

Abstract
Since the discovery of major histocompatibility complex (MHC) molecules, it took some 40 years to arrive at a coherent picture of how MHC class I and MHC class II molecules really work. This is a story of proteases and MHC-like chaperones that support the MHC class I and II molecules in presenting peptides to the immune system. We now understand that the MHC system shapes both the repertoire of presented peptides and the subsequent T cell responses, with important implications ranging from transplant rejection to tumor immunotherapies. Here we present an illustrated review on the ins and outs of MHC class I and MHC class II antigen presentation.
Keywords: Antigen Presentation, MHC class I, MHC class II, tumor immunology, transplantation, auto-immune diseases
Why Do We Need to Present Antigens?
T cells help eliminate pathogens present in infected cells and also help B cells make better and different kinds of antibodies to protect against extracellular microbes and toxic molecules. To accomplish these important functions, T cells have to interact intimately with other cells and then find and instruct or eliminate the ones that are harbouring or have been exposed to these pathogenic threats. However, T cells are unable to peek beneath the surface of cells to identify ones that have ingested bacteria or are synthesizing viral or mutant proteins. Instead, antigen presentation systems have evolved, that display on the cell surface information about the various antigens that cells are synthesizing or have ingested. These antigen presentation pathways monitor the major subcellular compartments wherein pathogens could be lurking and report their findings to the appropriate kinds of T cells. Endogenously synthesized antigens in the cytosol of all cells are presented to CD8+ T cells as peptides bound to MHC I molecules, thereby allowing the CD8+ lymphocytes to identify and eliminate virally infected cells or cancers. Antigens ingested into endocytic compartments of macrophages, dendritic cells or B cells are presented to CD4+ T cells as peptides bound to MHC II molecules. T cells have antigen receptors that recognize antigenic peptide, but only in the context of MHC I or MHC II molecules that are displaying the antigen on the cell surface. Consequently, T cells are directed to work with cells, while not being ‘distracted’ by free antigen, to which they would not be able to do anything. Moreover, the pattern of expression of MHC I and II molecules directs T cells to interact with exactly the right kind of cells. Here, we review the current understanding of the mechanisms of antigen presentation, as well as their implications in health and disease. The studies discussed here have paved the way for increasingly refined analyses of the biology of antigen presentation – in different physiologic or clinical contexts, different cells, different organs – that are the focus of this special issue.
The MHC Scaffold or How Did MHC-I and MHC-II Molecules Evolve?
MHC I and II molecules present protein fragments to CD8+ and CD4+ T cells, respectively. These molecules are essential for cell-mediated immunity and therefore appeared at the inception of the adaptive immune system, some 500 million years ago[1]. For their construction they used two Ig-domains topped by two parallel alpha helixes resting on a platform of beta-pleated sheets. This capital structure generated a peptide-binding groove between the alpha helixes[2, 3], which is ‘evolutionarily speaking’ likely borrowed from earlier chaperone structures. There are arguments for this: a. chaperones bind unfolded stretches of proteins and the prototypic unfolded structure is a peptide; b. various phylogenetically older chaperones have a somewhat similar structure[4]; c. there are various MHC molecule look-a-likes that act as specific chaperones in the process of MHC-restricted antigen presentation. These include tapasin[5], tapasin binding protein-related (TAPBPR)[6][7], DM [8,9] and DO[10] molecules, as we discuss later. Yet MHC I and MHC II molecules are unique in the proteome because of their extreme polymorphism (>10,000 different alleles of MHC I molecules have been identified thus far!). This has interesting consequences. Polymorphic residues on the top alpha helixes interact with the TCR and are the basis for the specificity of TCRs for both an antigen peptide plus a particular allelic form of an MHC molecule (a phenomenon called MHC restriction). Polymorphic residues in the MHC peptide binding groove change the nature and location of so-called pockets. These variable pockets are filled by complementary variable amino acid side chains of peptides (so-called anchor residues), with the effect that different fragments from a defined antigen are presented by different polymorphic MHC molecules[11,12] (Figure 1). Yet, next to the anchor residues, most other amino acids in a peptide fill a free space and can be (almost) any of the 20 amino acids[13,14]. By having pockets with specificity for only a few side chains and allowing the remaining 6–10 amino acids to vary between all possibilities, each kind of MHC molecules can present a very large repertoire of peptides. Moreover, by having 3 to 6 different MHC I as well as 3 to 12 different MHC II molecules (the exact number depending on how many different MHC alleles were inherited from one’s parents and how the MHC II subunits paired), cells can present a large fraction of the universe of peptides, although not all sequences. In theory then, MHC I molecules can present a peptidome of around 6 × 20(6–7) different peptides, and MHC II can display up to 12 × 20(10) peptides. In actuality, such a large array of peptides cannot all be presented because there are only around 200,000 MHC I and 20,000 MHC II molecules on cells such as B and T cells[15]. Moreover, since some peptides are present in high number (from highly expressed proteins), the real number of different peptides presented by one cell is likely less than 10,000. Importantly, when a pathogen alters a critical anchor residue in one of its antigenic epitopes, it may prevent presentation of this antigen in one individual but not in another person with different MHC molecules that will simply select different peptides from the same pathogen.[16] Therefore, MHC polymorphism is good for the survival of the population and not necessarily the individual. How is this extensive polymorphism maintained in the species? One possibility is based on evidence that females can distinguish by smell MHC allele differences in males and prefer as mates individuals with whom they do not share MHC alleles. The attendant consequence of such a preference would be to promote maximal expression of polymorphic MHC alleles in offspring and maintenance of diverse alleles in the population [17]. Perfumes may then mask natural scent of this basis of partner choice, with unknown effects in human species. The obvious modern disadvantage of MHC polymorphism is transplant rejection, but even this may serve a useful function in nature by preventing the seeding of cancer cells between individuals. This is illustrated by the fact that MHC-deficient oral cancers are currently being transferred between Tasmanian Devils through bites and decimating the population of these animals in the wild [18,19].
Figure 1. Of MHC Locus and Allelic Products with Polymorphism.
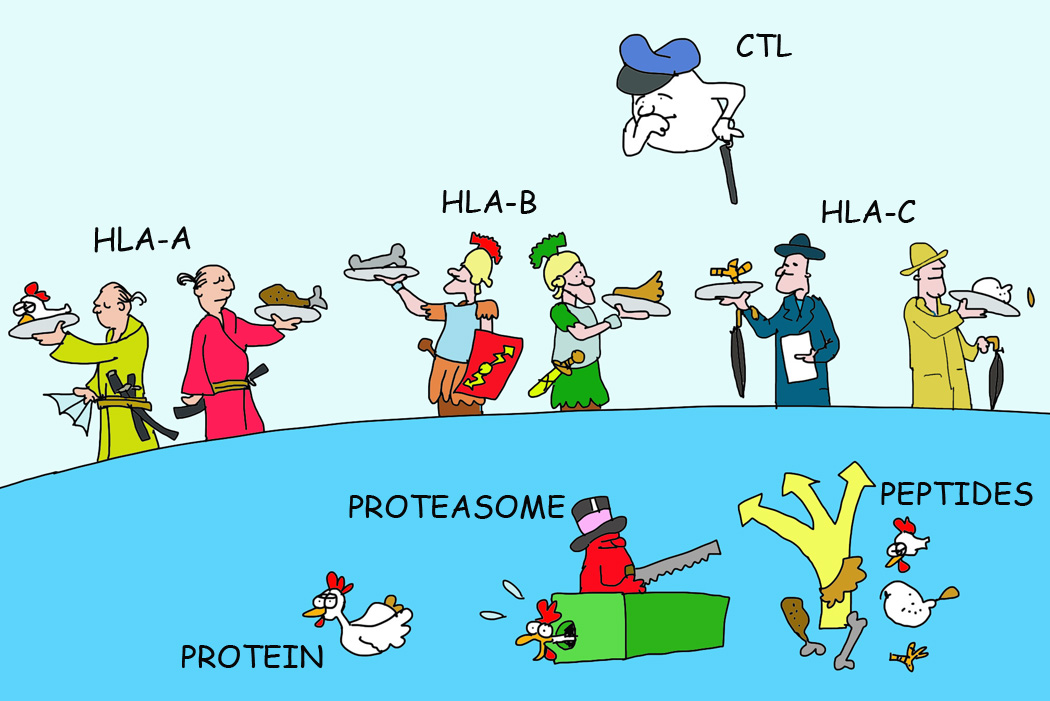
Most mammalian species express three different MHC I and three different MHC II molecules (shown here for the human MHC I HLA-A, HLA-B and HLA-C locus products). Since these are polymorphic and genetically encoded, a total of somewhere between 3 and 6 (depending on the differences between the inherited genes) alleles will be expressed on cells. These are polymorphic in the peptide-binding groove region of MHC molecules to present different peptides of a defined antigen.
