

Abstract
The present study was undertaken to evaluate the immediate and long-term effects of a single-day exposure to 5-Azacytidine (5-AzaC), a DNA methyltransferase inhibitor, on neurobehavioral abnormalities in mice. Our findings suggest that the 5-AzaC treatment significantly inhibited DNA methylation, impaired extracellular signal-regulated kinase (ERK1/2) activation and reduced expression of the activity-regulated cytoskeleton-associated protein (Arc). These events lead to the activation of caspase-3 (a marker for neurodegeneration) in several brain regions, including the hippocampus and cortex, two brain areas that are essential for memory formation and memory storage, respectively. 5-AzaC treatment of P7 mice induced significant deficits in spatial memory, social recognition, and object memory in adult mice and deficits in long-term potentiation (LTP) in adult hippocampal slices. Together, these data demonstrate that the inhibition of DNA methylation by 5-AzaC treatment in P7 mice causes neurodegeneration and impairs ERK1/2 activation and Arc protein expression in neonatal mice and induces behavioral abnormalities in adult mice. DNA methylation-mediated mechanisms appear to be necessary for the proper maturation of synaptic circuits during development, and disruption of this process by 5-AzaC could lead to abnormal cognitive function.
Keywords: Developing brain, Epigenetics, Methyltransferase, Neuronal loss, Synaptic plasticity, DNA methylation
Graphical abstract
I. Introduction
Adverse environmental exposure [1] (metals, anesthetics, alcohol, pesticides, nicotine, etc.) can interfere with brain development during critical periods and lead to a higher risk of developmental disabilities in adulthood [2–7]. According to recent estimates from the Centers for Disease Control and Prevention in the United States, approximately 15% (1 in 6) of children between the ages of 3 and 17 have one or more developmental disabilities [8]. Adults with developmental disabilities experience many health discrepancies, such as physical, mental, and social problems, that hamper their quality of life. Many drugs [1], alcohol [9–11] and environmental chemical agents [4] are shown to be teratogenic when administered during pregnancy, particularly during the period of organogenesis, neuronal growth and sprouting and synapse formation [12, 13]. Recent advancements in epigenetic research suggest that changes in DNA methylation and post-translational modification of DNA-associated histone proteins play a highly significant role in the teratogenic effects of many environmental agents [14–17], including alcohol [18].
In our earlier study, we established that the transient treatment of young mice with ethanol on postnatal day 7 (P7), which corresponds to the third trimester of human pregnancy, caused significant neurodegeneration, caspase-3 mediated degradation of DNA methyltransferases (DNMT1 and DNMT3A) and reduced DNA methylation [19]. At adulthood, the P7 alcohol-treated mice exhibited long-lasting synaptic and behavioral abnormalities [20–25]. DNA methylation is maintained by the combined function of DNMTs [26–30] and through the action of ten-eleven translocation (TET) proteins [31]. Methylated DNA functions during development to regulate gene expression [32]. Based on these observations, the present study was undertaken to assess whether administration of 5-azacytidine (5-AzaC), which is a DNMT inhibitor, to P7 mice inhibit DNA methylation and causes caspase-3 activation (a marker of neurodegeneration) in neonatal mice and behavioral abnormalities in adulthood. We administered 5-AzaC to P7 mice and evaluated DNA methylation, caspase-3 activation and signaling events in neonatal mice. To understand the mechanism of 5-AzaC, we also used cannabinoid receptor type 1 (CB1R) knockouts, histone methyltransferase (G9a) inhibitor or CB1R antagonist; all were shown to prevent caspase-3 activation in alcohol-treated P7 mice [22, 24]. At adulthood, we investigated long-term potentiation (LTP) and object, spatial and social memory behaviors. The findings suggest that 5-AzaC exposure in P7 mice induces DNA hypomethylation and neurodegeneration and impairs ERK1/2 activation and Arc expression in neonatal mice and causes long-lasting Arc deficits and behavioral abnormalities in adult mice. Our study provides novel insights and suggests that DNA methylation-mediated mechanisms appear to be essential for the maturation of synaptic events and that the disruption of this process, even transiently, delays the acquisition of mature cognitive processes.
2. Materials and Methods
2.1. Study animals
C57BL/6J mice were generated from a breeding colony at the Nathan Kline Institute (NKI). Cannabinoid receptor type 1 (CB1R) knockout (KO) and wild-type (WT) male and female littermates were obtained from a CB1R heterozygous [33] (C57BL/6J background) breeding colony at the NKI. All mice were housed (21–24°C with 40–60% humidity) in groups of four, with water and food available ad libitum on a 12 h light-dark cycle. The CB1R KO/WT mice were genotyped by polymerase chain reaction (PCR) of genomic DNA obtained from mouse tails, as described in our previous study [34]. The animal care and handling procedures followed NKI Institutional Animal Care and Use Committee and National Institutes of Health guidelines. For each treatment group, four to seven pups from 14 different litters were analyzed.
2.2. Drug treatments
The mouse pups were culled to four or six pups per litter. For each experimental group, six to ten pups from ten different litters were analyzed. On the day of treatment, half of the animals (male and female) from each litter were subjected to a subcutaneous (s. c.) injection of saline and the other half were injected with 5-AzaC at P7 (based on the day of birth). 5-AzaC (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) was dissolved in sterile saline solution. A 5-AzaC (0–10 mg/kg) solution was administered by s. c. injection in a volume of 5 μl/g body weight. Saline solution was injected as a control. To identify the events involved in the neurodegenerative effects of 5-AzaC, we used Bix, SR and CB1RKO mice in our study to rescue 5-AzaC-induced activation of caspase-3 in P7 mice. In our earlier studies [22, 24, 35], we showed that a 1 mg/kg (Bix or SR) pretreatment was more effective at preventing alcohol-induced caspase-3 activation in P7 mice. We evaluated whether Bix or SR was effective in preventing 5-AzaC-induced caspase-3 activation in P7 mice. Bix-01294 (2-(Hexahydro-4-methyl-1H-1,4-diazepin-1-yl)-6,7-dimethoxy-N-[1-(phenylmethyl)-4-piperidinyl]-4-quinazolinamine trihydrochloride) (histone methyltransferase or G9a/G9a-like protein (GLP) inhibitor (Bix) Cayman, Michigan, USA) (n = 6 pups/group) and SR141716A [N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole carboxamide] (CB1R antagonist, (SR)) (n = 6 pups/group) (gift from RBI, Natick, MA) were dissolved separately in alcohol (10 μl), followed by Tween 80 (10 μl), and the volume was made up with sterile saline solution. Bix or SR was administered (1 mg/kg) by s. c. injection at a volume of 5 μl/g body weight 30 min before the 5-AzaC (5 mg/kg) treatment. The vehicle [alcohol (10 μl) followed by Tween 80 (10 μl) and saline] was administered as a control. Pups remained with the dams until they were sacrificed, and their brains were removed 4–72 or 8 h after the 5-AzaC or Bix or SR injections, respectively, and processed for the various analyses as described below. After the P7 treatment with saline or 5-AzaC, three-month-old mice derived from different litters were used for long-term potentiation (LTP) and the learning and memory behavioral tests, as described below. Separate cohorts of mice were used for each behavioral analysis.
2.3. DNA methylation assay
For the DNA methylation assay, the pups were sacrificed by decapitation 8 h (the optimum time for maximum caspase-3 activation) after 5-AzaC injection (0–10 mg/kg; 6 pups from three different litters/group), and the hippocampal and neocortical tissues were dissected, flash frozen and stored at -80 °C. Genomic DNA was isolated from the hippocampus and neocortex using a Qiagen DNA Extraction Kit (Qiagen Sciences, Maryland, USA). The global DNA methylation levels were evaluated using an Epigentek (Farmingdale, NY, USA) MethylFlash DNA Methylation Quantification Kit (Colorimetric) according to the manufacturer’s instructions, as described previously [19]. The 5-methylcytosine (5-mC) fraction of the DNA was determined using capture and detection antibodies, and then absorbance was measured at 450 nm. DNA oligomers containing synthetic unmethylated (50 % of cytosine) and methylated (50 % of 5-mC) residues (Epigentek, Farmingdale, NY, USA) were included as negative and positive controls, respectively. The percentage of 5-mC was calculated using the formula provided in the kit and was normalized to the percentage of the saline (the graphs represent the DNA methylation levels multiplied by an arbitrary factor to set the saline to 100).
