

Introduction
Fetal echocardiography is most commonly used to diagnose fetal arrhythmias, but reveals only the mechanical consequences of normal and abnormal rhythms. The atrioventricular relationship and the atrial and ventricular rates can be measured, but cardiac waveform morphology and repolarization characteristics cannot be assessed.
In principle, waveform morphology and repolarization can be deduced by fetal ECG as early as 17 weeks of gestation (1, 2), but the electric signals from the fetal heart are attenuated by amniotic fluid and fetal vernix. In one series, although QRS complexes could generally be detected, only about 60% of subjects had interpretable p- and T-wave morphology (3). In general, diagnostic quality ECG signals are only obtained with direct fetal contact, such as with a scalp electrode placed after rupture of the membranes (4, 5).
Magnetocardiography, a non-invasive measurement of the magnetic fields of the heart, is uniquely suited for investigation of the unexplored territory of fetal arrhythmias. It is the only technique that has consistently and reliably provided a fetal ECG-like signal without direct fetal or maternal contact. Unlike the electrical fields of the heart, the magnetic field signals are accessible from 18 weeks onward using a SQUID magnetometer (6, 7) or, more recently, an optically-pumped magnetometer (8). Rhythm tracings, signal averaged waveforms, and simultaneous fetal heart rate/fetal movement tracings (actocardiograms) have provided both normative data throughout gestation and unique insight into the electrophysiological characteristics and natural histories of supraventricular arrhythmias, atrioventricular (AV) block, and arrhythmias associated with fetal rhabdomyomas, ventricular wall defects, and structural cardiac defects (6,7,9–12). The purpose of this paper is to summarize our experience with fMCG in the diagnosis, evaluation, and treatment of the fetus at risk for long QT syndrome (13.).
Clinical Features of LQTS
Long QT syndrome (LQTS) is an inherited channelopathy that may cause syncope, cardiac arrest or sudden death at any age as a result of life-threatening ventricular arrhythmias (14). The postnatal diagnosis of LQTS is straightforward and suggested by a prolonged QT interval on 12 lead ECG, a positive family history and/or characteristic LQTS arrhythmias, and can be confirmed by genetic testing. Diagnosing LQTS before symptoms is important because primary prevention is extremely effective in preventing life-threatening ventricular arrhythmias (15). Ideally, LQTS would be diagnosed before birth and primary prevention initiated during infancy, but unless LQTS is suspected because of a family history, prenatal diagnosis is difficult for several reasons. First, commercial genetic laboratories will only test for LQTS if there is a known family mutation. There is also risk to the pregnancy and the fetus with invasive testing. Second, although some fetuses present with arrhythmias easily recognized as LQTS (torsades de pointes (TdP) and/or 2:1 AV conduction), these rhythms are uncommon, occurring in <25% of fetal LQTS cases (16). Instead, the most common presentation of fetal LQTS is sinus bradycardia. Not only is sinus bradycardia a subtle rhythm disturbance frequently not recognized to be abnormal, but the obstetrical definition of sinus bradycardia (17) is inadequate in the ascertainment of LQTS fetuses (18, 19). Although sinus bradycardia has long been known to suggest fetal LQTS, the obstetrical definition of sinus bradycardia is a gestational age independent FHR of < 110 bpm (17). However, recent reports suggest that a FHR < 3rd percentile for gestational age may more accurately define LQTS sinus bradycardia (18, 19).
Thus, overall ascertainment of fetal LQTS includes those with: a family history of LQTS (at 50% risk of having the same family mutation), repeated FHR measures < 3rd percentile for gestational age, and the signature LQTS rhythms of 2:1 AV conduction and/or ventricular tachycardia. Although the data are limited, sinus bradycardia may begin as early as the second trimester, but TdP and 2:1 AV conduction often occur after ~30 weeks’ gestation (16, 20 – 21).