How to Present Your Inner Self? MHC Class I Molecules
MHC I molecules present peptides from the proteins that are synthesized by cells. In healthy cells, all of these proteins are autologous ones to which CD8+ T cells are tolerant. However, when cells are expressing mutant sequences (e.g. in cancers), microbial genes (e.g. from viral infections) or foreign polymorphic genes (e.g. from transplants), these ‘non-self’ antigenic peptides are included in the presented peptidome, allowing CD8+ T cells to detect and destroy these abnormal cells. How MHC I manages to present a blueprint of the intracellular proteome has been established over the last 25 years[20] (Figure 2). Briefly, normal and pathogenic proteins are degraded by the proteasome into peptide fragments [21][22]. These fragments are further trimmed and to a large extent destroyed by cytosolic peptidases[23] but some survive by escaping into the ER through a peptide transporter called transporter associated with antigen processing (TAP) that is embedded in the ER membrane[20]. In the ER, TAP forms the centre of a peptide loading complex that includes a dedicated chaperone, tapasin, empty MHC I molecules awaiting peptides and two common chaperones, calreticulin and protein disulfide isomerase ERp57[24]. TAP translocates peptides that then are considered for binding by MHC I molecules. These empty MHC I molecules are held in a peptide-receptive state by the chaperone tapasin and tapasin also promotes MHC I-binding of peptides with a slow-off rate, thereby helping to shape the repertoire of presented peptides[25]. In this reaction, MHC-I molecules test the binding of many peptides and subsequently release most of these until a proper (low off-rate) peptide is bound[26]. These are usually peptides of a very specific length of 8–10 amino acids with appropriate anchor residues. Peptides that are too long can be trimmed by an ER resident aminopeptidase, ERAP1 (and ERAP 2 in some species)[27–29] before consideration by MHC I molecules that are either in the peptide-loading complex or associating with another tapasin look-alike chaperone in the ER called TAPBPR. Like tapasin, TAPBPR also shapes the peptide repertoire on MHC I molecules 30,31]. Very interestingly, binding of peptides longer than 8–9 residues, but not shorter ones, triggers a conformational change in ERAP1 that activates its hydrolysis [32–34]. Through this mechanism, ERAP1 trims most peptides only down to 8–9 residues, corresponding to the size needed for optimal binding to MHC I molecules. In the end, the peptides that are available to be presented are the ones that have been cleaved to the right size and have somehow escaped further hydrolysis to a size that is too small to stably bind to MHC I molecules (Figure 3). Peptides that are unable to bind an MHC I molecule are ultimately translocated back into the cytosol for degradation[35]. Whether there are mechanisms that help to protect some of these peptides from destruction or release from the ER during the time before they bind MHC I molecules, is not entirely clear.
Figure 2. A Simple Illustration of MHC I Antigen Presentation.
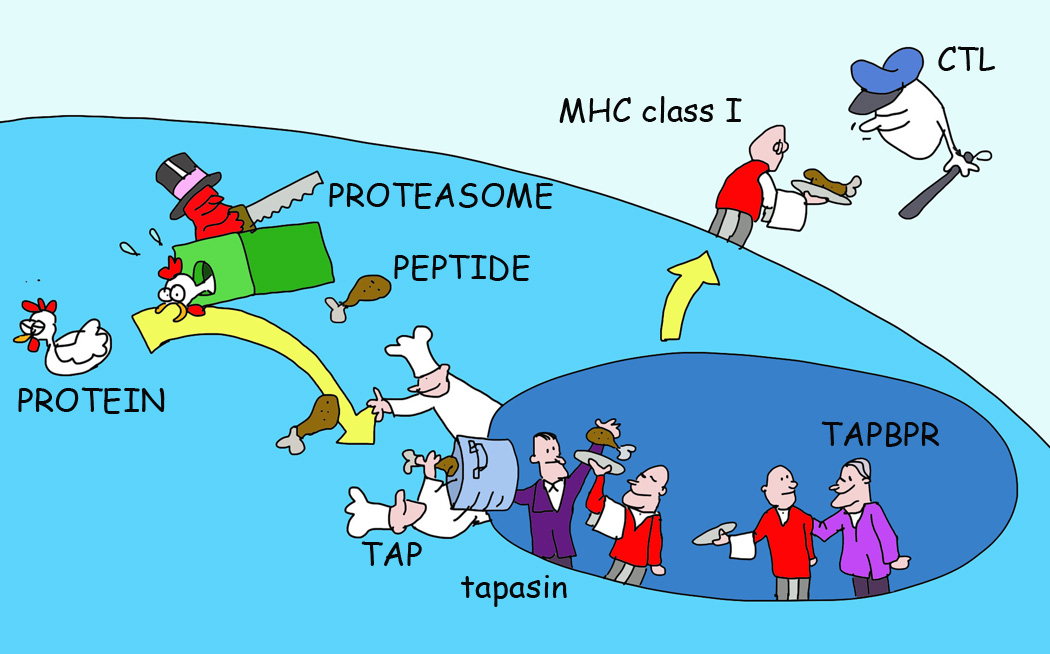
Antigens are degraded by the proteasome to yield peptide fragments. These peptides are then translocated from the cytosol into the endoplasmic reticulum (ER) lumen where MHC I in waiting for peptides is retained by a series of chaperones including a dedicated chaperone tapasin in the peptide-loading complex. A second dedicated chaperone (TAPBPR) can further optimize the peptides in MHC I. Only MHC I with optimal peptides is allowed to leave the ER to present the peptide fragments at the cell surface to CD8+ T cells.
Figure 3. Survival of The Fittest for MHC I Presentation and The Many Proteasomes.
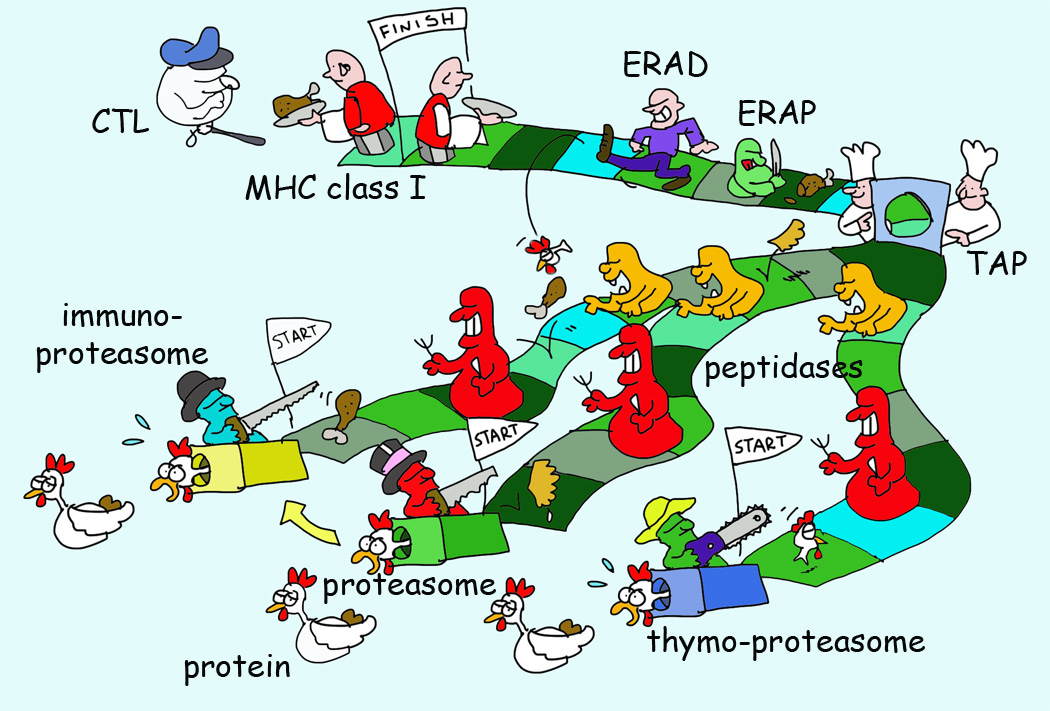
There are three types of proteasomes with unique tissue expression. These proteasomes have an altered cleavage specificity yielding (in part) different degradation fragments. These fragments are released in the hostile environment of the cytosol where the majority of peptides will be destroyed by peptidases. Few peptides survive this massacre through translocation in the ER by transporter associated with antigen processing (TAP). Here they can be further trimmed by ER resident aminopeptidase (ERAP) or translocated back into the cytosol by the ER associated degradation (ERAD) system. Only few peptides survive the chaperone-mediated survival selection for low off-rate peptides for a defined MHC I allele and these are ultimately presented.