2.4. DNA methylation dot blot
Genomic DNA was isolated from the P7 saline- and 5-AzaC-treated mouse hippocampi, neocortices, and cerebella (n= 8 pups from 4 different litters/group) using a Qiagen DNA Extraction Kit (Qiagen Sciences, Maryland, USA). 150 ng, 100 ng and 50ng of DNA were dot-blotted onto a 0.2 μm nitrocellulose (NC) membrane in a total volume of 1 μl per sample. The NC membranes were allowed to dry and then cross-linked by exposing the membrane to UV for 15 sec using a UV Staratalinker 1800. The NC membrane was blocked with 2.5% non-fat milk in Tris-buffered saline (1 h at room temperature). The NC membrane was incubated with a primary rabbit-anti-5-methylcytosine antibody (1:500, # BY-MECY-0100, AnaSpec Inc., Fremont, CA) at room temperature (3 h) or 4°C (overnight). The NC membrane was washed, incubated with a secondary antibody (1:1,000 for 30 min at room temperature), developed using ECL reagent, and processed as described by our laboratory [36]. The dot blots were normalized to the percentage of the saline.
2.5. Immunohistochemistry (IHC)
Eight hours (the optimum time for maximum caspase-3 activation) after the 5-AzaC treatment (5 mg/kg; 5 pups from four different litters/group), the pups were anesthetized with isoflurane and perfused with a cacodylate buffer (0.05 M) (pH 7.2) solution containing 4% paraformaldehyde and 4% sucrose. Serial brain sections (40 μm) were cut coronally throughout each whole brain. The free-floating sections (−2 to -2.6 mm from Bregma in the Paxinos atlas [37]) were chosen from treated and control groups for immunohistochemistry. In dual labeling studies, the free-floating brain sections obtained 8 h after saline or 5-AzaC treatment were subjected to a dual immunofluorescence method as described previously [24, 35]. To label methylated cytosine in the neurons, we used anti-mouse-5mC (1:1000; # Bi-MECY-0100, AnaSpec, Inc, Fremont, CA) and anti-rabbit-NeuN (1:500; # 24307, Cell Signaling Technology, Danvers, MA) in our experiments. The secondary antibodies conjugated with Alexa Fluor 488 and 568 (Invitrogen, NY, USA) were used in these studies. In anti-rabbit-5mC and anti-mouse-NeuN staining, both of the primary antibodies were omitted from the immunolabeling process to confer specificity of secondary antibodies. The amount of colocalization of 5-mC with NeuN positive neurons was measured quantitatively by Manders’ correlation coefficients as described previously [38] using LSM-880 with Airyscan and Zen Imaging software. Colocalization was analyzed by selecting CA1, retrosplenial cortex and cerebellum brain regions. For CC3 staining, free-floating coronal brain sections were immunostained with a anti-rabbit cleaved caspase-3 antibody (Asp175, CC3, polyclonal, #9961, 1:500, Cell Signaling Technology, Danvers, MA, USA). Staining was detected with ABC reagents (Vectastain ABC Elite Kit, Vector Labs, Burlingame, CA, USA) and a peroxidase substrate (DAB) Kit (Vector Labs) [24, 35]. As a control for secondary antibody specificity, the primary antibodies were omitted from the reactions. Three coronal sections/mice/group were used for counting. Counting was performed using stereological dissector [39] estimating mean numerical densities of CC-3 positive cells. An unbiased counting area (at least three non-adjacent fields in a section) (0.05 × 0.05 mm; dissector height, 0.07mm) and a high aperture objective was used for the sampling. Averages of 4 nonadjacent sections per pup were used to determine the number of CC-3 positive cells within the CA1 and RSC regions. A Nikon Eclipse TE2000 inverted microscope (DXM1200F, Morrell Instrument Company, Melville, NY, USA) with 2.5X or 40X objectives was used to capture all photomicrographs.
2.6. Protein extraction, electrophoresis and immunoblotting
For the Western blot analysis, the 10 pups/treatment group from five different litters were sacrificed by decapitation 4–72 h after 5-AzaC injection (0–10 mg/kg, n= 10 pups from five different litters/group or 5 mg/kg for 8 h, n = 10 pups from 5 different litters/group), and the hippocampal and neocortical tissues were dissected, flash frozen and stored at -80 °C. The hippocampal and neocortical homogenates were processed and subjected to immunoblotting [40, 41]. Ponceau S staining was used to verify equal protein loading in each blot. The blots were incubated with a primary antibody, including anti-rabbit cleaved caspase-3 (CC3, a marker for an apoptotic cell death or neurodegeneration) (Asp175, polyclonal, #9661, 1:1,000), anti-rabbit p44/42 MAPK (ERK1/2) (1:1,000, #9102), anti-rabbit phospho-p44/42 MAPK (1:1,000, #9101; Cell Signaling Technology, Danvers, MA, USA), anti-mouse activity regulated cytoskeleton-associated protein (Arc; #sc 17839, 1:1000; Santa Cruz Biotechnology) and anti-mouse-β-actin (monoclonal, #3700, 1:1,000, Cell Signaling Technology, Danvers, MA, USA) at room temperature (3 h) or 4°C (overnight) and processed as described by our laboratory [36]. The blots that were incubated with a secondary antibody (peroxidase-conjuated goat anti-mouse, #AP 124P, 1:5,000, Millipore; or goat anti-rabbit, #AP132P, 1:5000, Millipore) alone produced no bands.
2.7. Spatial recognition memory (SRM) using the Y-maze
SRM is based on the natural tendency of rodents to explore novel areas [42] and was performed [43] using the symmetrical Y-maze (Stoelting, Wood Dale, IL) exactly as we previously described [40, 41]. Briefly, the experiment included 3-month-old male and female mice that had been treated with saline or 5-AzaC (n = 8 mice from four different litters/group) at P7. During the training trials (10 min, training trial) the entry to one arm (the novel arm) was blocked using a sheet of opaque Perspex. After a 24 h intertrial interval, the mice were allowed to explore all three arms (3 min, preference trial). The number of entries, the time spent in each arm and the first choice of entry were noted manually and from video recordings by an observer who was blinded to the treatment groups. Preference for the novel arm was measured using the following formula [Preference for the novel arm over the familiar Other arm (Novel/Novel + Other)] as a ratio for discriminating both the time spent in the arms and number of arm entries [41]. A discrimination ratio of greater than 0.5 indicates a preference for the novel arms.
2.8. Social recognition memory
The social recognition memory procedure [44, 45] allows direct access between the experimental and the stimulus mice and is widely used to assess long-term memory in rodents [22, 23, 44, 46, 47]. Briefly, three-month-old male mice that had been treated with saline or 5-AzaC at P7 (n = 8 mice from four different litters/group) were subjected to a social recognition memory test that was performed as described previously [22]. An individual male mouse was placed in a cage in an observation room (dim light) and was allowed to habituate to the new environment for 15 min. The experimental cages were the same as those in which the animals were housed (plastic, 27 cm long × 16 cm wide × 12 cm high). After 15 min, a male juvenile mouse (3–4 weeks old) was immediately introduced into the cage with an adult for an initial contact trial of 2 min. After 24 h (intertrial delay), the same juvenile mouse was placed back in the adult’s cage for a 2-min test trial. The animals were returned to their home cages during the 24 h period between the initial and the test trials. A trained observer continuously timed the duration of the social investigation (with a hand-held stopwatch). The scored social investigative behaviors include direct contact with the juvenile while inspecting any part of the body (including grooming, licking, and pawing); sniffing of the mouth, ears, tail, or anogenital region; and close following (within 1 cm) of the juvenile [44, 45]. If the adult mouse did not investigate the young mouse for a minimum of 24 s during the initial trial (i.e., 20% of the test time), they were retested once with another juvenile. The trials with initial investigation times of less than 24 s were excluded from the analysis. Any aggressive encounter between the animals was an immediate call for the termination of the experiment, and these data were excluded from the statistical analysis. The percentage of time spent in social investigation was evaluated by dividing the investigation time during the second exposure by the initial investigation time × 100.
2.9. Object recognition memory (ORM)
ORM is based on the natural tendency of rodents to explore novel objects [48] and is widely used as a measure of memory in rodents [40, 41, 49–51]. ORM was evaluated in a 40 X 40 cm open field surrounded by 35-cm-high walls made of Plexiglas (Stoelting, Wood Dale, IL) [24]. Twenty-four hours after habituation, three-month-old male mice that had been treated with saline or 5-AzaC at P7 (n = 8 mice from four different litters/group) were subjected to the ORM task [24, 41, 48]. Briefly, each mouse was subjected to a habituation session (allowed to freely explore the open field for 5 min twice a day for two days without any objects in the box). Twenty-four hours after habituation, the training (T1) session was performed by placing individual mice in the open field for 5 min with two identical objects (objects a1 and a2) placed in two adjacent corners at a 10 cm distance from the walls. Twenty-four hours (retention) after training (T2), the mice were allowed to explore the open field with one familiar (a1) and one novel object (b1) for 5 min. Although all objects had similar textures and sizes, they had distinct shapes and colors. Between trials, the objects were rinsed with a 10% alcohol solution. Typical exploration was defined as directing the nose toward the object at a distance of no more than 2 cm and touching the object with the nose. Sitting on the object was not treated as exploratory behavior. The total exploration time for both objects at T1 and T2 (24 h retention) was represented as e1 and e2, respectively. The discrimination index (d2) was considered a means of discriminating between familiar and novel objects and seems to be independent of the total exploration time [52]. The time spent exploring each object during T1 and T2 was recorded by an observer who was blinded to the treatment.