Why LQTS diagnosis before birth is important
The diagnosis of LQTS before birth is important for several reasons. First, the disease burden is high: LQTS is the most common cause of arrhythmic death in children, and causative in at least 8–10% of unexplained intrauterine fetal demise and victims of Sudden Infant Death syndrome (22–24). Second, if the fetus is recognized to have LQTS, cascade testing of family members can identity those at risk for sudden cardiac death before symptoms. Third, withholding or limiting QT prolonging maternal medications common in obstetrics such as Pitocin or ondansetron, and optimizing maternal magnesium, vitamin D and calcium levels could reduce risk of TdP to the LQTS fetus. Fourth, if it is known that the fetus has LQTS, fetal bradycardia due to 2° AV block or a non-reactive fetal heart rate tracing will be attributed to the LQTS phenotype and not fetal distress, and preterm delivery can be avoided. Fifth, anticipatory postnatal care from a prenatal diagnosis improves outcome has been shown to improve outcome (25). Lastly, screening every newborn with a 12 lead ECG is controversial, and will not detect all cases of LQTS (26).
fMCG diagnosis of LQTS
Once suspected, the diagnosis of fetal LQTS can be confirmed by fMCG in two ways. During sinus rhythm, signal averaged fMCG demonstrates a gestational age independent QTc interval > 95th percentile or ≥ 490 ms (13). Signal averaing is performed by detecting each QRS complex precisely using autocorrection and time-aligning the beats. This will be sufficient to diagnose LQTS when the fetus manifests only sinus bradycardia (Figure 1). Supporting evidence for an LQTS diagnosis comes from identifying polymorphic ventricular tachycardia during episodes of tachycardia with AV dissociation, and confirming the echo diagnosis of 2:1 AV conduction secondary to prolonged ventricular repolarization. Using only QTc duration measured during sinus rhythm, LQTS was successfully recognized with a sensitivity and specificity of 89% (13).
Figure 1.
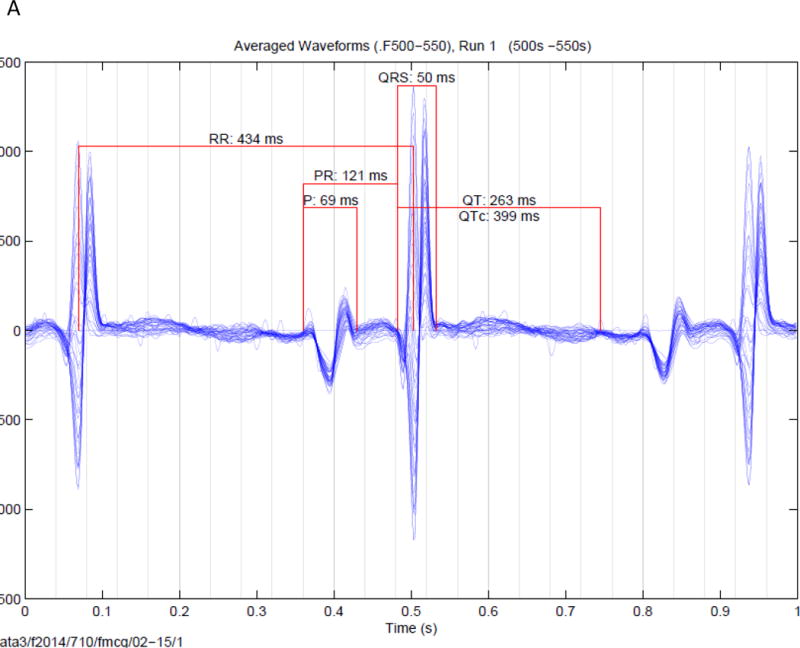
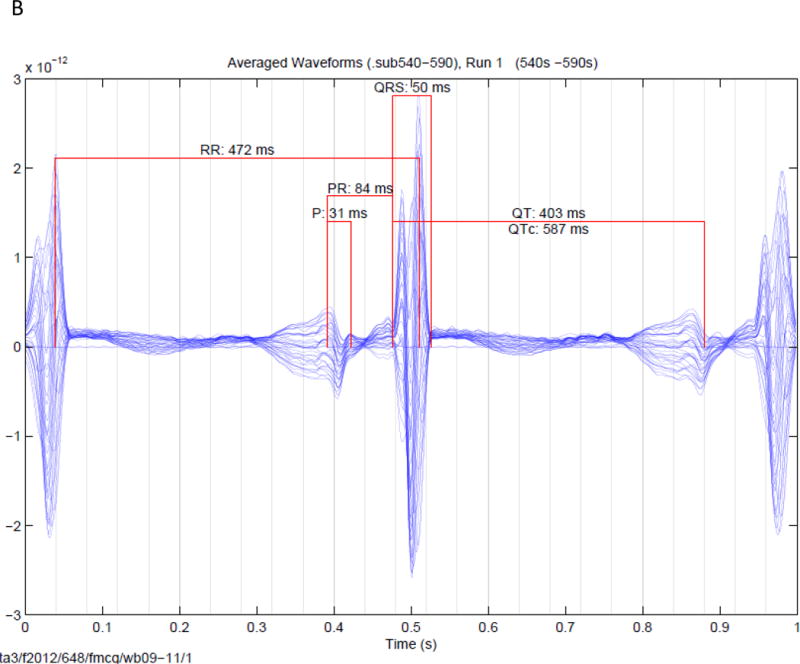
