA, as hyperkalemia is often accompanied by slight elevations in aldosterone. The entities are distinct because patients with type 4 hyperkalemic RTA maintain their ability to acidify urine in response to acidemia.3 Although the electrochemical mechanism and serum K+ differ, the causes and laboratory findings of voltage-dependent RTA are essentially the same as for classic distal RTA (Figure 2). Amiloride, a K+-sparing diuretic, is an important exception that generates voltage-dependent RTA but does not induce classic distal RTA.21
Figure 2.

Suggested algorithm for suspected renal tubular acidosis (RTA) in patients with non–anion gap metabolic acidosis and hyperkalemia.3
CONCLUSION
RTA is a complex and poorly understood problem that encompasses a range of kidney disorders characterized by deranged acid-base balance. RTA is an underrecognized condition that may be inherited or acquired. Diagnosis is difficult but can be verified with a thoughtful workup. RTA should be considered for any patient with otherwise unexplained hyperchloremic metabolic acidosis. RTA is often a biochemical disease, but some patients present with features such as poor growth or vomiting and dehydration. Despite electrolyte disturbance and acid-base imbalance, the GFR should be approximately normal. Proximal and distal RTA are best diagnosed by demonstrating maladaptive changes in urinary acidification following trial administration of HCO3− or acid in the form of NH4Cl. Hyperkalemia is a classic hallmark of RTA associated with hypoaldosteronism.
ACKNOWLEDGMENTS
The authors have no financial or proprietary interest in the subject matter of this article.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care, Medical Knowledge, and Practice-Based Learning and Improvement.
Renal Tubular Acidosis (RTA) Diagnostic Loading Tests
Bicarbonate Loading Test12,13
Commence an intravenous infusion of sodium bicarbonate at 1 mmol/kg/h. Serum bicarbonate (HCO3−), serum creatinine, urine pH, urine HCO3−, and urine creatinine levels should be measured at the beginning of the test, followed by hourly measurements until serum HCO3− approaches the normal range. The test is complete once serum HCO3− is within or near normal limits. A urine pH >7.5 or fractional excretion of HCO3− >15% is diagnostic of proximal RTA. Urine pH will be unchanged in normal patients or those with distal RTA. A fractional excretion of HCO3− <5% excludes proximal RTA, and a value of 5%-15% is indeterminate. The calculation of fractional HCO3− excretion is as follows:
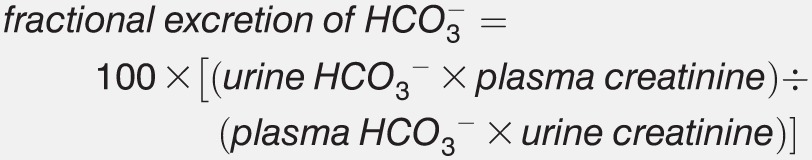 |
Ammonium Loading Test14,15
Administer 100 mg/kg oral ammonium chloride (NH4Cl), ingested slowly with a meal. Urine pH and urine anion gap (UAG) should be measured at the beginning of the test and after 6 hours. Achieving a urinary pH <5.3 at 6 hours is indicative of a normal study or the presence of proximal RTA. If urine pH remains >5.3, distal RTA is likely. At 6 hours following NH4Cl ingestion, normal patients or those with proximal RTA will develop a negative UAG. If the UAG remains positive at 6 hours, this result is diagnostic of distal RTA. The UAG is calculated as follows:
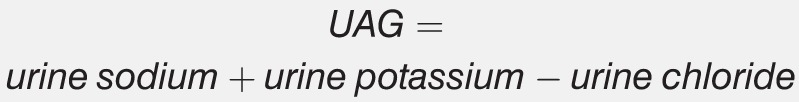 |
REFERENCES
- 1. Rodriguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002. August; 13 8: 2160- 2170. [DOI] [PubMed] [Google Scholar]
- 2. Caruana RJ, Buckalew VM., Jr The syndrome of distal (type 1) renal tubular acidosis. Clinical and laboratory findings in 58 cases. Medicine (Baltimore). 1988. March; 67 2: 84- 99. [DOI] [PubMed] [Google Scholar]