Peptides should be considered the third subunit of MHC I molecules as they are required to stabilize these complexes when the MHC I molecules are not bound to chaperones in the ER[36]. Peptides also allow the MHC I molecules to be released from the ER quality control system (the various chaperones) for transport to the cell surface for presentation to CD8+ T cells[37,38]. This system of low off-rate peptide selection, exporting only peptide-loaded MHC I complexes may help prevent healthy cells from easily replacing their bound endogenous peptides for exogenous antigenic peptides, an event that would lead to the presentation of peptides that do not reflect the status of a given cell and possible execution by CD8+ T cells.
Complexity in the MHC Class I Antigen Presentation Pathway
The general scheme outlined above, is somewhat more complex when taking into consideration the diversity of the MHC I family. Indeed, the different loci expressed (in humans, HLA-A, -B and –C) and the many polymorphic allelic forms behave somewhat differently with respect to expression, peptide binding and stability[39,40]. Peptides may also be generated from multiple sources. This is because the proteasome not only degrades proteins as part of normal protein turn over, but also degrades abnormal ones that arise from errors in translation, folding and/or pairing. The degradation of these disabled proteins prevents their aggregation and also potentially more directly couples protein translation to antigen presentation[41–43]. Such antigens are called defective ribosomal products (DRiPs) and may be produced in greater amounts during high protein synthesis conditions, such as occur during viral infection. The degradation of DRiPs would quickly generate peptides after initial translation of the antigen and this may allow rapid detection of infected cells [44,45].
While the source of antigens can be different, so can be the cleavage of these antigens by different proteasomes. Many presented peptides can be generated through the phylogenetically older ‘conventional’ proteasome. However, the development of the immune system coincided with the evolution of alternate forms of active site subunits for this particle, leading to the assembly of an immunoproteasome. A set of these subunits (β1i, β2i, β5i) [46] is constitutively expressed in dendritic cells and lymphocytes and can be induced in all other cells by interferons, for example during viral infections [47,48]. When these subunits are expressed, they preferentially incorporate into newly assembling particles to form immunoproteasomes, that generate a distinct set of peptides during protein degradation[49]. This shift from constitutive to immunoproteasome in cells often enhances the generation of peptides presented by MHC I molecules, including many unique ones[50]. Generation of new peptides will be at the cost of other peptide fragments that are cleaved to make the new fragment [51]. Another set of alternate active site subunits (β1i, β2i, β5t) are expressed uniquely only in cortical thymic epithelial cells (cTECs), where they incorporate into thymoproteasome particles[52]. Among the peptides generated by thymoproteasomes, a number are unique and these play a critical role in the auditioning of developing CD8+ T cells during positive selection and also in allowing many of these cells to avoid subsequent negative selection [50]. Proteasomes but also the cytosolic and ER associated peptidases are variable in content and numbers. From this stew of proteolytic activities an estimated 0.02% of the peptides generated by the proteasome survive for presentation to the immune system [53](Figure 3).
Not How to Present… but What to Present
MHC I molecules present peptides from a cell’s expressed genes and thereby allow the immune system to monitor the proteins synthesized in a cell. Yet, cells may also alter signalling in response to transformation or infection, resulting in an altered phosphoproteome[54], acetylome, glycome[55] or any other (small) post-translational modifications[56–58]. The peptide transporter TAP allows peptides with these small modifications to enter the ER and some of these can bind to MHC I molecules for presentation to CD8+ T cells. These modified peptides are in fact not genetically-encoded but neo-epitopes to which the immune system may not be tolerant[59,60]. This can allow the immune system to detect cells in abnormal states (e.g. transformed ones) for elimination. Other non-genetically encoded antigenic peptides arise by peptide splicing by the proteasome, where the proteasome in fact performs the opposite reaction, linking two peptide fragments into a new one[61]. Whether this is just a consequence of the reverse proteolysis reaction of the proteasome or is influenced by particular cellular states is unclear, but these peptides can be presented and stimulate CD8+ T cell responses. Through these various mechanisms the repertoires of MHC I-presented peptide (the “presentome”) is expanded beyond the genetically-encoded sequences and add additional options for the detection of abnormal cells, but also provide risks for auto-immune reactions.
How to Hide One’s Inner Self? Pathogen and Tumor Escape of Antigen Presentation
While the MHC class I pathway evolved to allow detection and elimination of the nidi of viruses in an infected host, some viruses have co-evolved cloaking mechanisms to avoid such detection (Figure 4). A large majority of the human species – for example- is chronically infected with cytomegalovirus (CMV) and Herpes Simplex Virus (HSV). It is clear that these viruses have evolved ways to tamper with the process of antigen presentation [62]. CMV encodes proteins that inhibit the peptide transporter TAP or that induce the degradation of MHC I molecules in the ER or plasma membrane[63,64]. Other viruses shut down genomic MHC class I expression[65], produce a peptide mimic that blocks TAP or block the transport of MHC I molecules to the cell surface[66]. In fact, any step in the MHC class I antigen presentation pathway not interfering with cell viability can be expected to be manipulated by viruses to prevent their presentation.
Figure 4. Various Viral Immune Evasion Strategies for MHC I Antigen Presentation.
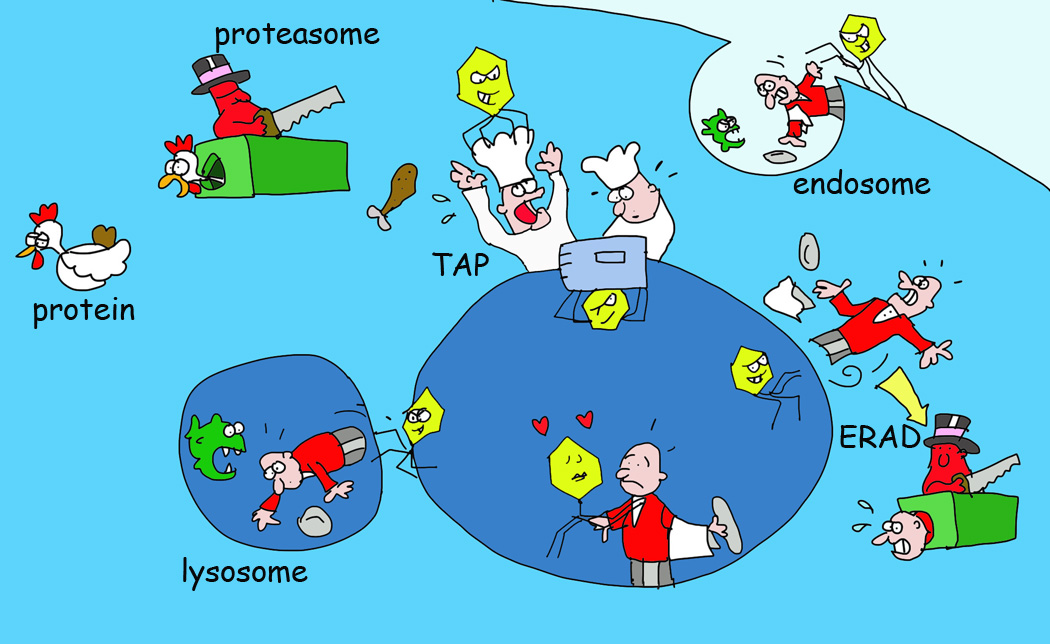
Pathogens have evolved different ways to obstruct processes selective in the antigen presentation pathway. Examples include viral proteins inhibiting peptide transport by TAP, retaining MHC I in the ER or recognizing MHC I in the ER for targeting these back into the cytosol for degradation by the proteasome (the ERAD system). Other viral proteins recognize MHC I at the cell surface for internalization and destruction in lysosomes.
Since most of the steps in the MHC class I pathway are not essential for viability and because cancer cells are often genetically unstable, tumors can, under the selection pressure imposed by CD8+ T cells, generate variants that have lost key components of the MHC class I pathway and escape control by CD8+ T cells [67]. In fact, a reduction of MHC I molecule expression in human tumors is often detected by pathologists[68]. This is one of the reasons for assuming an important role of the immune system in the control of particular tumors, especially those with more neo-antigens (from mutated proteins) such as melanoma and lung cancer[69,70]. Understanding how a given tumor can evade detection by CD8+ T cells could help determine the immunotherapies that are most likely to succeed against that tumor.