Signal averaged fMCG tracings of fetuses. (A). Signal averaged tracing (over 50 seconds from a fetus a normal QTc (399 ms) found after birth to be negative for the family KCNQ1 mutation. (B). Signal averaged tracing (over 50 seconds) from a fetus with a prolonged QTc found after birth to be positive for the family KCNH2 mutation. In both figures the fetal heart rates are constant, and vary by ~ 10 ms. Since 10 ms is < the QRS interval, widening of the QRS complex and reduction in QRS amplitude is minimal.
Genotype/Phenotype correlations and risk stratification of fetal LQTS
In those 39 fetuses studied by fMCG (13), fetal QTc duration correlated directly with the likelihood of pre- and postnatal TdP. Those with TdP had fetal QTc of 659.8 ± 31.0 ms, while those with no TdP had a QTc of 549.1 ± 55.2 ms (P =0.003). This correlation was also seen in LQTS infants: postnatal QTc of those with fetal/neonatal TdP was 664.7± 24.9 compared to 483.1 ±13.7 ms of those with only sinus bradycardia (16). Furthermore, the fetal rhythm phenotype was suggestive of genotype: subjects with 2:1 conduction and/or TdP were more likely to have de novo and sporadic mutations including SCN5A R1623Q mutations, KCNH2 mutations in the pore region, or CALM 2 mutations (13,16) and fetuses with isolated sinus bradycardia had familial (mostly) or de novo KCNQ1 mutations (13,16). In fact, of 28 subjects with TdP or aborted cardiac arrest as fetuses, infants or children (13, 20) 5 had a family history of LQTS familial LQTS (). The rhythms and genotypes of the 39 fetuses with LQTS studied by fMCG (13) are shown in Figure 2
Figure 2.
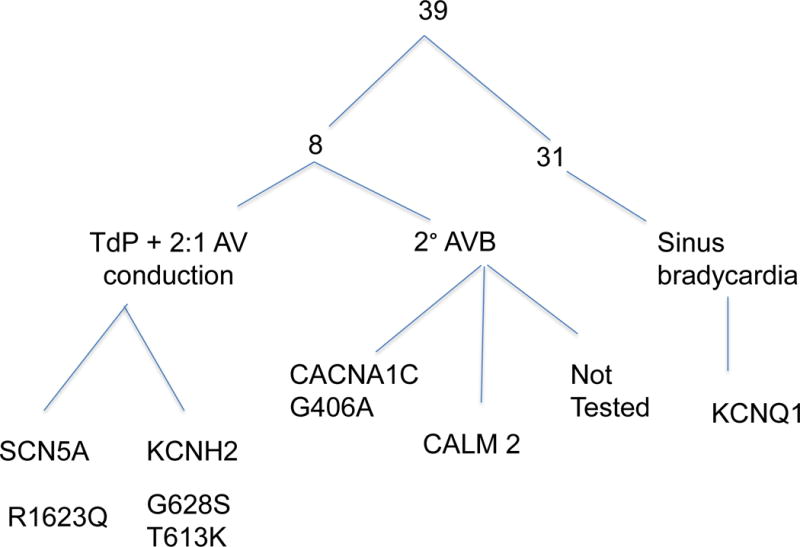
Long QT mutations and rhythms of subjects undergoing fMCG with either a family history of LQTS or an LQTS rhythm (sinus bradycardia, 2° AV block or torsades de pointes).
fMCG evaluation of fetuses at risk for LQTS
We studied 39 fetuses by fMCG (n=13) at 19–38 (29.5 ± 5.2) weeks: 27 with a positive family history and 12 with LQTS rhythms. The LQTS rhythms included repeated measures of FHR < 3rd percentile for gestational age, 2:1 AV conduction and/or ventricular tachycardia diagnosed by fetal echocardiography. When compared to FHR and fetal QTc intervals, neonatal values were not statistically different (13).