- 3. Kurtzman NA. Renal tubular acidosis syndromes. South Med J. 2000. November; 93 11: 1042- 1052. [PubMed] [Google Scholar]
- 4. Bökenkamp A, Bökenhauer D, Cheong HI, et al. Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr. 2009. July; 155 1: 94- 99. [DOI] [PubMed] [Google Scholar]
- 5. Rose BD, Post TW. Clinical Physiology of Acid-Base and Electrolyte Disorders. 5th ed. New York, NY: McGraw-Hill; 2001. [Google Scholar]
- 6. Kovesdy CP. Metabolic acidosis and kidney disease: does bicarbonate therapy slow the progression of CKD? Nephrol Dial Transplant. 2012. August; 27 8: 3056- 3062. [DOI] [PubMed] [Google Scholar]
- 7. Welbourne T, Weber M, Bank N. The effect of glutamine administration on urinary ammonium excretion in normal subjects and patients with renal disease. J Clin Invest. 1972. July; 51 7: 1852- 1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Warnock DG. Uremic acidosis. Kidney Int. 1988. August; 34 2: 278- 287. [DOI] [PubMed] [Google Scholar]
- 9. Gargan RA, Hamilton-Miller JM, Brumitt W. Effect of pH and osmolality on in vitro phagocytosis and killing by neutrophils in urine. Infect Immun. 1993. January; 61 1: 8- 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DuBose TD, Jr, Good DW. Chronic hyperkalemia impairs ammonium transport and accumulation in the inner medulla of the rat. J Clin Invest. 1992. October; 90 4: 1443- 1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karet FE. Mechanisms in hyperkalemic renal tubular acidosis. J Am Soc Nephrol. 2009. February; 20 2: 251- 254. [DOI] [PubMed] [Google Scholar]
- 12. Hirshman GH, Rao DD, Oyemade O, Chan JC. Renal tubular acidosis: practical guides to diagnosis and treatment. Clin Pediatr (Phila). 1976. July; 15 7: 645- 650. [DOI] [PubMed] [Google Scholar]
- 13. Santos F, Ordóñez FA, Claramunt-Taberner D, Gil-Peña H. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol. 2015. December; 30 12: 2099- 2107. [DOI] [PubMed] [Google Scholar]
- 14. Smulders YM, Frissen PH, Slaats EH, Silberbusch J. Renal tubular acidosis: pathophysiology and diagnosis. Arch Intern Med. 1996. August 12-26; 156 15: 1629- 1636. [PubMed] [Google Scholar]
- 15. Hood W. A-Z of Clinical Chemistry: A Guide for the Trainee. New York, NY: Springer Publishing; 1980. [Google Scholar]
- 16. Bagga A, Sinha A. Evaluation of renal tubular acidosis. Indian J Pediatr. 2007. July; 74 7: 679- 686. [DOI] [PubMed] [Google Scholar]
- 17. Crocetti M, Barone MA. Oski's Essential Pediatrics. 2nd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2004. [Google Scholar]
- 18. Goswami RP, Mondal S, Karmakar PS, Ghosh A. Type 3 renal tubular acidosis. Indian J Nephrol. 2012. November; 22 6: 466- 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McSherry E, Sebastian A, Morris R. Renal tubular acidosis in infants: the several kinds, including bicarbonate-wasting, classic renal tubular acidosis. J Clin Invest. 1972. March; 51 3: 499- 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schleuter W, Keilani T, Hizon M, Kaplan B, Batlle DC. On the mechanism of impaired distal acidification in hyperkalemic renal tubular acidosis: evaluation with amiloride and bumetadine. J Am Soc Nephrol. 1992. October; 3 4: 953- 964. [DOI] [PubMed] [Google Scholar]
- 21. Kurtzman NA. Disorders of distal acidification. Kidney Int. 1990. October; 38 4: 720- 727. [DOI] [PubMed] [Google Scholar]