Some Exceptions on Self-Presentation by MHC Class I Molecules
The MHC class I antigen presentation system is constructed in such a way that most cells exclusively present their own antigens. Because of this, it was thought that CD8+ T cells selectively eliminate infected cells without destroying neighbouring ‘innocent bystanders’. However, this concept is challenged by the fact that the cytosol of many healthy cells (unlike cancer cells) are connected by so-called gap junctions. These gap junctions allow peptide fragments to pass into their direct connected neighbouring cells for entry in their antigen presentation pathway and presentation through the neighbour’s MHC I molecules to CD8+ T cells[71,72]. It is likely that cytosolic peptidases will limit the spread of such peptides beyond the most proximal neighbouring cells. However, since the proximal cells are at high risk of viral invasion, gap junctions may allow their elimination even before real entry of the pathogen.
A more intensely studied system where the MHC-I presents antigens are different than those made by the cell itself is cross-presentation[73,74] (Figure 5). This pathway operates in dendritic cells and other phagocytes and is quite important because it plays a central role in immune surveillance 74–76]. It allows dendritic cells to acquire antigens from other infected cells and cancers in the periphery and then report their presence to naive CD8+ T cells in lymphoid organs in ways that initiate an immune response. Phagocytes acquire these antigens when they ingest them by phagocytosis (e.g. eating cell debris [77–79] or possibly even by taking a “bite” of living cells[80]) or via receptor-mediated endocytosis (e.g. of glycan modified proteins through lectin receptors or of antibody-bound antigens through Fc-receptors[81,82]). There is more than one mechanism by which ingested antigens can be cross-presented but one likely involves cytosolic transfer of antigen from the phagosome into the cytosol for degradation by the proteasome[83,84]. The fragments may then be loaded on MHC I molecules in the ER or translocated back into the phagosome for local MHC class I peptide loading[78]. In another mechanism, some antigens are degraded by lysosomal proteases and loaded onto recycling MHC class I molecules in a pathway similar to MHC class II molecules[85–87]. TLR signalling can induce the accumulation of MHC I in recycling endosomes to promote cross-presentation[88]. A potential limitation of this latter mechanism is that it could result in priming CD8+ T cells to different peptides than the ones the T cells will encounter in infected cells (as the priming peptides are generated by proteases different from the proteasome). However, this may also be a problem in the phagosome-to-cytosol mechanism of cross presentation because in the absence of inflammation, immunoproteasomes in dendritic cells may also generate different fragments than the constitutive proteasome in most peripheral (and notably, tumor) cells [89]. Thus, whether and how cross-presentation allows activation of CD8+ T cells recognizing the full range of antigenic peptides presented by peripheral cells is unclear. Regardless of the exact mechanism, cross-presentation allows MHC I molecules to present peptides from antigens that usually are handled by MHC II molecules. The system employed by MHC II molecules for antigen presentation is understood in considerable more molecular detail and may share elements used for cross-presentation by MHC I molecules.
Figure 5. Crossing Boundaries; Various Models of Cross-Presentation by MHC I.
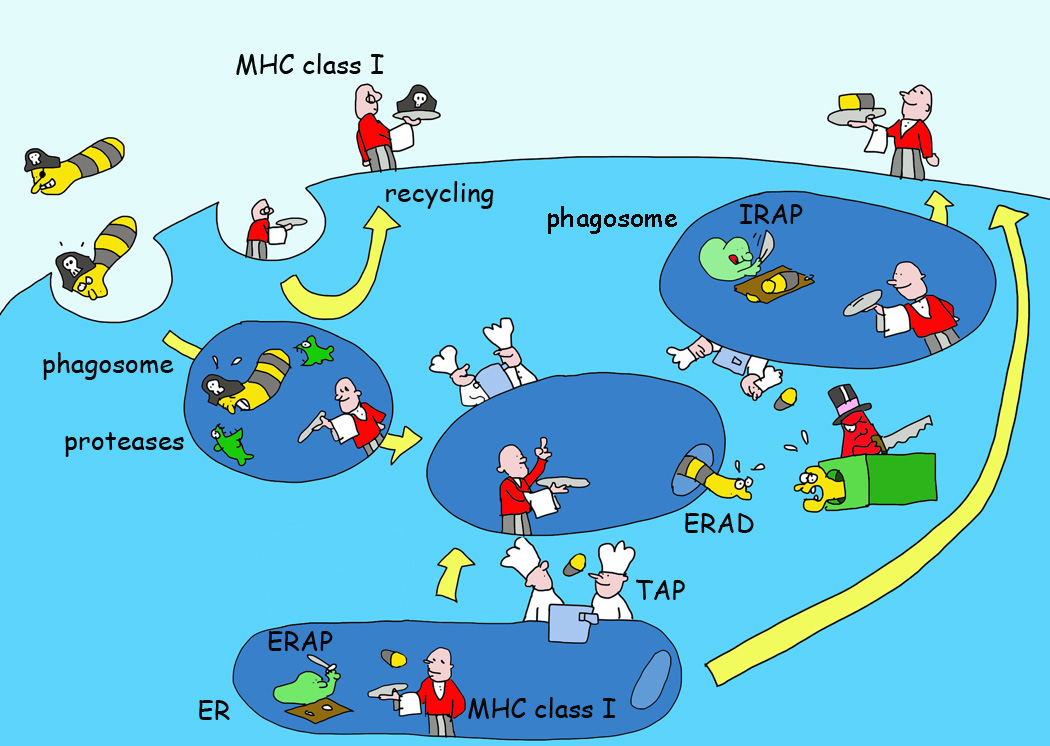
MHC I molecules can present exogenous antigens and antigens delivered in apoptotic bodies and other types of cell debris. For dendritic cells, this process can result in cross-priming of CD8+ T cells. MHC I can recycle through the endosomal pathway to acquire antigen fragments made by proteases such as insulin regulated aminopeptidase (IRAP)[129] for cross-presentation. Endosomes may also acquire TAP and other ER molecules that may help export antigens into the cytosol for proteasomal hydrolysis and the resulting peptides may be reimported into the endosomes for MHC I loading and recycling and/or be delivered in the normal antigen presentation pathway as shown in Figure 2.
MHC class II Molecules
MHC II molecules are both similar and different from MHC I molecules, and so are their mechanisms of presentation. MHC II molecules are expressed on immune cells such as B cells, monocytes, macrophages and dendritic cells and on epithelial cells following inflammatory signals, while MHC I molecules are expressed more ubiquitously. MHC II molecules on dendritic cells present antigen to naïve CD4+ T cells to activate them, and later MHC II molecules participate in the interaction of B cells and macrophages with these specific CD4+ effector T cells[90]. This is a critical function as exemplified by patients with deficits in MHC class II expression (bare lymphocyte syndrome), which results in extreme susceptibility to infections to a variety of microorganisms and death at young age[91]. The structure of MHC class II resembles that of MHC class I and they are both polymorphic proteins (and thus transplantation antigens)[90, 92]. Interestingly, Gadus morhua fish (atlantic cod) lack MHC II but express an MHC I molecule containing endocytosis signals, that effectively takes over MHC II function, illustrating the strong relationship and conserved function of these two MHC classes [93]. However, the nature of the presented peptides usually differ and so does the underlying biology of MHC class II antigen presentation.
How to Present the Outside World?
MHC class II presents peptide fragments that are generally larger than those presented by MHC class I, because the peptide-binding groove of MHC class II is open, allowing peptides to extend out of this site[94]. The MHC class II associated peptides are derived from extracellular proteins and from self-proteins that are degraded in the endosomal pathway (Figure 6)[95]. MHC II molecules associate during their assembly in the ER with the invariant chain Ii that acts as a pseudopeptide by filling the MHC class II peptide-binding groove and in addition targets MHC II molecules into the endosomal pathway through its cytosolic dileucine motif[20, 96]. In a compartment commonly named MIIC[97], MHC II molecules then meet antigenic fragments generated by resident proteases. In order for these peptides to bind MHC II molecules, the invariant chain has to be degraded by the same mix of proteases, especially cathepsin L and S[90]. This leaves an invariant chain fragment (called CLIP) inaccessible for proteases and remaining in the peptide-binding groove of MHC II molecules[98]. This CLIP fragment has to be exchanged for higher affinity peptides with the help of a dedicated MHC class II-like chaperone called DM (in human, HLA-DM)[99]. The structure of HLA-DM in association with MHC-II (HLA-DR1) reveals that DM locally opens the groove to release low-affinity peptides such as CLIP. DM release from MHC class II then locks the proper peptide fragments in the MHC class II peptide-binding groove[100]. After some residence in MIIC, MHC II molecules move to the plasma membrane either via vesicular transport or in the form of tubules[101–103]. Since the targeting information in the invariant chain has been removed after its degradation in MIIC, MHC II molecules can stably reside on the plasma membrane.