LQTS was correctly diagnosed in 37/39 fetuses studied by fMCG. There were 2 false results: one false positive and one false negative. The subject with false negative results (Figure 3A) had a family history of an SCN5A E1784K mutation. This subject had no fetal LQTS arrhythmias; in fact, all FHRs were > 3rd % for gestational age and the fMCG QTc was 423 ms. At birth, the infant’s QTc was 430 ms, but genetic testing was positive for the family mutation. The subject with the false positive result (Figure 3B) had no family history of LQTS but repeated FHR measures < 3rd% for gestational age and fMCG QTc intervals of >500 ms at 31 and 38 weeks. The subject’s postnatal ECG QTc was 440 ms after birth and at one month of age and genetic testing was negative for a mutation in a known LQTS gene. In retrospect, the mother had very low vitamin D (25-OH) and magnesium levels during her pregnancy, which may have contributed to the prolonged fetal QTc. We now check vitamin D and magnesium levels on all mothers with suspected fetal LQTS and treat to normalize deficiencies.
Figure 3.



Fetal MCG rhythm tracings. (A) False negative study of a 27 week fetus with an fMCG QT interval of 423 ms. At birth the ECG QTc was 430 ms, but genetic testing was positive for the family SCN5A E1784K mutation. (B): False positive studies at 31 and 38 weeks of a fetus with a prolonged fCMG QTc who had a normal ECG QTc (440 ms) at birth and no LQTS mutation.
FMCG detects evanescent rhythms in LQTS fetuses
Fetuses referred for fMCG with an LQTS family history had all been monitored by serial fetal echocardiograms from ~20 weeks of gestation. Nonetheless, fMCG uncovered rhythms not detected during echocardiography in 3 subjects, which altered pre- and postnatal management (13, 27). One subject with a KCNH2 family mutation had multiple short episodes of TdP which were treated in utero with magnesium and beta adrenergic blocking agents. Because of the findings, we correctly anticipated non-sustained TdP after birth and the infant was sent home with an automatic external defibrillator. A 2nd subject, also with a KCNH2 family mutation, had monomorphic premature ventricular contractions which prompted increased surveillance and an earlier delivery. The 3rd subject, with a de novo CALM 2 mutation, had a brief episode of 2:1 AV conduction which recurred after birth and preceded an aborted cardiac arrest (27). Detecting these transient rhythms was possible because fMCG records each fetal heart beat in a ~ 1 hour period.
FHR variability/reactivity and repolarization in LQTS fetuses
Magnetocardiography is an ideal technique to assess beat-to-beat FHR variability, which typically shows a prominent increase at > 28 weeks’ gestation. Decreased FHR variability is known to be a feature of LQTS subjects attributed to sympathetic imbalance, and may lead to the mistaken diagnosis of fetal distress. In our cohort, 36% of subjects demonstrated low FHR reactivity and variability in sinus rhythm (13).
Gross T-wave abnormalities, such as macroscopic T-wave alternans, have been associated with postnatal LQTS, but until fMCG, these abnormalities had not been known to be presenting in utero. Fetal T-wave alternans was seen in all subjects with pre- or postnatal TdP. Electrical alternans was not limited to T-waves: QRS and even P-wave alternans was demonstrated subjects with the longest QTc intervals in the beats preceding TdP.
Torsades de pointes
As previously noted in both fetal and postnatal studies, TdP occurred in subjects with the longest QTc intervals (13, 16). Fetal MCG studies demonstrate that the duration of TdP episodes varied from 1 sec to almost 8 minutes; the number of episodes ranged from 15–45 during a 48 minute period (13). Torsade was both fast and slow with cycle lengths varying from 238 to 351 ms. All fetuses with TdP diagnosed by fMCG in utero were born alive. One subject had depressed ventricular function and hydrops. All others, including the subject with the longest and most frequent episodes of TdP, had normal ventricular function and no signs of congestive heart failure.