3.0. Long-term potentiation (LTP)
LTP is a lasting increase in synaptic strength, and it establishes the crucial cellular mechanism of learning and memory [53]. Three-month-old male mice (n = 5 mice from four different litters/group) that had been treated with saline and 5-AzaC at P7 were sacrificed by cervical dislocation, followed by decapitation. Hippocampal slices (400 μm) were prepared [24] and recorded using standard procedures, as previously described [22, 24, 40, 41]. After sectioning, the slices were placed in a recording chamber (29°C) and continuously perfused with artificial cerebrospinal fluid (ACSF in mM: 4.4 KCl, 124.0 NaCl, 25.0 NaHCO3, 1.0 Na2HPO4, 2.0 CaCl2, 2.0 MgSO4, and 10.0 glucose, osmolarity 290–300) bubbled with 95% O2 and 5% CO2. CA1 field excitatory post-synaptic potentials (fEPSPs) were recorded by placing both the stimulating and the recording electrodes in the CA1 stratum radiatum. Basal synaptic transmission (BST) was measured by plotting the fiber volley amplitude against the slopes of the fEPSPs, as previously described in detail [24]. Before theta-burst stimulation (TBS) (4 pulses at 100 Hz, with the bursts repeated at 5 Hz and each tetanus including 3x 10-burst trains separated by 15 seconds), a baseline was recorded for 10 min at an intensity that evokes a response that is approximately 35% of the maximum evoked response. LTP was induced by TBS of the Schaeffer collateral pathway. The responses were recorded for 2 hrs after TBS and measured as fEPSP slopes, which were expressed as a percentage of the baseline.
3.10. Statistical analysis
The experiments were evaluated with an equal number of mice/treatment. All of the data are presented as the mean ± SEM. The data were statistically analyzed by one- or two-way analysis of variance (ANOVA) with Bonferroni’s post hoc test. Statistical significance was set at P < 0.05. The statistical analyses were performed using Prism software (GraphPad, San Diego, CA).
4. Results
4.1. 5-AzaC treatment of P7 mice inhibits DNA methylation and activates caspase-3
Previous studies report that the administration of the DNA methylation inhibitor 5-AzaC to pregnant animals causes apoptotic cell death in the embryonic brain [54]. However, it is not known whether the inhibition of DNA methylation by 5-AzaC for a short period during synaptogenesis (a very sensitive stage for apoptotic cell death [55–58]) causes caspase-3 activation. The results showed that 5-AzaC inhibits DNA methylation in a dose-dependent manner (Fig. 1A; hippocampus, F4, 35 = 112, p < 0.01; neocortex, F4, 35 = 30, p < 0.01; cerebellum, F4, 35 = 28, p < 0.01) (two-way ANOVA). We also observed a significant reduction in the amount of 5-mC in the genomic DNA from the 5-AzaC-treated P7 mice HP (F1, 46 = 25, p < 0.01), NC (F1, 46 = 20, p < 0.01) and CB (F1, 46 = 15, p < 0.01) (one-way ANOVA with Bonferroni’s post hoc tests) compared to the saline groups (1B). The dot blots were normalized to the percentage of the saline (the graphs represent the 5-mC levels multiplied by an arbitrary factor to set the saline to 100). Manders coefficient analysis suggests that 5-AzaC-treatment significantly (p < 0.01) reduced the 5-mC positive NeuN neurons in hippocampus (CA1 region), retrosplenial cortex (RSC) and cerebellum (CB) brain regions (Fig. 1C) when compared to saline treatment groups.
Fig. 1.

5-AzaC treatment of P7 mice inhibits DNA methylation in the mouse brain. (A) The global DNA methylation levels were quantified using DNA from the hippocampus (HP), neocortex (NC) and cerebellum (CB) tissues. HP, NC and CB of P7 mice after treatment with saline or various doses of 5-AzaC (0–10 mg/kg body weight, s. c. 8 h). *p < 0.01 vs. 0 mg (n= 6 pups/group). (B) Global 5-mC-specific dot-blot intensities of genomic DNA prepared from the HP, NC and CB (150 ng, 100 ng and 5 ng of genomic DNA) of P7 mice after treatment with saline or 5-AzaC (5 mg/kg body weight, s. c. 8 h). *p < 0.01 vs. saline. The amounts of 5-mC are shown as a percentage of the 5-mC-specific dot-blot intensities compared to the saline controls (n = 8 pups/group). *p < 0.01 vs. Saline. (C) Free-floating coronal brain sections (hippocampus, retrosplenial cortex and cerebellum) from both the groups (saline and 5-AzaC) were subjected to immunohistochemistry with anti-mouse-5mC and anti-rabbit-NeuN to label methylated cytosine in neurons. Manders coefficient analysis was used to evaluate 5-mC positive NeuN neurons in the hippocampus (CA1 region), retrosplenial cortex (RSC) and cerebellum (CB) (cerebellar folia region). Error bars, SEM (n = 6 pups/group). *p < 0.01 vs. Saline. Scale bars = 10 μm.
5-AzaC also induces the formation of CC3 in the hippocampus (F4, 37 = 55, p < 0.01) and neocortex (F4, 37 = 58, p < 0.01) (Fig. 2A) (two-way ANOVA) in a dose-dependent manner but did not induce the formation of CC3 in the cerebellum (p > 0.05) at any of the doses examined. 5-AzaC also induces the formation of CC3 in the hippocampus, (F1, 46 = 45, p < 0.01) and neocortex, (F1, 46 = 38, p < 0.01) (one-way ANOVA with Bonferroni’s post hoc tests) of neonatal mice at 8 and 24 h. The CC3 levels returned to normal levels by 48 h after the 5-AzaC treatment (Fig. 2B). We then examined CC3 IHC after 8 h of 5-AzaC exposure. We counted the CC3-positive cells (4 sections/brain) in the hippocampus and cortex [hippocampus (F1, 11 = 54, p<0.01) and cortex (F1, 11 = 80, p<0.01) regions] (one-way ANOVA). The results suggest that the 5-AzaC treatment enhanced caspase-3 activation, as indicated by the increased number of CC3-positive cells in the 5-AzaC-exposed brain sections (5 mg/kg, s. c. at 8 h) (Fig. 2C).
Fig. 2.

The 5-AzaC treatment of P7 mice induces neurodegeneration. (A) Western blot analysis of CC3 HP, NC and CB in cytosolic extracts from the saline- and 5-AzaC-treated (0–10 mg/kg, s. c. 8 h) groups. (B) Western blot analysis of CC3 using cytosolic extracts of the HP and NC from the saline- and 5-AzaC-treated (5 mg/kg, s. c.) groups at various time points. Ponceau S staining confirmed equal loading of the protein, and β-actin was used as a loading controls Error bars, SEM (n = 10 pups/group). (C) Free-floating coronal brain sections (hippocampus and retrosplenial cortex) from the saline- and 5-AzaC-treated (5 mg/kg, s. c. 8 h) animals were immunostained with an anti-rabbit CC3 antibody. The white arrows indicate the CC3-positive neurons in the hippocampus and retrosplenial cortex. Scale bars = 200 μm. The hippocampal region was enlarged to show the CC3-positive cells (*). The scale bars represent 50 μm. The CC3-positive cells in the hippocampus and retrosplenial cortex were counted. Error bars, SEM (n = 5 pups/group). *p < 0.01 vs. 0 h (saline).
4.2. 5-AzaC treatment of P7 mice inhibits ERK1/2 phosphorylation and Arc protein expression
To further evaluate the participation of intracellular signaling events in the effects of 5-AzaC on the developing brain, we determined the levels of both pERK1/2 (activated), ERK1/2 (total) and Arc proteins. The 5-AzaC treatment significantly reduced the pERK1/2 and Arc protein levels in the hippocampus (pERK1/2, F1, 37 = 34, p<0.01; Arc, F1, 37 = 54, p<0.01) and neocortex (pERK1/2, F1, 37 = 44, p<0.01; Arc, F1, 37 = 59, p<0.01) (one-way ANOVA) at 8 and 24 h. 5-AzaC treatment failed to alter the total ERK1/2 protein levels at any time point (Fig. 3).
Fig. 3.

5-AzaC inhibits ERK1/2 phosphorylation and Arc protein expression in the neonatal brain. Western blot analysis of pERK1/2, ERK1/2 and Arc in cytosolic extracts from the hippocampus (HP) and neocortex (NC) of the saline- or 5-AzaC-treated groups; β-actin was used as a loading control. Error bars, SEM (n = 10 pups/group) (*p < 0.01 vs. 0 h (saline)).
