

Abstract
In addition to maternal genes and environmental exposures, variation in fetal imprinted genes could also affect maternal blood pressure during pregnancy. Our objective was to test the associations between polymorphic variants in 16 imprinted genes and maternal mean arterial blood pressures in 1,160 DNA trios from two established birth cohorts (the Cambridge Baby Growth and Wellbeing Studies), and seek replication in 1,367 Hyperglycemia and Adverse Pregnancy Outcome Study participants. Significant univariate associations, all independent of fetal sex, were observed in the Cambridge cohorts, including: FAM99A rs1489945 transmitted from the mother (p=2×10−4), DLK1 rs10139403 (mother; p=9×10−4), DLK1 rs12147008 (mother; p=1×10−3), H19 rs217222 (father; p=1×10−3), SNRPN rs1453556 (father; p=1×10−3), IGF2 rs6356 (father; p=1×10−3) and NNAT rs6066671 (father; p=1×10−3). In meta-analysis including additional independent Hyperglycemia and Adverse Pregnancy Outcome Study data, the association with maternally-transmitted fetal DLK1 rs10139403 reached genome-wide significance (p=6.3×10−10). With the exception of fetal rs1489945 and rs217222, all of other associations were unidirectional and most were statistically significant. To further explore the significance of these relationships we developed an allele score based on the univariate findings. The score was strongly associated with maternal blood pressure at 31 weeks (p=4.1×10−8; adjusted r2=5.6%) and 37 weeks of pregnancy (p=1.1×10−4; r2=3.6%), and during the last 2 weeks prior to parturition (p=1.1×10−10; r2=8.7%). It was also associated with gestational hypertension (odds ratio 1.54 (1.14, 2.09) per allele; p=0.005; 45 cases and 549 controls). These data support the concept that fetal imprinted genes are related to the development of gestational hypertension.
Keywords: pregnancy-induced hypertension, preeclampsia, gestational hypertension, placenta, meta-analysis
Introduction
Epidemiological studies such as the Swedish Twin Register and Medical Birth Register suggest that pregnancy-induced hypertension (raised blood pressure which originates in pregnancy and encompasses both gestational hypertension and preeclampsia) has both genetic and environmental aetiological components1. Genetic variation may have a key role in the overall control of blood pressure in pregnant women, but the maternal genetic variants linked or associated with gestational hypertension, preeclampsia or changes in blood pressure2 only account for a small fraction of the total genetic risk of pregnancy-induced hypertension3. The reasons for this may relate to a the lack of sensitivity of even large genome wide association studies (GWAS) to detect associations with small effect sizes, effects of rare variants with large effect sizes, and gene-gene and gene-environmental interactions4, like those observed for non-gravid hypertension5.
An additional explanation, only pertinent to pregnancy, is that some of the genetic risk could relate to the fetal rather than maternal genotype. It has been suggested that fetal genes may influence maternal metabolism and physiology6 and we hypothesised that fetal growth genes in particular, may influence a mother’s risk of developing gestational hypertension and gestational diabetes7,8. Consistent with this, in the Swedish Birth and Multi-Generation Registries 20% of the variability in the risk for preeclampsia could be attributed to fetal genetic effects9. Of fetal growth genes, imprinted genes, with their role in regulating fetal growth10 and their parent of origin-specific effects, are thought to mediate part of the “separate evolutionary reproductive needs” of the father and the mother11. Paternally-expressed imprinted genes tend to enhance fetal growth whereas maternally-expressed genes tend to restrain it. It has been suggested that the mechanism behind these processes arise from changes in fetal demand and supply12. Consistent with the hypothesis, in pregnancies affected by Beckwith Wiedemann syndrome, a fetal overgrowth syndrome caused by deletion or mutation of fetal imprinted genes within the 11p15 chromosomal region, the mother is at increased risk of developing gestational hypertension and preeclampsia13.
In this study we explored this hypothesis further by investigating associations between polymorphic variation in fetal imprinted genes and maternal blood pressure in pregnancy.