The patterns of initiation and termination of TdP as seen during fMCG varied between subjects, between episodes in the same subject, and within the same LQTS type. The long-short pattern of initiation was rarely seen. Much more frequently, TdP initiated from sinus rhythm with wide complex aberrantly conducted beats, QRS alternans, or aberrantly conducted QRS complexes at the same coupling interval as narrow QRS complexes preceding them (Figure 4). Torsades de pointes also followed periods of 2:1 AV conduction. Termination patterns included 2:1 conduction followed by sinus rhythm with QRS alternans, transient 3:1 AV conduction and a long pause (1.2 s) followed by a narrow complex regular rhythm. T-wave alternans frequently occurred during the recovery rhythms after TdP.
Figure 4.
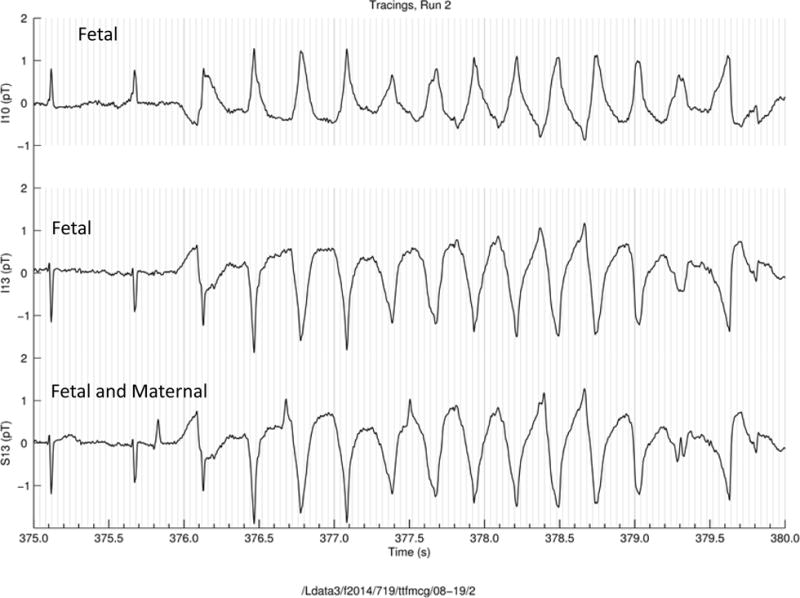
FMCG rhythm tracing of TdP in a subject with an LQT-8 mutation (CACNA1CG406A). Torsades de pointes begins following a wide complex beat (arrow) of the same coupling interval as the preceding narrow complex beat. The top 2 tracings are the fetal rhythm the 3rd is a tracing of both maternal and fetal rhythms.
Conclusions
The natural history of fetal LQTS depends on the mutation type. Fetuses with a familial or de novo KCNQ1 mutation most commonly present with a subtle bradycardia often not even recognized to be abnormal. On the other hand, fetuses with a KCNH2 mutation are more likely to present with either sinus bradycardia or 2:1 AV conduction. Torsades de pointes as a manifestation of a KCNH2 mutation can occur with a family or a de novo mutation. The most likely diagnosis of a fetus with 2:1 AV conduction and TdP is either an SCN5A R1623Q or a de novo KCNH2 mutation in the pore region of the gene. As LQTS occurs in 1/2000–2500 persons, but has been detected in only 1/8658 fetuses undergoing routine ultrasounds (28), ascertainment during fetal life is poor. Fetal MCG is diagnostic of LQTS and can elucidate the spectrum of rhythms that occur in the LQT fetus, but only after a referral by a clinician who suspects the fetus has LQTS. Appreciating the manifestations of LQTS before birth can improve management of this group at risk for ventricular arrhythmias by provide primary rather than secondary prevention for sudden arrhythmic death at all ages.
Acknowledgments
Dr. Wakai and Dr. Strasburger were supported by the National Institutes of Health (grant number R01 HL63174).