4.3. Bix or SR pre-administration or CB1RKO does not provide protection against 5-AzaC-induced caspase-3 activation
The administration of the maximum dose of Bix (1 mg/kg) 30 min before the 5-AzaC treatment failed to prevent caspase-3 activation (Fig. 4A) in P7 mice (p > 0.05). Furthermore, in our previous studies, both a CB1R antagonist (SR) and the CB1RKO mice prevented alcohol-induced caspase-3 activation in P7 mice [24]. Pre-administration of SR 30 min before the 5-AzaC treatment in P7 mice failed to prevent caspase-3 activation. Similarly, 5-AzaC-induced caspase-3 activation was not prevented in the P7 CB1RKO mice compared to the WT littermates (p > 0.05) (Fig. 4B). Consistent with our previous observations, Bix or SR alone failed to activate caspase-3 in P7 mice. These observations suggest that alcohol-induced inhibition of DNA methylation is possibly initiated upstream of DNMTs, and therefore the neurodegeneration induced by the direct inhibition of DNMTs by 5-AzaC cannot be rescued by Bix or SR treatment or as a consequence of genetic ablation of CB1R in mice.
Fig. 4.

Pharmacological inhibition of G9a/CB1R or genetic ablation of CB1R fails to rescue 5-AzaC-induced caspase-3 activation in P7 mice. (A) The mice were pre-treated with Bix (1 mg/kg) or vehicle for 30 min and then exposed to 5-AzaC (5 mg/kg). The CC3 levels in cytosolic extracts of the hippocampus (HP) and neocortex (NC) were determined by Western blot analysis. (B) P7 CB1RWT mice were pre-treated with SR (1 mg/kg) or vehicle for 30 min, and P7 CB1RKO mice were exposed to 5-AzaC (5 mg/kg). The CC3 levels in cytosolic extracts of the hippocampus (HP) were determined by Western blot analysis. β-actin was used as a loading control. Error bars, SEM (n = 10 pups/group) (*p < 0.01 vs. vehicle).
4.4. 5-AzaC-exposed P7 mice exhibit normal ERK activation but impaired Arc expression in adulthood
To further examine whether impaired ERK activation and Arc expression persist in adulthood, we determined the levels of pERK1/2 (activated), ERK1/2 (total) and Arc proteins by Western blot analysis using specific protein antibodies. The 5-AzaC treatment significantly reduced the Arc protein levels but not the pERK1/2 levels in the hippocampus (Arc, F1, 37 = 34, p<0.01; pERK1/2, F1, 37 = 2, p >0.05) (One-way ANOVA) and NC (data not shown) of adult mice. The 5-AzaC treatment failed to alter the total ERK1/2 protein levels at any time points (Fig. 5).
Fig. 5.

5-AzaC-induced deficits in Arc protein expression are long-lasting and persist into adulthood but not ERK1/2 phosphorylation deficits. Western blot analysis of pERK1/2, ERK1/2 and Arc in cytosolic extracts of the hippocampus (HP) from the P7 saline- or 5-AzaC-treated adult mice; β-actin was used as a loading control. Error bars, SEM (n = 10 pups/group) (*p < 0.01 vs. 0 h (saline)).
4.5. Exposure of P7 mice to 5-AzaC induces learning and memory deficits in adult mice
In the spatial recognition memory test, the P7 saline-treated adult male and female mice more frequently entered the novel, previously unvisited arm of the Y-maze (Arm Entry: [F1,20 = 36, p < 0.01]) and spent more time within the novel arm (Time in arm: [F1,20 = 32, p < 0.01]). In contrast, the P7 5-AzaC-treated adult male and female mice showed a reduced preference for the novel arm (p<0.01) and spent less time (p<0.01) in the novel arm compared to the P7 saline-treated adult mice (Fig. 6A and B). We did not observe a significant difference (F1, 20 = 1.6, p > 0.05, one-way ANOVA) in the spatial recognition memory performance of the adult male and female mice that had been treated with or without 5-AzaC at P7. Although the saline-treated mice (male and female) selected the novel arm as the first choice, the P7 5-AzaC-treated adult mice (male and female) showed a reduced preference for the novel arm (Fig. 6C) (F1, 20 = 40, p < 0.01, one-way ANOVA). The other behavioral experiments were carried out with male mice only due to the lack of a significant influence of gender in the Y-maze behavioral studies.
Fig. 6.

5-AzaC treatment of P7 mice induces long-lasting impairments in spatial memory and social recognition memory in adulthood. Spatial memory performance was measured using the Y-maze. (A) Discrimination index [preference for the novel arm over the familiar other arm (Novel/Novel + Other)] for arm entries and (B) dwell time of the saline- and 5-AzaC-treated male and female mice at 24 h after the first encounter with the partially opened maze. The dashed line denotes chance performance (0.5). (C) The percentage of saline- and 5-AzaC-treated animals (male and female combined) that selected the novel arm as the first choice 24 h after the first encounter with the partially open maze is shown. (D) Exposure to 5-AzaC at P7 induces a loss of social recognition memory in adult male mice. The percent of time spent in social investigation at 24 h after the first encounter with the same juvenile mouse is shown for the saline- and 5-AzaC-treated mice. Error bars, SEM (n = 8 mice/group). *p < 0.01 vs. saline.
In the social recognition memory performance task, the P7 5-AzaC-treated adult male mice exhibited a significant reduction in social recognition memory performance compared to the saline-treated mice (F1,20 = 48, p < 0.01, one-way ANOVA; Fig. 6D). The 5-AzaC treatment of P7 mice for a short period impaired social recognition memory in adult mice.
In the object recognition test, the 5-AzaC treatment at P7 had no significant effect on the total exploration times [e1; F1, 20 = 2.0, p > 0.05; e2; F1, 20 = 1.7, p > 0.05] (Fig. 7A) (one-way ANOVA) but impaired ORT performance (F1, 20 = 49, p < 0.01, one-way ANOVA; Fig. 7B) in adult male mice. Together, these behavioral tests suggest that the exposure of P7 mice to 5-AzaC during an active synaptogenesis period dramatically impaired learning and memory behavior in adulthood that are dependent on the limbic system without affecting the overall mice movement that is mediated by the cerebellum.
Fig. 7.
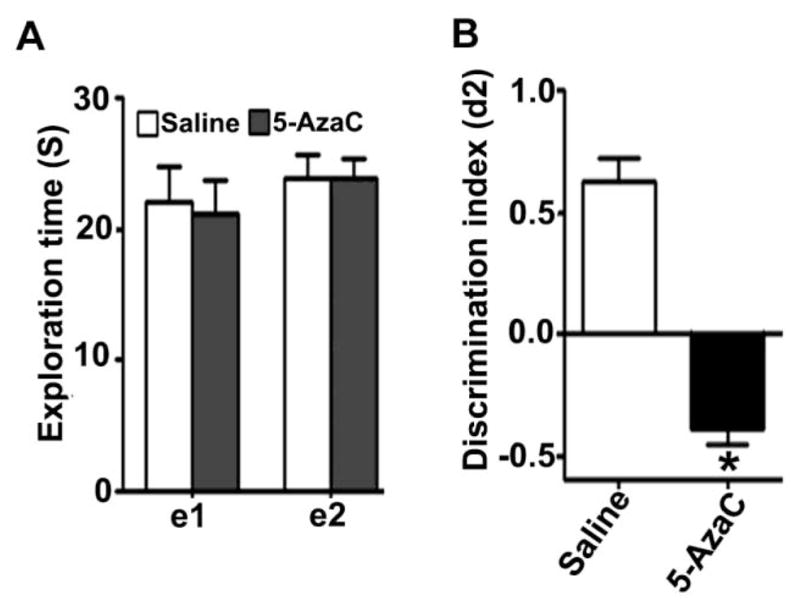
Treatment of P7 mice with 5-AzaC causes objects recognition memory deficits in adulthood. (A) The level of exploration was measured at el and e2 (24 h), namely the time the saline- and 5-AzaC-treated male mice spent exploring the two objects in T1 and T2 (24 h). (B) Discrimination indices (d2) obtained from the saline- and 5-AzaC-treated mice after 24 h retention intervals. e1 and e2 are measures of the total time spent exploring both objects during T1 and T2, respectively. d2 is an index measuring discrimination between the new and the familiar objects. Error bars, SEM (n = 8 mice/group). *p < 0.01 vs. saline.
4.6. The pharmacological inhibition of DNA methylation by 5-AzaC in P7 mice induces LTP deficits in adult mice
To understand whether the treatment of P7 mice with 5-AzaC for a short period produces long-lasting LTP deficits, we performed in vitro recordings in the Schaffer collateral pathway of hippocampal slices (Fig. 8A) prepared from adult male mice (P90) that had been treated with saline or 5-AzaC at P7. Increasing the stimulus intensity evoked robust I/O responses of the fEPSPs in both groups. The fEPSP I/O curve was not affected by 5-AzaC treatment (p > 0.05; Fig. 8B). The baseline fEPSP was recorded at 60-s intervals for ten mins with stimulation at an intensity equivalent to ~35% of the maximum evoked response. The TBS elicited a typical fEPSP LTP [40, 41] (Fig. 8C) in slices from adult mice that had been treated with saline or 5-AzaC at P7. These responses were stable over 120 min. However, TBS evoked a significantly reduced fEPSP slope in the slices (n = 10 slices/5 mice/group) prepared from the P7 5-AzaC-treated adult animals compared with slices from the saline-treated adult animals (F1, 20 = 33, p < 0.01, one-way ANOVA) (Fig. 8D). These findings suggest that the treatment of P7 mice with 5-AzaC for a short period during active synaptogenesis dramatically impairs LTP in adult mice.