Figure 6. A Simple Illustration of MHC II Antigen Presentation.
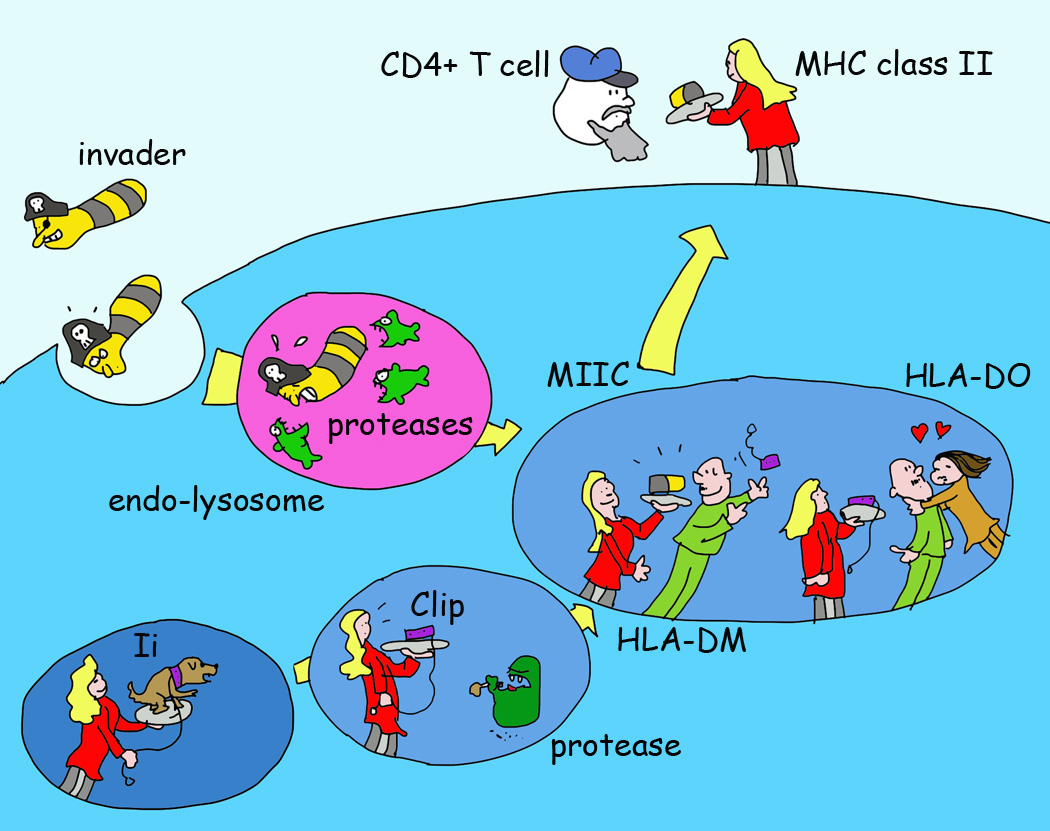
MHC II proteins are made in the ER where they pair with a third chain, the invariant chain or Ii. Ii fills (through a peptide sequence called CLIP) the MHC II peptide-binding groove and allows efficient exit of MHC II from the ER. Ii also guides MHC II through the cells to a late endosomal compartment, MIIC. Ii is degraded by endosomal proteases, as are antigens taken up by endocytosis or phagocytosis. The CLIP segment is protected from destruction and exchanged for an antigenic peptide with the help of a dedicated chaperone called DM (HLA-DM in human). Another chaperone expressed in a few immune cell types (immature B cells, some DC forms). called DO (HLA-DO) can compete for DM binding to MHC II and thereby affect the peptide repertoire on MHC II that is ultimately presented at the cell surface to CD4+ T cells.
Variations in MHC class II presentation
Like for MHC I molecules, there are also different MHC II loci in most species (in man three named HLA-DR, HLA-DQ and HLA-DP). Also, MHC II molecules are polymorphic (>3,000 alleles known) [90] and their polymorphic amino acids similarly cluster in and around the peptide-binding groove, shaping the peptide-binding pockets. Consequently, different MHC II alleles bind different peptides by virtue of their different anchor residues[104]. This likely explains why different MHC II alleles link strongly to defined auto-immune diseases [105] and immune responses against external environmental and food antigens[90]. This is well illustrated for the link between gluten sensitivity (Celiac disease) and the MHC class II allele HLA-DQ2. HLA-DQ2 is able to present a peptide from gluten after de-amination by tissue transglutaminase to activate CD4+ T cells and drives the disease[106]. The dominant antigens for auto-immune diseases are often not clear but there are some suggestions. For example, insulin has been suspected to mediate HLA-DR3/4 associated Type I Diabetes and myelin basic protein to participate in HLA-DR1 related Multiple Sclerosis[107, 108]. One additional mechanism involves the presentation of an atypical conformation of a peptide [109] as the result of peptide-loading of MHC II molecules in compartments that lack DM molecules, such as may occur in recycling endocytic compartments [110] or in the ER [111,112]. In the absence of DM’s function, the non-optimal peptide conformation bound to MHC II is not corrected. A different conformation of a self-peptide can be recognized as non-self by the CD4+ T cells that may drive the induction of auto-antibodies[109]. Yet, the fact that many people with these MHC class II alleles never develop auto-immune diseases and that for most of these conditions well less than half of identical twins are concordant for disease, indicates that epigenetic and other factors must also be involved and these are as of yet unclear.
Next to genetic variation resulting in polymorphism of MHC II molecules, another variation in the life of MHC class II lies with the associated invariant chain, that is actually not so invariant. In fact, there are multiple splice variants including one (p44) that contains an additional protease inhibitor (cystatin) domain[90]. This variant can be expected to modify the protease activities involved in antigen preparation for MHC II molecules. The proteases (cathepsins in the endosomal pathway) also vary in the different MHC class II-expressing immune cells, as do natural inhibitors for these cathepsins (called cystatins)[90,96]. Likely, antigens are degraded after denaturation (which involves the reduction of their disulphide bonds by the enzyme GILT[113], and acidic pH[114]) by a swarm of proteases that will generate and destroy potential peptides for a defined MHC II allele.
Peptides are selected for presentation by MHC II with the help of the chaperone DM. DM can be further controlled by a dedicated co-chaperone called DO (HLA-DO in man) that strongly pairs with DM[115]. DO associates to DM at the same interface as occupied by MHC II molecules [100] and thus inhibits DM-assisted peptide loading of MHC II molecules unless the DM-DO complex enters very acidic endosomes along with MHC II molecules[116,117]. DO thus shapes the peptide repertoire by preventing peptide binding in earlier vesicular compartments. Deleting DO induces type I diabetes in mouse models and possibly other autoimmune diseases[118].
Other variations entail the transport and surface half-life of MHC II molecules. MHC II transport from MIIC to the plasma membrane is not constitutive but controlled in dendritic cells, monocytes and B cells. Activation of dendritic cells promotes MHC II transport to the cell surface and strongly enhances the half-life of these molecules on the plasma membrane[119, 120]. As a result, activated dendritic cells have high numbers (around 2 million per cell) of MHC II molecules on the cell surface that continue to present antigens for long periods. However, there are also other ways to control MHC II expression. For example, in human monocytes IL-10 increases the expression of an ubiquitin ligase membrane associated RING-CH1 (MARCH-1) that ubiquitinates the tail of cell surface MHC II molecules initiating their rapid internalization and destruction[121]. Genome-wide analyses have identified many factors that control the complex process of control of MHC II expression and transport in dendritic cells[122]. Many pathogens, especially those residing in MHC II-containing phagosomes, also inhibit MHC II expression or peptide loading[123, 124]. Such immune evasion by pathogens may be caused by manipulation of DM interactions with MHC II molecules, manipulation of pH levels, alteration of protein networks, and induction of MHC ubiquitination by MARCH homologs [125], or other processes required for optimal antigen presentation by MHC II molecules.