References
- 1.Taylor MJ, Smith MJ, Thomas M, et al. Non-invasive fetal electrocardiography in singleton and multiple pregnancies. BJOG. 2003;110(7):668–678. [PubMed] [Google Scholar]
- 2.Taylor MJ, Thomas MJ, Smith MJ, et al. Non-invasive intrapartum fetal ECG: preliminary report. BJOG. 2005;112(8):1016–1021. doi: 10.1111/j.1471-0528.2005.00643.x. [DOI] [PubMed] [Google Scholar]
- 3.Gardiner HM, Belmar C, Pasquini L, et al. Fetal ECG: a novel predictor of atrioventricular block in anti-Ro positive pregnancies. Heart. 2007;93(11):1454–1460. doi: 10.1136/hrt.2006.099242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ojala K, Vääräsmäki M, Mäkikallio K, Valkama M, Tekay A. A comparison of intrapartum automated fetal electrocardiography and conventional cardiotocography—a randomised controlledstudy. BJOG. 2006;113(4):419–423. doi: 10.1111/j.1471-0528.2006.00886.x. [DOI] [PubMed] [Google Scholar]
- 5.Westerhuis ME, Visser GH, Moons KG, et al. Cardiotocography plus ST analysis of fetal electrocardiogram compared with cardiotocography only for intrapartum monitoring: arandomized controlled trial. Obstet Gynecol. 2010;115(6):1173–1180. doi: 10.1097/AOG.0b013e3181dfffd6. [DOI] [PubMed] [Google Scholar]
- 6.Horigome H, Takahashi MI, Asaka M, Shigemitsu S, Kandori A, Tsukada K. Magnetocardiographic determination of the developmental changes in PQ, QRS and QT intervals in the foetus. Acta Paediatr. 2000;89(1):64–67. doi: 10.1080/080352500750029086. [DOI] [PubMed] [Google Scholar]
- 7.Leuthold A, Wakai RT, Martin CB. Noninvasive in utero assessment of PR and QRS intervals from the fetal magnetocardiogram. Early Hum Dev. 1999;54(3):235–243. doi: 10.1016/s0378-3782(98)00100-5. [DOI] [PubMed] [Google Scholar]
- 8.Shah VK, Wakai RT. A compact, high performance atomic magnetometer for biomedical applications. Phys Med Biol. 2013;58(22):8153–61. doi: 10.1088/0031-9155/58/22/8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters C, Wacker-Gussmann A, Strasburger JF, Cuneo BF, Gotteiner NL, Gulecyuz M, Wakai RT. Electrophysiologic features of fetal ventricular aneurysms and diverticula. Prenat Diagn. 2015;35(2):129–36. doi: 10.1002/pd.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wacker-Gussmann A, Strasburger JF, Cuneo BF, Wiggins DL, Gotteiner NL, Wakai RT. Fetal arrhythmias associated with cardiac rhabdomyomas. Heart Rhythm. 2014;11(4):677–83. doi: 10.1016/j.hrthm.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao H, Cuneo BF, Strasburger JF, Huhta JC, Gotteiner NL, Wakai RT. Electrophysiological characteristics of fetal atrioventricular block. J Am Coll Cardiol. 2008;51(1):77–84. doi: 10.1016/j.jacc.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakai RT, Strasburger JF, Li Z, Deal BJ, Gotteiner NL. Magnetocardiographic rhythm patterns at initiation and termination of fetal supraventricular tachycardia. Circulation. 2003;107(2):307–12. doi: 10.1161/01.cir.0000043801.92580.79. [DOI] [PubMed] [Google Scholar]
- 13.Cuneo BF, Strasburger JF, Yu S, Horigome H, Hosono T, Kandori A, Wakai RT. In utero diagnosis of long QT syndrome by magnetocardiography. Circulation. 2013;128(20):2183–91. doi: 10.1161/CIRCULATIONAHA.113.004840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moss AJ, Schwartz PJ, Crampton RS, Locati E, Carleen E. The long QT syndrome: a prospective international study. Circulation. 1985;71(1):17–21. doi: 10.1161/01.cir.71.1.17. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz PJ, Ackerman MJ, George AL, Jr, Wilde AA. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013 Jul 16;62(3):169–80. doi: 10.1016/j.jacc.2013.