Fig. 8.

Exposure of P7 mice to 5-AzaC induces LTP deficits in adult mice. (A) A schematic drawing showing the positions of the stimulating and recording electrodes in the CA1 region of the hippocampus. (B) A summary graph showing the field input/output relationships for the P7 saline- and 5-AzaC-treated adult mice. Insert: An example of traces taken from representative experiments showing the input/output relationships for the saline group. Although it is not shown, the other groups also exhibited comparable patterns. (C) The time course of the average fEPSP slope obtained from the P7 saline- and 5-AzaC-treated adult mice. The fEPSP slopes from each LTP recording were normalized to the mean value of 10 min recordings obtained before stimulation. The arrows indicate the time of TBS (4 pulses at 100 Hz, with bursts repeated at 5 Hz, and each tetanus including 3 different 10-burst trains separated by 15 s). Representative traces of the fEPSPs before (trace 1) and after (trace 2) the induction of LTP in hippocampal slices from P7 saline- and 5-AzaC-treated adult mice. (C) A combined plot of the average of the fEPSP slopes at several time points. Error bars, SEM (n = 5 mice/group; 10 slices/group). *p < 0.01 vs. saline.
5. Discussion
In this study, we demonstrate for the first time that 5-AzaC-treatment for a short period during postnatal brain development (active synaptogenesis period, equivalent to the third trimester in humans) induces DNA hypomethylation and neurodegeneration in P7 mice and long-lasting synaptic plasticity and learning and memory deficits in adulthood. Although we found a similar reduction of DNA methylation in all the brain regions measured, the cerebellum is less susceptible to 5-AzaC-induced neurodegeneration at P7 than the neocortex and hippocampus. This is possibly because the cerebellum is more sensitive to external agents, including ethanol, at P4 but not at P7 in the neonatal period [59–61]. Our findings suggest that the mechanism by which 5-AzaC induces caspase-3 activation differs significantly from that of alcohol-induced caspase-3 activation in P7 mice [9]. This is partly because pre-administration of a G9a/GLP inhibitor (Bix) [22, 35] or CB1R antagonist (SR) [19, 23, 24], which are known to prevent alcohol-induced caspase-3 activation, failed to rescue 5-AzaC-induced caspase-3 activation in neonatal mice. Furthermore, the CB1RKO, which provides protection against alcohol-induced caspase-3 activation [19, 23, 24], also failed to rescue 5-AzaC-induced caspase-3 activation in neonatal mice. Although future studies are required to identify the DNA hypomethylation-induced regulation of apoptosis-related genes in P7 mice, it should be noted that the DNA methylation inhibitor activity of 5-AzaC involves its incorporation into cellular DNA/RNA, with subsequent sequestration of DNA methyltransferases (DNMTs) through the formation of a covalent bond between C6 of 5-AzaC and the cysteine thiolate of DNMTs [62]. Both DNMT1 and DNMT3A are highly expressed during early brain development compared to adulthood [19] and are involved in the regulation of DNA methylation in both embryonic and adult brain neurons [63–65]. Moreover, extensive apoptosis was observed in the brains of DNMT1 knockout (KO) embryos just after gastrulation [66]. Furthermore, inhibition of DNMT1 activities, either via genetic ablation or pharmacological treatment, protects injured neurons, suggesting that a balanced level of DNA methylation is essential for neuronal survival [67]. Conditional DNMT3A KO mice (lacking DNMT3A throughout the entire brain) are normal at birth but die in young adulthood [63]. DNMT3A was shown to contribute to the survival of motor neurons and the maintenance of the neuromuscular endplate structure [63]. Interestingly, the conditional deletion of both DNMT1 and DNMT3A in postnatal post-mitotic neurons (P14) at the end of synaptogenesis failed to induce neuronal loss but caused synaptic defects in adulthood [68]. Together, these findings suggest that DNA methylation levels during embryonic and synaptogenesis are important for neuronal survival and the proper maturation of neuronal circuits.
The DNA methylation pattern observed in adult cells is established during gametogenesis and early embryonic development through a sequential process of demethylation and de novo methylation [69]. Although our study demonstrates that 5-AzaC inhibits DNA methylation, our study does not exclude other possible effects of 5-AzaC, such as incorporation into RNA and the inhibition of DNA, RNA, and protein synthesis, and these events can also affect chromatin organization and gene expression. Together, these studies suggest that the hypomethylation induced by short-term 5-AzaC treatment during periods of active synaptogenesis (P7) may lead to increased genomic instability and, in turn, neurodegeneration. Hence, DNA methylation is crucial for the function of postnatal neurons in the brain. These observations are consistent with previous studies demonstrating that 5-AzaC-induced changes in DNA methylation could alter chromosomal architecture, leading to chromosome instability [70] and changes in gene expression [71–74].
The present caspase-3 activation results are consistent with previous studies in which 5-AzaC was administered to pregnant animals as a single dose at the 11th [75] or 13th day [76] of gestation (equivalent to the first and second trimesters in humans, respectively), and this treatment evoked significant apoptosis in the brain of fetuses 12–24 h after treatment. Together, these findings suggest that exposure to 5-AzaC during the first, second or third trimester of pregnancy is deleterious to the developing fetus. Our results are also consistent with several in vitro studies showing that 5-AzaC treatment caused apoptotic cell death not only in cancerous cells [77–79] but also in many normal cells [75, 76, 80–82].
The present study is the first to demonstrate that postnatal administration of 5-AzaC down-regulates ERK1/2 phosphorylation and Arc expression in both the neocortex and the hippocampus. Activated ERK1/2 signaling is critical in mediating the response to neurotrophic stimuli, such as nerve growth factor and brain-derived neurotrophic factor, to support neuronal survival; the disruption of activated ERK1/2 signaling has been associated with neurodegeneration, synaptic deficits [20, 24, 83] and abnormal brain development, leading to cognitive dysfunction [84]. The roles of Arc proteins in the mature neural network [85–88] have been thoroughly studied, but their roles during development are less clear. Moreover, both ERK1/2 activation and Arc expression are induced by neural activity or are increased by experimental stimuli (glutamate or electrical stimulation) or by natural stimulation through behavioral learning or experiencing novel conditions [89–91].
Previous studies have identified the roles of DNA methylation and demethylation in neural plasticity (LTP) and memory formation within the adult hippocampus [68, 92–95]. We have used three memory behaviors that are dependent on the integrity of the hippocampus-entorhinal cortex [48, 96] (limbic system) [97, 98] (spatial memory) as well as the olfactory system [99] (social behavior). In humans, recognition memory, a subtype of declarative memory, is crucial for recalling various events, objects and people [100] and can be tested in animals through memory for an object (what), its spatial location (where) and the temporal element (when) of an event [49, 101–104]. Our findings suggest that transient inhibition of DNA methylation by 5-AzaC for a short period at P7 also produced long-lasting deficits in object, spatial and social recognition memory and LTP abnormalities in adulthood. LTP has been implicated in the activity-dependent refinement of neural circuits during development and the formation of dendritic spines [105]. Therefore, suppression of both ERK1/2 phosphorylation and Arc expression may disrupt critical events, such as synaptic plasticity, during postnatal development, leading to impairments in object, spatial and social recognition memory in P7 5-AzaC-treated adult mice. Furthermore, in our previous studies, alcohol exposure at P7 resulted in DNA hypomethylation in neonatal mice [19] and also caused long-lasting abnormalities object, spatial and social recognition memory and synaptic plasticity in adult mice [22–24]. In addition, adult neurogenesis generates various functional neural cell types from multipotent neural stem cells (NSCs) in the particular zones of the mammalian brain throughout the life time of an organism and may correlate with complex neuronal activities, such as learning and memory [106–108]. The observed deficits in learning and memory behaviors might also arise from the lack of protection of the newly generated functional neuronal cells from NSCs or impairments in the neurogenesis process in adult mice treated with 5-AzaC at P7 and should be investigated in future studies.