Concluding Remarks and Future Perspectives
The work of many labs over the last decades has informed our understanding of the function of MHC class I and MHC class II molecules at the immunological, cell biological, genetic and atomic level. Many of the major mechanisms of both the MHC I and MHC II pathways are understood and we can look forward to an even more comprehensive understanding of the antigen presentation mechanisms in the coming years. With such knowledge, we can look forward to better understanding how these processes help to maintain health and/or contribute to disease pathogenesis. We anticipate that there will be the strong potential to translate this knowledge into clinical medicine. One important area is in cancer immunotherapy. While the role of MHC class I and -II antigen presentation in tumor immunology was initially restricted to curing cancer in mouse models, the recent development of checkpoint inhibitor antibodies has translated this into clinical responses in human melanoma, lung cancer and other tumors[126]. Characterizing the defects in antigen presentation in such tumors might in theory help identify patients who can or cannot respond to such interventions. Moreover, if methods can be developed to reverse such defects, they might be able to improve efficacy of these immunotherapies. Similarly, manipulating antigen presentation pathways might overcome the inhibition of antigen presentation induced by certain microbes, and then help eliminate these chronic infections. The rules for antigen presentation by MHC I molecules can be used to predict neo-epitopes detected by CD8+ T cells[127] and this information might be exploited to actively immunize cancer patients in conjunction with removal of checkpoint control and also to monitor patient responses. Targeting antigens into dendritic cells and in particular into specific presentation pathways has the potential to generate more robust and effective kinds of responses to vaccines and immunotherapies. On the other hand, antigen presentation might be manipulated in the opposite way to dampen autoimmune diseases, e.g. with toleragenic peptides or toleragenic antigen presenting cells. Despite the remarkable advances in our knowledge about antigen presentation, the picture is still incomplete but should continue to improve. For example, forward genetic screens are uncovering unsuspected new components in the pathways [122,128]. Filling in these gaps (see Outstanding Questions) should provide a higher resolution understanding of the pathways and their contribution to disease pathogenesis, as well as increasing the opportunities to exploit these pathways to develop better immunotherapies to prevent and/or treat disease.
Outstanding questions.
While antigen presentation by MHC molecules has been intensively studied for over almost 40 years, many aspects are still unclear. For example (and note that this is only a partial listing):
Why can peptides presented by MHC molecules not be more accurately predicted? What are we missing?
What are the many proteases involved in generating and destroying presented peptides, and what is their relative contribution and specificity?
How do the antigen presentation pathways so successfully present a broad peptidome from very large numbers of different proteins of very different abundances and in the face of robust peptide destruction?
Are there mechanisms that protect peptides from destruction before they bind to MHC molecules?
The components in the MHC antigen presentation pathway have remarkable heterogeneity in expression and activity in different tissues. Does that lead to different presentation of self-antigens and contribute to auto-immune responses?
Why do we have three different proteasomes?
Why do we express only three MHC I and three MHC II locus products and not more to cover all possible peptides for presentation?
What is the major mechanism(s) that generate DRIPS? - Inaccurate transcription, translation, folding or assembly?
Is antigen cross-presentation the result of many different systems or are there dominant systems?
For cross-presentation, how are antigens transported from endosomes into the cytosol?
For cross-presentation, how are MHC I molecules kept stable during transport to and after arrival in endosomes?
Are there mechanisms that promote the loading of peptides in endosomes for cross presentation?
Do dendritic cells or specific subsets of dendritic cells have unique components that promote cross presentation?
How are hydrophobic peptides delivered in the peptide binding groove of MHC I and MHC II molecules? Are there unique chaperones for such peptides?
Is there an MHC class II Peptide-Loading Complex?
Supplementary Material
Trends.
MHC molecules are critical in transplantation, auto-immunity, infections and tumor immunotherapy.
The biology of antigen presentation by MHC I and MHC II molecules provides targets for manipulation of these diseases.
This biology also explains the fragments presented to the immune system and the cellular evolutionary race to escape immune control of infected and transformed cells.
Many of the players determining antigen degradation and subsequent peptide-loading on MHC molecules are defined and are helping to improve the accuracy of predicting presented fragments.
The combined understanding of antigen presentation by MHC molecules allows exploitation to improve the responses of the cellular arm of the immune system to vaccinations and immunotherapies.
Acknowledgments
The work was supported by grants from NWO-TOP and an ERC Advanced grant to JN and NIH grants RO1AI114495 and RO1AI110374 to KLR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nature reviews Genetics. 2010;11:47–59. doi: 10.1038/nrg2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown JH, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 3.Bjorkman PJ, et al. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987;329:512–518. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- 4.Zhang P, et al. Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PloS one. 2014;9:e103518. doi: 10.1371/journal.pone.0103518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong G, et al. Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity. 2009;30:21–32. doi: 10.1016/j.immuni.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teng MS, et al. A human TAPBP (TAPASIN)-related gene, TAPBP-R. European journal of immunology. 2002;32:1059–1068. doi: 10.1002/1521-4141(200204)32:4<1059::AID-IMMU1059>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 7.Boyle LH, et al. Tapasin-related protein TAPBPR is an additional component of the MHC class I presentation pathway. Proc Natl Acad Sci U S A. 2013;110:3465–3470. doi: 10.1073/pnas.1222342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosyak L, et al. The structure of HLA-DM, the peptide exchange catalyst that loads antigen onto class II MHC molecules during antigen presentation. Immunity. 1998;9:377–383. doi: 10.1016/s1074-7613(00)80620-2. [DOI] [PubMed] [Google Scholar]
- 9.Fremont DH, et al. Crystal structure of mouse H2-M. Immunity. 1998;9:385–393. doi: 10.1016/s1074-7613(00)80621-4. [DOI] [PubMed] [Google Scholar]
- 10.Guce AI, et al. HLA-DO acts as a substrate mimic to inhibit HLA-DM by a competitive mechanism. Nature structural & molecular biology. 2013;20:90–98. doi: 10.1038/nsmb.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falk K, et al. Pool sequencing of natural HLA-DR, DQ, and DP ligands reveals detailed peptide motifs, constraints of processing, and general rules. Immunogenetics. 1994;39:230–242. doi: 10.1007/BF00188785. [DOI] [PubMed] [Google Scholar]
- 12.Falk K, et al. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 13.Madden DR, et al. The antigenic identity of peptide-MHC complexes: a comparison of the conformations of five viral peptides presented by HLA-A2. Cell. 1993;75:693–708. doi: 10.1016/0092-8674(93)90490-h. [DOI] [PubMed] [Google Scholar]
- 14.Smith KJ, et al. Bound water structure and polymorphic amino acids act together to allow the binding of different peptides to MHC class I HLA-B53. Immunity. 1996;4:215–228. doi: 10.1016/s1074-7613(00)80430-6. [DOI] [PubMed] [Google Scholar]
- 15.Walz S, et al. The antigenic landscape of multiple myeloma: mass spectrometry (re)defines targets for T-cell-based immunotherapy. Blood. 2015;126:1203–1213. doi: 10.1182/blood-2015-04-640532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmid BV, et al. Quantifying how MHC polymorphism prevents pathogens from adapting to the antigen presentation pathway. Epidemics. 2010;2:99–108. doi: 10.1016/j.epidem.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Chaix R, Cao C, Donnelly P. Is mate choice in humans MHC-dependent? PLoS Genet. 2008;4:e1000184. doi: 10.1371/journal.pgen.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siddle HV, et al. Characterization of major histocompatibility complex class I and class II genes from the Tasmanian devil (Sarcophilus harrisii) Immunogenetics. 2007;59:753–760. doi: 10.1007/s00251-007-0238-2. [DOI] [PubMed] [Google Scholar]
- 19.Siddle HV, et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc Natl Acad Sci U S A. 2013;110:5103–5108. doi: 10.1073/pnas.1219920110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neefjes J, et al. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nature reviews. Immunology. 2011;11:823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 21.Michalek MT, et al. A role for the ubiquitin-dependent proteolytic pathway in MHC class I-restricted antigen presentation. Nature. 1993;363:552–554. doi: 10.1038/363552a0. [DOI] [PubMed] [Google Scholar]
- 22.Rock KL, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 23.Reits E, et al. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity. 2003;18:97–108. doi: 10.1016/s1074-7613(02)00511-3. [DOI] [PubMed] [Google Scholar]