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuneo BF, Etheridge SP, Horigome H, Sallee D, Moon-Grady A, Weng HY, Ackerman MJ, Benson DW. Arrhythmia phenotype during fetal life suggests long-QT syndrome genotype: risk stratification of perinatal long-QT syndrome. Circ Arrhythm Electrophysiol. 2013;6(5):946–51. doi: 10.1161/CIRCEP.113.000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.ACOG Committee on Practice Bulletins. Intrapartum fetal heart rate monitoring: nomenclature, interpretation and general management principals. Obstet Gynecol. 2009;114:192–202. doi: 10.1097/AOG.0b013e3181aef106. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell JL, Cuneo BF, Etheridge SP, Horigome H, Weng HY, Benson DW. Fetal heart rate predictors of long QT syndrome. Circulation. 2012;126(23):2688–95. doi: 10.1161/CIRCULATIONAHA.112.114132. [DOI] [PubMed] [Google Scholar]
- 19.Winbo A, Fosdal I, Lindh M, Diamant UB, Persson J, Wettrell G, Rydberg A. Third trimester fetal heart rate predicts phenotype and mutation burden in the type 1 long QT syndrome. Circ Arrhythm Electrophysiol. 2015;8(4):806–14. doi: 10.1161/CIRCEP.114.002552. [DOI] [PubMed] [Google Scholar]
- 20.Horigome H, Nagashima M, Sumitomo N, Yoshinaga M, Ushinohama H, Iwamoto M, Shiono J, Ichihashi K, Hasegawa S, Yoshikawa T, Matsunaga T, Goto H, Waki K, Arima M, Takasugi H, Tanaka Y, Tauchi N, Ikoma M, Inamura N, Takahashi H, Shimizu W, Horie M. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: a nationwide questionnaire survey in Japan. Circ Arrhythm Electrophysiol. 2010;3(1):10–7. doi: 10.1161/CIRCEP.109.882159. [DOI] [PubMed] [Google Scholar]
- 21.Cuneo BF, Ovadia M, Strasburger JF, Zhao H, Petropulos T, Schneider J, Wakai RT. Prenatal diagnosis and in utero treatment of torsades de pointes associated with congenital long QT syndrome. Am J Cardiol. 2003;91(11):1395–8. doi: 10.1016/s0002-9149(03)00343-6. [DOI] [PubMed] [Google Scholar]
- 22.Miller TE, Estrella E, Meyerburg RJ, et al. Recurrent third-trimester fetal loss and maternal mosaicism for Long-QT Syndrome. Circulation. 2004;109:3029–34. doi: 10.1161/01.CIR.0000130666.81539.9E. [DOI] [PubMed] [Google Scholar]
- 23.Crotti L, Tester DJ, White WM, et al. Long QT syndrome and intrauterine fetal death. JAMA. 2013;309(14):1473–82. doi: 10.1001/jama.2013.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz Peter J. Stillbirths, Sudden Infant Deaths, and Long-QT Syndrome : Puzzle or Mosaic, the Pieces of the Jigsaw Are Being Fitted Together. Circulation. 2004;109:2930–2. doi: 10.1161/01.CIR.0000133180.77213.43. [DOI] [PubMed] [Google Scholar]
- 25.Greene AE, Berul CI, Donorio MT. Prenatal diagnosis of long QT syndrome: implications for delivery room and neonatal management. Cardiology in the Young. 2012;23(1):141–5. doi: 10.1017/S1047951112000583. [DOI] [PubMed] [Google Scholar]
- 26.Skinner JR, Van Hare GF. Routine ECG screening in infancy and early childhood should not be performed. Heart Rhythm. 2014;11(12):2322–7. doi: 10.1016/j.hrthm.2014.09.046. [DOI] [PubMed] [Google Scholar]
- 27.Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, Papagiannis J, Feldkamp MD, Rathi SG, Kunic JD, Pedrazzini M, Wieland T, Lichtner P, Beckmann BM, Clark T, Shaffer C, Benson DW, Kääb S, Meitinger T, Strom TM, Chazin WJ, Schwartz PJ, George AL., Jr Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127(9):1009–17. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flock A, Herberg U, Gembruch U, Mertz WM. Clinical spectrum of fetal long QT syndrome: A single center experience. Journal Maternal-Fetal Neonatal Medicine. 2015;28:1731–1735. doi: 10.3109/14767058.2014.967205. [DOI] [PubMed] [Google Scholar]