Conditional deletion of DNMT1 in mouse brain precursor cells in vivo leads to global DNA hypomethylation and postnatal death of neurons in several brain regions [109]. Targeted deletion of DNMT1 in the dorsal forebrain causes the severe and progressive degeneration of cortical and hippocampal neurons due to hypomethylation-induced neurodegeneration, both pre- and postnatally [110]. In addition, deletion of DNMT1 leads to long-lasting deregulation of neuronal gene expression and deficits in LTP, learning, and memory in adult mice [110, 111]. In mice with a brain-specific deletion of DNMT3A, global DNA methylation is not altered, but these mice exhibit abnormal motor neurons, neuromuscular junctions and premature death [63]. Although individual conditional genetic ablation of DNMT1 or DNMT3A after P14 failed to alter adult LTP, learning, or memory, the double KO (deletion of both DNMT1 and DNMT3A) induced significant impairments in synaptic plasticity, learning and memory in adult mice [68].
Although most developmental studies are limited to the genetic deletion of one or more DNA methylating enzymes (DNMTs), many adult studies determine the function of DNA methylation in learning and memory using 5-AzaC. For example, an infusion of 5-AzaC directly into the CA1 region immediately after contextual fear conditioning blocks memory formation [112]. DNMT inhibitors applied acutely to adult brain slices in vitro or the adult hippocampus in vivo can inhibit synaptic plasticity and learning and memory [112–114]. In addition, impaired DNMT activity and the resulting reduction in DNA methylation levels and DNA binding proteins were observed for in several genes associated with neurodegenerative disorders in adults (for references see [115]). Recent studies suggest that DNA demethylation could be achieved also through the teneleven translocation (Tet) family of methylcytosine dioxygenases (Tet1, Tet2, and Tet3) [31]. Genetic deletion of Tet1 impairs several synaptic plasticity-related genes, including Arc, and induces abnormal hippocampal synaptic plasticity and impaired spatial [116] and contextual fear memory [117]. Although future studies are required to understand the brain region specific epigenome changes and their relationship to observed neurobehavioral deficits in adult animals, our studies clearly suggest that the impairment of DNA methylation during early brain development affects the expression of survival factors [118] such as Arc [23] expression, inducing a delay in neuronal maturation [119]. Moreover, such impairment may be responsible for the observed object, spatial and social recognition memory deficits and synaptic plasticity abnormalities in adult mice.
6. Conclusions
In summary, the experimental evidence from the current study reveals that 5-AzaC treatment at P7 inhibits DNA methylation, ERK1/2 activation, and Arc expression, leading to neurodegeneration in neonatal mice and subsequently to impaired Arc expression and behavioral abnormalities in adult mice. The current findings also suggest that the mechanism by which 5-AzaC induces caspase-3 activation is significantly independent of many upstream targets such as CB1R and G9a which are known to be upregulated by alcohol in P7 mice [19, 22–24, 35, 120, 121], though both produced a similar reduction in DNA methylation, Arc expression along with long-lasting synaptic deficits. Understanding the function of DNA methylation during this distinct period of brain development may shed light on the developmental origin of many adult disabilities.
HIGHLIGHTS.
5-AzaC treatment at P7 inhibits DNA methylation and activates caspase-3.
Loss of pERK1/2 and Arc were observed after 5-AzaC treatment in P7 mice.
5-AzaC treatment at P7 impairs synaptic plasticity in adult hippocampal slices.
5-AzaC in P7 mice causes persistent Arc and neurobehavioral deficits in adult mice.
5-AzaC acts as a teratogenic in the immature brain and causes synaptic dysfunction.
Acknowledgments
This research was supported by a grant from the National Institute of Alcohol and Alcoholism (RO1-AA019443) to B.S.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross EJ, Graham DL, Money KM, Stanwood GD. Developmental consequences of fetal exposure to drugs: what we know and what we still must learn. Neuropsychopharmacology. 2015;40:61–87. doi: 10.1038/npp.2014.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldman LR, Koduru S. Chemicals in the environment and developmental toxicity to children: a public health and policy perspective. Environ Health Perspect. 2000;108(Suppl 3):443–8. doi: 10.1289/ehp.00108s3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldschmidt L, Day NL, Richardson GA. Effects of prenatal marijuana exposure on child behavior problems at age 10. Neurotoxicol Teratol. 2000;22:325–36. doi: 10.1016/s0892-0362(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 4.Koger SM, Schettler T, Weiss B. Environmental toxicants and developmental disabilities: a challenge for psychologists. Am Psychol. 2005;60:243–55. doi: 10.1037/0003-066X.60.3.243. [DOI] [PubMed] [Google Scholar]
- 5.Mattson SN, Roesch SC, Fagerlund A, Autti-Ramo I, Jones KL, May PA, et al. Toward a neurobehavioral profile of fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2010;34:1640–50. doi: 10.1111/j.1530-0277.2010.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schettler T. Toxic threats to neurologic development of children. Environ Health Perspect. 2001;109(Suppl 6):813–6. doi: 10.1289/ehp.01109s6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stein J, Schettler T, Wallinga D, Valenti M. In harm’s way: toxic threats to child development. J Dev Behav Pediatr. 2002;23:S13–22. doi: 10.1097/00004703-200202001-00004. [DOI] [PubMed] [Google Scholar]
- 8.Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, Yeargin-Allsopp M, et al. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics. 2011;127:1034–42. doi: 10.1542/peds.2010-2989. [DOI] [PubMed] [Google Scholar]
- 9.Basavarajappa BS. Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain sciences. 2015;5:456–93. doi: 10.3390/brainsci5040456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mantha K, Laufer BI, Singh SM. Molecular changes during neurodevelopment following second-trimester binge ethanol exposure in a mouse model of fetal alcohol spectrum disorder: from immediate effects to long-term adaptation. Dev Neurosci. 2014;36:29–43. doi: 10.1159/000357496. [DOI] [PubMed] [Google Scholar]
- 11.Norman AL, O’Brien JW, Spadoni AD, Tapert SF, Jones KL, Riley EP, et al. A functional magnetic resonance imaging study of spatial working memory in children with prenatal alcohol exposure: contribution of familial history of alcohol use disorders. Alcohol Clin Exp Res. 2013;37:132–40. doi: 10.1111/j.1530-0277.2012.01880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brent RL. Environmental causes of human congenital malformations: the pediatrician’s role in dealing with these complex clinical problems caused by a multiplicity of environmental and genetic factors. Pediatrics. 2004;113:957–68. [PubMed] [Google Scholar]
- 13.Brent RL. Teratology in the 20th century environmental causes of congenital malformations in humans and how they were established. Neurotoxicol Teratol. 2004;26:1–12. doi: 10.1016/j.ntt.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–51. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardenas A, Houseman EA, Baccarelli AA, Quamruzzaman Q, Rahman M, Mostofa G, et al. In utero arsenic exposure and epigenome-wide associations in placenta, umbilical artery, and human umbilical vein endothelial cells. Epigenetics: official journal of the DNA Methylation Society. 2015;10:1054–63. doi: 10.1080/15592294.2015.1105424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin VW, Baccarelli AA, Burris HH. Epigenetics-a potential mediator between air pollution and preterm birth. Environmental epigenetics. 2016;2 doi: 10.1093/eep/dvv008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen A, Cingolani P, Senut MC, Land S, Mercado-Garcia A, Tellez-Rojo MM, et al. Lead exposure induces changes in 5-hydroxymethylcytosine clusters in CpG islands in human embryonic stem cells and umbilical cord blood. Epigenetics: official journal of the DNA Methylation Society. 2015;10:607–21. doi: 10.1080/15592294.2015.1050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basavarajappa BS, Subbanna S. Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits. Brain sciences. 2016;6 doi: 10.3390/brainsci6020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagre NN, Subbanna S, Shivakumar M, Psychoyos D, Basavarajappa BS. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J Neurochem. 2015;132:429–42. doi: 10.1111/jnc.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Izumi Y, Kitabayashi R, Funatsu M, Izumi M, Yuede C, Hartman RE, et al. A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-D-aspartate receptor function and ethanol sensitivity. Neuroscience. 2005;136:269–79. doi: 10.1016/j.neuroscience.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 21.Noel M, Norris EH, Strickland S. Tissue plasminogen activator is required for the development of fetal alcohol syndrome in mice. Proc Natl Acad Sci U S A. 2011;108:5069–74. doi: 10.1073/pnas.1017608108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subbanna S, Basavarajappa BS. Pre-administration of G9a/GLP inhibitor during Synaptogenesis Prevents Postnatal Ethanol-induced LTP Deficits and Neurobehavioral Abnormalities in Adult Mice. Exp Neurol. 2014;261:34–43. doi: 10.1016/j.expneurol.