- 24.Cresswell P, et al. The nature of the MHC class I peptide loading complex. Immunological reviews. 1999;172:21–28. doi: 10.1111/j.1600-065x.1999.tb01353.x. [DOI] [PubMed] [Google Scholar]
- 25.Wearsch PA, Cresswell P. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nature immunology. 2007;8:873–881. doi: 10.1038/ni1485. [DOI] [PubMed] [Google Scholar]
- 26.Garstka MA, et al. The first step of peptide selection in antigen presentation by MHC class I molecules. Proc Natl Acad Sci U S A. 2015;112:1505–1510. doi: 10.1073/pnas.1416543112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saveanu L, et al. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nature immunology. 2005;6:689–697. doi: 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- 28.Saric T, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nature immunology. 2002;3:1169–1176. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- 29.Serwold T, et al. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 30.Morozov GI, et al. Interaction of TAPBPR, a tapasin homolog, with MHC-I molecules promotes peptide editing. Proc Natl Acad Sci U S A. 2016;113:E1006–E1015. doi: 10.1073/pnas.1519894113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hermann C, et al. TAPBPR alters MHC class I peptide presentation by functioning as a peptide exchange catalyst. eLife. 2015;4 doi: 10.7554/eLife.09617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang SC, et al. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a "molecular ruler" mechanism. Proc Natl Acad Sci U S A. 2005;102:17107–17112. doi: 10.1073/pnas.0500721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.York IA, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nature immunology. 2002;3:1177–1184. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen TT, et al. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nature structural & molecular biology. 2011;18:604–613. doi: 10.1038/nsmb.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roelse J, et al. Trimming of TAP-translocated peptides in the endoplasmic reticulum and in the cytosol during recycling. J. Exp. Med. 1994;180:1591–1597. 127. doi: 10.1084/jem.180.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elliott T, et al. Peptide-induced conformational change of the class I heavy chain. Nature. 1991;351:402–406. doi: 10.1038/351402a0. [DOI] [PubMed] [Google Scholar]
- 37.Schumacher TN, et al. Direct binding of peptide to empty MHC class I molecules on intact cells and in vitro. Cell. 1990;62:563–567. doi: 10.1016/0092-8674(90)90020-f. [DOI] [PubMed] [Google Scholar]
- 38.Kelly A, et al. Assembly and function of the two ABC transporter proteins encoded in the human major histocompatibility complex. Nature. 1992;355:641–644. doi: 10.1038/355641a0. [DOI] [PubMed] [Google Scholar]
- 39.Neefjes JJ, Ploegh HL. Allele and locus-specific differences in cell surface expression and the association of HLA class I heavy chain with beta 2-microglobulin: differential effects of inhibition of glycosylation on class I subunit association. European journal of immunology. 1988;18:801–810. doi: 10.1002/eji.1830180522. [DOI] [PubMed] [Google Scholar]
- 40.Rammensee H, et al. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 41.Schubert U, et al. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci U S A. 2000;97:13057–13062. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Princiotta MF, et al. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity. 2003;18:343–354. doi: 10.1016/s1074-7613(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 43.Reits EA, et al. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature. 2000;404:774–778. doi: 10.1038/35008103. [DOI] [PubMed] [Google Scholar]
- 44.Yewdell JW. DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends in immunology. 2011;32:548–558. doi: 10.1016/j.it.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rock KL, et al. Re-examining class-I presentation and the DRiP hypothesis. Trends in immunology. 2014;35:144–152. doi: 10.1016/j.it.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monaco JJ, Nandi D. The genetics of proteasomes and antigen processing. Annual review of genetics. 1995;29:729–754. doi: 10.1146/annurev.ge.29.120195.003501. [DOI] [PubMed] [Google Scholar]
- 47.Gaczynska M, et al. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature. 1993;365:264–267. doi: 10.1038/365264a0. [DOI] [PubMed] [Google Scholar]
- 48.Groettrup M, et al. A third interferon-gamma-induced subunit exchange in the 20S proteasome. European journal of immunology. 1996;26:863–869. doi: 10.1002/eji.1830260421. [DOI] [PubMed] [Google Scholar]
- 49.Toes RE, et al. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. The Journal of experimental medicine. 2001;194:1–12. doi: 10.1084/jem.194.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kincaid EZ, et al. Specialized proteasome subunits have an essential role in the thymic selection of CD8(+) T cells. Nat. Immunol. 2016;17:938–945. doi: 10.1038/ni.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ossendorp F, et al. A single residue exchange within a viral CTL epitope alters proteasome-mediated degradation resulting in lack of antigen presentation. Immunity. 1996;5:115–124. doi: 10.1016/s1074-7613(00)80488-4. [DOI] [PubMed] [Google Scholar]
- 52.Tomaru U, et al. Exclusive expression of proteasome subunit beta5t in the human thymic cortex. Blood. 2009;113:5186–5191. doi: 10.1182/blood-2008-11-187633. [DOI] [PubMed] [Google Scholar]
- 53.Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003;3:952–961. doi: 10.1038/nri1250. [DOI] [PubMed] [Google Scholar]
- 54.Meyer VS, et al. Identification of natural MHC class II presented phosphopeptides and tumor-derived MHC class I phospholigands. Journal of proteome research. 2009;8:3666–3674. doi: 10.1021/pr800937k. [DOI] [PubMed] [Google Scholar]
- 55.Haurum JS, et al. Presentation of cytosolic glycosylated peptides by human class I major histocompatibility complex molecules in vivo. The Journal of experimental medicine. 1999;190:145–150. doi: 10.1084/jem.190.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petersen J, et al. Post-translationally modified T cell epitopes: immune recognition and immunotherapy. Journal of molecular medicine. 2009;87:1045–1051. doi: 10.1007/s00109-009-0526-4. [DOI] [PubMed] [Google Scholar]
- 57.Gromme M, et al. The rational design of TAP inhibitors using peptide substrate modifications and peptidomimetics. European journal of immunology. 1997;27:898–904. doi: 10.1002/eji.1830270415. [DOI] [PubMed] [Google Scholar]
- 58.Andersen MH, et al. Phosphorylated peptides can be transported by TAP molecules, presented by class I MHC molecules, and recognized by phosphopeptide-specific CTL. Journal of immunology. 1999;163:3812–3818. [PubMed] [Google Scholar]
- 59.Mohammed F, et al. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nature immunology. 2008;9:1236–1243. doi: 10.1038/ni.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zarling AL, et al. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. The Journal of experimental medicine. 2000;192:1755–1762. doi: 10.1084/jem.192.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berkers CR, et al. Definition of Proteasomal Peptide Splicing Rules for High-Efficiency Spliced Peptide Presentation by MHC Class I Molecules. Journal of immunology. 2015;195:4085–4095. doi: 10.4049/jimmunol.1402455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ploegh HL. Viral strategies of immune evasion. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- 63.Ploegh HL. Trafficking and assembly of MHC molecules: how viruses elude the immune system. Cold Spring Harbor symposia on quantitative biology. 1995;60:263–266. doi: 10.1101/sqb.1995.060.01.030. [DOI] [PubMed] [Google Scholar]
- 64.van Hall T, et al. The varicellovirus-encoded TAP inhibitor UL49.5 regulates the presentation of CTL epitopes by Qa-1b1. Journal of immunology. 2007;178:657–662. doi: 10.4049/jimmunol.178.2.657. [DOI] [PubMed] [Google Scholar]
- 65.Lichtenstein DL, Wold WS. Experimental infections of humans with wild-type adenoviruses and with replication-competent adenovirus vectors: replication, safety, and transmission. Cancer gene therapy. 2004;11:819–829. doi: 10.1038/sj.cgt.7700765. [DOI] [PubMed] [Google Scholar]
- 66.Ziegler H, et al. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. The EMBO journal. 2000;19:870–881. doi: 10.1093/emboj/19.5.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mittal D, et al. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Current opinion in immunology. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Lora A, et al. MHC class I antigens, immune surveillance, and tumor immune escape. Journal of cellular physiology. 2003;195:346–355. doi: 10.1002/jcp.10290. [DOI] [PubMed] [Google Scholar]
- 69.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. The New England journal of medicine. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smit EF, Baas P. Lung cancer in 2015: Bypassing checkpoints, overcoming resistance, and honing in on new targets. Nature reviews. Clinical oncology. 2016;13:75–76. doi: 10.1038/nrclinonc.2015.223. [DOI] [PubMed] [Google Scholar]
- 71.Saccheri F, et al. Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Science translational medicine. 2010;2:44ra57. doi: 10.1126/scitranslmed.3000739. [DOI] [PubMed] [Google Scholar]
- 72.Neijssen J, et al. Cross-presentation by intercellular peptide transfer through gap junctions. Nature. 2005;434:83–88. doi: 10.1038/nature03290. [DOI] [PubMed] [Google Scholar]
- 73.Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. The Journal of experimental medicine. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rock KL, et al. Presentation of exogenous antigen with class I major histocompatibility complex molecules. Science. 1990;249:918–921. doi: 10.1126/science.2392683. [DOI] [PubMed] [Google Scholar]