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subbanna S, Nagaraja NN, Umapathy NS, Pace BS, Basavarajappa BS. Ethanol Exposure Induces Neonatal Neurodegeneration by Enhancing CB1R Exon1 Histone H4K8 Acetylation and Up-regulating CB1R Function causing Neurobehavioral Abnormalities in Adult Mice. International Journal of Neuropsychopharmacology. 2015 doi: 10.1093/ijnp/pyu028. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subbanna S, Shivakumar M, Psychoyos D, Xie S, Basavarajappa BS. Anandamide-CB1 Receptor Signaling Contributes to Postnatal Ethanol-Induced Neonatal Neurodegeneration, Adult Synaptic and Memory Deficits. Journal of neuoscience. 2013;33:6350–66. doi: 10.1523/JNEUROSCI.3786-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson DA, Peterson J, Basavaraj BS, Saito M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcoholism Clin and Exp Res. 2011;35:1974–84. doi: 10.1111/j.1530-0277.2011.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown SE, Weaver IC, Meaney MJ, Szyf M. Regional-specific global cytosine methylation and DNA methyltransferase expression in the adult rat hippocampus. Neurosci Lett. 2008;440:49–53. doi: 10.1016/j.neulet.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 27.Feng J, Chang H, Li E, Fan G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res. 2005;79:734–46. doi: 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- 28.Meadows JP, Guzman-Karlsson MC, Phillips S, Brown JA, Strange SK, Sweatt JD, Hablitz JJ. Dynamic DNA methylation regulates neuronal intrinsic membrane excitability. Sci Signal. 2016;9(442):ra83. doi: 10.1126/scisignal.aaf5642. http://dx.doi.org/10.1126/scisignal.aaf5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–8. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simmons RK, Stringfellow SA, Glover ME, Wagle AA, Clinton SM. DNA methylation markers in the postnatal developing rat brain. Brain Res. 2013;1533:26–36. doi: 10.1016/j.brainres.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nabel CS, Kohli RM. Molecular biology. Demystifying DNA demethylation. Science. 2011;333:1229–30. doi: 10.1126/science.1211917. [DOI] [PubMed] [Google Scholar]
- 32.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 33.Steiner H, Bonner TI, Zimmer AM, Kitai ST, Zimmer A. Altered gene expression in striatal projection neurons in CB1 cannabinoid receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5786–90. doi: 10.1073/pnas.96.10.5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basavarajappa BS, Saito M, Cooper TB, Hungund BL. Chronic ethanol inhibits the anandamide transport and increases extracellular anandamide levels in cerebellar granule neurons. Eur J Pharmacol. 2003;466:73–83. doi: 10.1016/s0014-2999(03)01557-7. [DOI] [PubMed] [Google Scholar]
- 35.Subbanna S, Shivakumar M, Umapathy NS, Saito M, Mohan PS, Kumar A, et al. G9a-Mediated Histone Methylation Regulates Ethanol-Induced Neurodegeneration in the Neonatal Mouse Brain. Neurobiol Dis. 2013;54:475–85. doi: 10.1016/j.nbd.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basavarajappa BS, Ninan I, Arancio O. Acute Ethanol Suppresses Glutamatergic Neurotransmission through Endocannabinoids in Hippocampal Neurons. J Neurochem. 2008;107:1001–13. doi: 10.1111/j.1471-4159.2008.05685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paxinos G, Franklin K. The mouse Brain in Stereotaxic Coordinates. Academic press; San Diego, USA: 2004. [Google Scholar]
- 38.Emi Y, Harada Y, Sakaguchi M. Involvement of a di-leucine motif in targeting of ABCC1 to the basolateral plasma membrane of polarized epithelial cells. Biochem Biophys Res Commun. 2013;441:89–95. doi: 10.1016/j.bbrc.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 39.West MJ. Stereological studies of the hippocampus: a comparison of the hippocampal subdivisions of diverse species including hedgehogs, laboratory rodents, wild mice and men. Prog Brain Res. 1990;83:13–36. doi: 10.1016/s0079-6123(08)61238-8. [DOI] [PubMed] [Google Scholar]
- 40.Basavarajappa BS, Nagre NN, Xie S, Subbanna S. Elevation of endogenous anandamide impairs LTP, learning, and memory through CB1 receptor signaling in mice. Hippocampus. 2014;24:808–18. doi: 10.1002/hipo.22272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basavarajappa BS, Subbanna S. CB1 Receptor-Mediated Signaling Underlies the Hippocampal Synaptic, Learning and Memory Deficits Following Treatment with JWH-081, a New Component of Spice/K2 Preparations. Hippocampus. 2014;24:178–88. doi: 10.1002/hipo.22213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dellu F, Mayo W, Cherkaoui J, Le Moal M, Simon H. A two-trial memory task with automated recording: study in young and aged rats. Brain Res. 1992;588:132–9. doi: 10.1016/0006-8993(92)91352-f. [DOI] [PubMed] [Google Scholar]
- 43.Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc Natl Acad Sci U S A. 2000;97:14731–6. doi: 10.1073/pnas.97.26.14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56. doi: 10.1002/(SICI)1098-1063(2000)10:1<47::AID-HIPO5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 45.Thor DH, Wainwright KL, Holloway WR. Persistence of attention to a novel conspecific: some developmental variables in laboratory rats. Dev Psychobiol. 1982;15:1–8. doi: 10.1002/dev.420150102. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG, et al. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci. 2011;31:8786–802. doi: 10.1523/JNEUROSCI.3257-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terranova JP, Storme JJ, Lafon N, Perio A, Rinaldi-Carmona M, Le Fur G, et al. Improvement of memory in rodents by the selective CB1 cannabinoid receptor antagonist, SR 141716. Psychopharmacology (Berl) 1996;126:165–72. doi: 10.1007/BF02246352. [DOI] [PubMed] [Google Scholar]
- 48.Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- 49.Dere E, Huston JP, De Souza Silva MA. Episodic-like memory in mice: simultaneous assessment of object, place and temporal order memory. Brain Res Brain Res Protoc. 2005;16:10–9. doi: 10.1016/j.brainresprot.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Gilbert PE, Kesner RP. Memory for objects and their locations: the role of the hippocampus in retention of object-place associations. Neurobiol Learn Mem. 2004;81:39–45. doi: 10.1016/s1074-7427(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 51.van Goethem NP, Rutten K, van der Staay FJ, Jans LA, Akkerman S, Steinbusch HW, et al. Object recognition testing: rodent species, strains, housing conditions, and estrous cycle. Behav Brain Res. 2012;232:323–34. doi: 10.1016/j.bbr.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 52.Sik A, van Nieuwehuyzen P, Prickaerts J, Blokland A. Performance of different mouse strains in an object recognition task. Behav Brain Res. 2003;147:49–54. doi: 10.1016/s0166-4328(03)00117-7. [DOI] [PubMed] [Google Scholar]
- 53.Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 54.Nakayama H, Kajikawa S, Shinozuka J, Su WP, Doi K. Possible involvement of DNA methylation in 5-azacytidine-induced neuronal cell apoptosis. Histol Histopathol. 1999;14:143–50. doi: 10.14670/HH-14.143. [DOI] [PubMed] [Google Scholar]
- 55.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 56.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 57.Olney JW. New insights and new issues in developmental neurotoxicology. Neurotoxicology. 2002;23:659–68. doi: 10.1016/S0161-813X(01)00092-4. [DOI] [PubMed] [Google Scholar]
- 58.Olney JW. Focus on apoptosis to decipher how alcohol and many other drugs disrupt brain development. Frontiers in pediatrics. 2014;2:81. doi: 10.3389/fped.2014.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney JW. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Res Dev Brain Res. 2005;155:1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 60.Saito M, Chakraborty G, Hegde M, Ohsie J, Paik SM, Vadasz C. Involvement of ceramide in ethanol-induced apoptotic neurodegeneration in the neonatal mouse brain. J Neurochem. 2010;115:168–77. doi: 10.1111/j.1471-4159.2010.06913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siler-Marsiglio KI, Madorsky I, Pan Q, Paiva M, Neeley AW, Shaw G, et al. Effects of acute ethanol exposure on regulatory mechanisms of Bcl-2-associated apoptosis promoter, bad, in neonatal rat cerebellum: differential effects during vulnerable and resistant developmental periods. Alcohol Clin Exp Res. 2006;30:1031–8. doi: 10.1111/j.1530-0277.2006.000126.x. [DOI] [PubMed] [Google Scholar]
- 62.Broday L, Lee YW, Costa M. 5-azacytidine induces transgene silencing by DNA methylation in Chinese hamster cells. Mol Cell Biol. 1999;19:3198–204. doi: 10.1128/mcb.19.4.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen S, Meletis K, Fu D, Jhaveri S, Jaenisch R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. 2007;236:1663–76. doi: 10.1002/dvdy.21176. [DOI] [PubMed] [Google Scholar]
- 64.Siedlecki P, Zielenkiewicz P. Mammalian DNA methyltransferases. Acta Biochim Pol. 2006;53:245–56. [PubMed] [Google Scholar]
- 65.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature reviews Genetics. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 66.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 67.Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–81. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–30. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99:371–82. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 70.Matarazzo MR, Boyle S, D’Esposito M, Bickmore WA. Chromosome territory reorganization in a human disease with altered DNA methylation. Proc Natl Acad Sci U S A. 2007;104:16546–51. doi: 10.1073/pnas.0702924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dasmahapatra AK, Khan IA. DNA methyltransferase expressions in Japanese rice fish (Oryzias latipes) embryogenesis is developmentally regulated and modulated by ethanol and 5-azacytidine. Comparative biochemistry and physiology Toxicology & pharmacology: CBP. 2015;176–177:1–9. doi: 10.1016/j.cbpc.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 72.Dasmahapatra AK, Khan IA. Modulation of DNA methylation machineries in Japanese rice fish (Oryzias latipes) embryogenesis by ethanol and 5-azacytidine. Comparative biochemistry and physiology Toxicology & pharmacology: CBP. 2016;179:174–83. doi: 10.1016/j.cbpc.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 73.Laverriere JN, Muller M, Buisson N, Tougard C, Tixier-Vidal A, Martial JA, et al. Differential implication of deoxyribonucleic acid methylation in rat prolactin and rat growth hormone gene expressions: a comparison between rat pituitary cell strains. Endocrinology. 1986;118:198–206. doi: 10.1210/endo-118-1-198. [DOI] [PubMed] [Google Scholar]
- 74.Zagris N, Podimatas T. 5-Azacytidine changes gene expression and causes developmental arrest of early chick embryo. Int J Dev Biol. 1994;38:741–4. [PubMed] [Google Scholar]
- 75.Hossain MM, Nakayama H, Goto N. Apoptosis in the central nervous system of developing mouse fetuses from 5-azacytidine-administered dams. Toxicol Pathol. 1995;23:367–72. doi: 10.1177/019262339502300313. [DOI] [PubMed] [Google Scholar]
- 76.Ueno M, Katayama K, Nakayama H, Doi K. Mechanisms of 5-azacytidine (5AzC)-induced toxicity in the rat foetal brain. Int J Exp Pathol. 2002;83:139–50. doi: 10.1046/j.1365-2613.2002.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang HW, Wang HC, Chen CY, Hung TW, Hou MF, Yuan SS, et al. 5-azacytidine induces anoikis, inhibits mammosphere formation and reduces metalloproteinase 9 activity in MCF-7 human breast cancer cells. Molecules. 2014;19:3149–59. doi: 10.3390/molecules19033149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Frontiers in genetics. 2012;3:249. doi: 10.3389/fgene.2012.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kiziltepe T, Hideshima T, Catley L, Raje N, Yasui H, Shiraishi N, et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol Cancer Ther. 2007;6:1718–27. doi: 10.1158/1535-7163.MCT-07-0010. [DOI] [PubMed] [Google Scholar]
- 80.Hossain MM, Nakayama H, Takashima A, Goto N, Doi K. 5-Azacytidine (5Az) induces apoptosis in PC12 cells: a model for 5Az-induced apoptosis in developing neuronal cells. Histol Histopathol. 1997;12:439–45. [PubMed] [Google Scholar]
- 81.Hossain MM, Takashima A, Nakayama H, Doi K. 5-Azacytidine induces toxicity in PC12 cells by apoptosis. Exp Toxicol Pathol. 1997;49:201–6. doi: 10.1016/S0940-2993(97)80008-5. [DOI] [PubMed] [Google Scholar]
- 82.Zhao FY, Shao CP, Li Y, Ma WY, Tian N, Zheng JH. 5-Azacytidine induces early stage apoptosis and promotes in vitro maturation by changing chromosomal construction in murine oocytes. Reprod Toxicol. 2013;37:56–61. doi: 10.1016/j.reprotox.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 83.Young C, Straiko MM, Johnson SA, Creeley C, Olney JW. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol Dis. 2008;31:355–60. doi: 10.1016/j.nbd.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Samuels IS, Saitta SC, Landreth GE. MAP’ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–7. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Korb E, Finkbeiner S. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci. 2011;34:591–8. doi: 10.1016/j.tins.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li L, Carter J, Gao X, Whitehead J, Tourtellotte WG. The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol Cell Biol. 2005;25:10286–300. doi: 10.1128/MCB.25.23.10286-10300.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Messaoudi E, Kanhema T, Soule J, Tiron A, Dagyte G, da Silva B, et al. Sustained Arc/Arg3. 1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci. 2007;27:10445–55. doi: 10.1523/JNEUROSCI.2883-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.West AE, Greenberg ME. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chotiner JK, Nielson J, Farris S, Lewandowski G, Huang F, Banos K, et al. Assessment of the role of MAP kinase in mediating activity-dependent transcriptional activation of the immediate early gene Arc/Arg3. 1 in the dentate gyrus in vivo. Learn Mem. 2010;17:117–29. doi: 10.1101/lm.1585910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, et al. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2:1120–4. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- 92.Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013;20:274–81. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- 93.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–51. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szyf M. DNA methylation, behavior and early life adversity. Journal of genetics and genomics = Yi chuan xue bao. 2013;40:331–8. doi: 10.1016/j.jgg.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 95.Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Logothetis NK, Sheinberg DL. Visual object recognition. Annu Rev Neurosci. 1996;19:577–621. doi: 10.1146/annurev.ne.19.030196.003045. [DOI] [PubMed] [Google Scholar]
- 97.Baron-Cohen S, Ring H, Moriarty J, Schmitz B, Costa D, Ell P. Recognition of mental state terms. Clinical findings in children with autism and a functional neuroimaging study of normal adults. Br J Psychiatry. 1994;165:640–9. doi: 10.1192/bjp.165.5.640. [DOI] [PubMed] [Google Scholar]
- 98.Brothers L, Ring B, Kling A. Response of neurons in the macaque amygdala to complex social stimuli. Behav Brain Res. 1990;41:199–213. doi: 10.1016/0166-4328(90)90108-q. [DOI] [PubMed] [Google Scholar]
- 99.Sanchez-Andrade G, Kendrick KM. The main olfactory system and social learning in mammals. Behav Brain Res. 2009;200:323–35. doi: 10.1016/j.bbr.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 100.Clayton NS, Dickinson A. Episodic-like memory during cache recovery by scrub jays. Nature. 1998;395:272–4. doi: 10.1038/26216. [DOI] [PubMed] [Google Scholar]
- 101.Eacott MJ, Easton A, Zinkivskay A. Recollection in an episodic-like memory task in the rat. Learn Mem. 2005;12:221–3. doi: 10.1101/lm.92505. [DOI] [PubMed] [Google Scholar]
- 102.Eacott MJ, Norman G. Integrated memory for object, place, and context in rats: a possible model of episodic-like memory? J Neurosci. 2004;24:1948–53. doi: 10.1523/JNEUROSCI.2975-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fortin NJ, Wright SP, Eichenbaum H. Recollection-like memory retrieval in rats is dependent on the hippocampus. Nature. 2004;431:188–91. doi: 10.1038/nature02853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kart-Teke E, De Souza Silva MA, Huston JP, Dere E. Wistar rats show episodic-like memory for unique experiences. Neurobiol Learn Mem. 2006;85:173–82. doi: 10.1016/j.nlm.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 105.Lin B, Kramar EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–9. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 107.Hsieh J, Eisch AJ. Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol Dis. 2010;39:73–84. doi: 10.1016/j.nbd.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–97. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hutnick LK, Golshani P, Namihira M, Xue Z, Matynia A, Yang XW, et al. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. 2009;18:2875–88. doi: 10.1093/hmg/ddp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Golshani P, Hutnick L, Schweizer F, Fan G. Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus & related systems. 2005;3:227–33. doi: 10.1017/S1472928807000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–69. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 113.Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–73. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- 114.Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lagali PS, Picketts DJ. Matters of life and death: the role of chromatin remodeling proteins in retinal neuron survival. Journal of ocular biology, diseases, and informatics. 2011;4:111–20. doi: 10.1007/s12177-012-9080-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rudenko A, Dawlaty MM, Seo J, Cheng AW, Meng J, Le T, et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79:1109–22. doi: 10.1016/j.neuron.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, et al. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79:1086–93. doi: 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kokubo M, Nishio M, Ribar TJ, Anderson KA, West AE, Means AR. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. J Neurosci. 2009;29:8901–13. doi: 10.1523/JNEUROSCI.0040-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen Y, Ozturk NC, Zhou FC. DNA methylation program in developing hippocampus and its alteration by alcohol. PloS one. 2013;8:e60503. doi: 10.1371/journal.pone.0060503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Subbanna S, Nagre NN, Shivakumar M, Umapathy NS, Psychoyos D, Basavarajappa BS. Ethanol Induced Acetylation of Histone at G9a Exon1 and G9a-Mediated Histone H3 Dimethylation leads to Neurodegeneration in Neonatal Mice. Neuroscience. 2014;258C:422–32. doi: 10.1016/j.neuroscience.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Subbanna S, Psychoyos D, Xie S, Basavarajappa BS. Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol lipase does not cause neurodegeneration in neonatal mice. J Neurochem. 2015;134:276–87. doi: 10.1111/jnc.13120. [DOI] [PMC free article] [PubMed] [Google Scholar]