- 75.Kurts C, et al. Cross-priming in health and disease. Nature reviews Immunology. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 76.Amigorena S. Y in X priming. Nature immunology. 2003;4:1047–1048. doi: 10.1038/ni1103-1047. [DOI] [PubMed] [Google Scholar]
- 77.Segura E, Amigorena S. Cross-Presentation in Mouse and Human Dendritic Cells. Advances in immunology. 2015;127:1–31. doi: 10.1016/bs.ai.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 78.Guermonprez P, et al. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 2003;425:397–402. doi: 10.1038/nature01911. [DOI] [PubMed] [Google Scholar]
- 79.Zitvogel L, et al. Immunological aspects of anticancer chemotherapy. Bulletin de l'Academie nationale de medecine. 2008;192:1469–1487. [PubMed] [Google Scholar]
- 80.Kovacsovics-Bankowski M, et al. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc Natl Acad Sci U S A. 1993;90:4942–4946. doi: 10.1073/pnas.90.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matheoud D, et al. Cross-presentation by dendritic cells from live cells induces protective immune responses in vivo. Blood. 2010;115:4412–4420. doi: 10.1182/blood-2009-11-255935. [DOI] [PubMed] [Google Scholar]
- 82.Amigorena S. Fc gamma receptors and cross-presentation in dendritic cells. The Journal of experimental medicine. 2002;195:F1–F3. doi: 10.1084/jem.20011925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schuette V, Burgdorf S. The ins-and-outs of endosomal antigens for cross-presentation. Current opinion in immunology. 2014;26:63–68. doi: 10.1016/j.coi.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 84.Joffre OP, et al. Cross-presentation by dendritic cells. Nature reviews Immunology. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 85.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 86.Gromme M, et al. Recycling MHC class I molecules and endosomal peptide loading. Proc Natl Acad Sci U S A. 1999;96:10326–10331. doi: 10.1073/pnas.96.18.10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dorsey BD, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. Journal of medicinal chemistry. 2008;51:1068–1072. doi: 10.1021/jm7010589. [DOI] [PubMed] [Google Scholar]
- 88.Shen L, et al. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity. 2004;21:155–165. doi: 10.1016/j.immuni.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 89.Nair-Gupta P, et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell. 2014;158:506–521. doi: 10.1016/j.cell.2014.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Unanue ER, et al. Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annual review of immunology. 2016;34:265–297. doi: 10.1146/annurev-immunol-041015-055420. [DOI] [PubMed] [Google Scholar]
- 91.Waldburger JM, et al. Lessons from the bare lymphocyte syndrome: molecular mechanisms regulating MHC class II expression. Immunological reviews. 2000;178:148–165. doi: 10.1034/j.1600-065x.2000.17813.x. [DOI] [PubMed] [Google Scholar]
- 92.Jones EY. MHC class I and class II structures. Current opinion in immunology. 1997;9:75–79. doi: 10.1016/s0952-7915(97)80162-8. [DOI] [PubMed] [Google Scholar]
- 93.Malmstrom M, et al. Unraveling the Evolution of the Atlantic Cod’s (Gadus morhua L.) Alternative Immune Strategy. PLoS ONE. 2013;8:e74004. doi: 10.1371/journal.pone.0074004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stern LJ, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 95.Suri A, et al. The wide diversity and complexity of peptides bound to class II MHC molecules. Current opinion in immunology. 2006;18:70–77. doi: 10.1016/j.coi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 96.Cresswell P, Roche PA. Invariant chain-MHC class II complexes: always odd and never invariant. Immunology and cell biology. 2014;92:471–472. doi: 10.1038/icb.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Neefjes J. CIIV, MIIC and other compartments for MHC class II loading. European journal of immunology. 1999;29:1421–1425. doi: 10.1002/(SICI)1521-4141(199905)29:05<1421::AID-IMMU1421>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 98.Ghosh P, et al. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature. 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 99.Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 100.Pos W, et al. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell. 2012;151:1557–1568. doi: 10.1016/j.cell.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wubbolts R, et al. Direct vesicular transport of MHC class II molecules from lysosomal structures to the cell surface. The Journal of cell biology. 1996;135:611–622. doi: 10.1083/jcb.135.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boes M, et al. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–988. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- 103.Kleijmeer M, et al. Reorganization of multivesicular bodies regulates MHC class II antigen presentation by dendritic cells. The Journal of cell biology. 2001;155:53–63. doi: 10.1083/jcb.200103071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nielsen M, et al. NetMHCIIpan-2.0 - Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome research. 2010;6:9. doi: 10.1186/1745-7580-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fernando MM, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS genetics. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sollid LM, Jabri B. Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Current opinion in immunology. 2011;23:732–738. doi: 10.1016/j.coi.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clinical chemistry. 2011;57:176–185. doi: 10.1373/clinchem.2010.148221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pette M, et al. Myelin autoreactivity in multiple sclerosis: recognition of myelin basic protein in the context of HLA-DR2 products by T lymphocytes of multiple-sclerosis patients and healthy donors. Proc Natl Acad Sci U S A. 1990;87:7968–7972. doi: 10.1073/pnas.87.20.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wan X, Thomas JW, Unanue ER. Class-switched anti-insulin antibodies originate from unconventional antigen presentation in multiple lymphoid sites. J. Exp. Med. 2016;213:967–978. doi: 10.1084/jem.20151869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sinnathamby G, Eisenlohr LC. Presentation by recycling MHC class II molecules of an influenza hemagglutinin-derived epitope that is revealed in the early endosome by acidification. J Immunol. 2003;170:3504–3513. doi: 10.4049/jimmunol.170.7.3504. [DOI] [PubMed] [Google Scholar]
- 111.Tewari MK, et al. A cytosolic pathway for MHC class II-restricted antigen processing that is proteasome and TAP dependent. Nat Immunol. 2005;6:287–294. doi: 10.1038/ni1171. [DOI] [PubMed] [Google Scholar]
- 112.van Luijn MM, et al. Alternative Ii-independent antigen-processing pathway in leukemic blasts involves TAP-dependent peptide loading of HLA class II complexes. Cancer Immunol Immunother. 2010;59:1825–1838. doi: 10.1007/s00262-010-0908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maric M, et al. Defective antigen processing in GILT-free mice. Science. 2001;294:1361–1365. doi: 10.1126/science.1065500. [DOI] [PubMed] [Google Scholar]
- 114.Ziegler HK, Unanue ER. Decrease in macrophage antigen catabolism caused by ammonia and chloroquine is associated with inhibition of antigen presentation to T cells. Proc Natl Acad Sci U S A. 1982;79:175–178. doi: 10.1073/pnas.79.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liljedahl M, et al. HLA-DO is a lysosomal resident which requires association with HLA-DM for efficient intracellular transport. The EMBO journal. 1996;15:4817–4824. [PMC free article] [PubMed] [Google Scholar]
- 116.Denzin LK, et al. Negative regulation by HLA-DO of MHC class II-restricted antigen processing. Science. 1997;278:106–109. doi: 10.1126/science.278.5335.106. [DOI] [PubMed] [Google Scholar]
- 117.van Ham SM, et al. HLA-DO is a negative modulator of HLA-DM-mediated MHC class II peptide loading. Current biology. 1997;7:950–957. doi: 10.1016/s0960-9822(06)00414-3. [DOI] [PubMed] [Google Scholar]
- 118.Yi W, et al. Targeted regulation of self-peptide presentation prevents type I diabetes in mice without disrupting general immunocompetence. The Journal of clinical investigation. 2010;120:1324–1336. doi: 10.1172/JCI40220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cella M, et al. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 120.Pierre P, et al. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 121.Thibodeau J, et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. European journal of immunology. 2008;38:1225–1230. doi: 10.1002/eji.200737902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Paul P, et al. A Genome-wide multidimensional RNAi screen reveals pathways controlling MHC class II antigen presentation. Cell. 2011;145:268–283. doi: 10.1016/j.cell.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 123.Mitchell EK, et al. Inhibition of cell surface MHC class II expression by Salmonella. European journal of immunology. 2004;34:2559–2567. doi: 10.1002/eji.200425314. [DOI] [PubMed] [Google Scholar]
- 124.Zwart W, et al. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity. 2005;22:221–233. doi: 10.1016/j.immuni.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 125.van Niel G, et al. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity. 2006;25:885–894. doi: 10.1016/j.immuni.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 126.Pennock GK, Chow LQ. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. The oncologist. 2015;20:812–822. doi: 10.1634/theoncologist.2014-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 128.Jongsma ML, Berlin I, et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell. 2016;166:152–166. doi: 10.1016/j.cell.2016.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Saveanu L, et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science. 2009;325:213–217. doi: 10.1126/science.1172845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.