Methods
Cohort 1: Cambridge Baby Growth Study
The prospective and longitudinal Cambridge Baby Growth Study recruited mothers (and their partners and offspring) attending ultrasound clinics during early pregnancy at the Rosie Maternity Hospital, Cambridge, U.K. between 2001–200914. Study participant characteristics are shown in Table 1A. Of the 2,229 mothers initially recruited to the study, all routinely performed blood pressure measurements in the pregnancy that were recorded in the hospital notes were collected retrospectively. In total, 845 DNA trios were collected from the families of the mothers. Blood and/or mouth swab samples for DNA extraction were collected from the father and the offspring after birth. In this cohort 96.9% of the offspring were of Caucasian ethnicity, 0.8% were of mixed race, 0.6% were Black (African or Caribbean), 0.8% were East-Asian and 0.9% were Indo-Asian.
Table 1.
Study participant demographics for the (A) 845 family trios in the Cambridge Baby Growth Study, (B) 315 family trios in the Cambridge Wellbeing Study and (C) 1,367 mother/baby dyads from HAPO17. Data are mean (SD) or percentages. OGTT = oral glucose tolerance test.
| (A) | |
|---|---|
| Maternal Age at Delivery (years) | 33.5 (4.3) |
| Maternal Height (cm) | 165.9 (7.2) |
| Maternal Pre-pregnancy Weight (kg) | 66.3 (13.5) |
| Maternal Pre-pregnancy Body Mass Index (kg/m2) | 24.1 (4.6) |
| Pre-pregnancy Gravidity | 1.8 (0.9) |
| Pre-pregnancy Parity | 0.4 (0.8) |
| Twin Pregnancies (%) | 2.0 |
| Maternal Smoking in Pregnancy (%) | 5.4 |
| Gestational Age at Delivery (weeks) | 39.8 (1.6) |
| Offspring Ethnicity (% Caucasian) | 96.9 |
| Offspring Sex (% male) | 51.6 |
| Offspring Birth Weight (g) | 3479 (533) |
| Paternal Age at Delivery (years) | 35.4 (5.3) |
| (B) | |
|---|---|
| Maternal Age at Delivery (years) | 33.1 (4.1) |
| Pre-pregnancy Parity | 0.8 (0.8) |
| Twin Pregnancies (%) | 0 (inclusion criteria) |
| Gestational Age at Delivery (weeks) | 39.6 (1.3) |
| Offspring Ethnicity (% Caucasian) | 100 (inclusion criteria) |
| Offspring Sex (% male) | 50.8 |
| Offspring Birth Weight (g) | 3562 (495) |
| (C) | |
|---|---|
| Maternal Age at OGTT (years) | 31.3 (5.2) |
| Body Mass Index at OGTT (kg/m2) | 28.5 (4.8) |
| Primiparous births (%) | 86.3 |
| Twin Pregnancies (%) | 0 (inclusion criteria) |
| Maternal smoking during pregnancy (%) | 13.5 |
| Gestational Age at Delivery (weeks) | 39.6 (1.1) |
| Offspring Ethnicity (% Caucasian) | 100 (inclusion criteria) |
| Offspring Sex (% male) | 49.9 |
Cohort 2: Cambridge Wellbeing Study
The Cambridge Wellbeing Study is a retrospective study that recruited mothers, fathers and children where the pregnant mother had delivered a full term, singleton baby at the Rosie Maternity Hospital between 1999–200014. Exclusion criteria were maternal chronic hypertension and treatment for diabetes during pregnancy. Study participant characteristics are shown in Table 1B. In this cohort all the offspring were of Caucasian ethnicity. We sought permission from the mother’s General Practitioner to approach the mother, partner and child to collect their DNA samples by postal mouth swab kits. In total 315 DNA trios were collected out of 563 women who consented.
Cohort 3: Hyperglycemia and Adverse Pregnancy Outcome Study
To validate our seven most significant findings in the Cambridge cohorts we tested the same associations between maternal mean arterial blood pressures and the fetal imprinted gene alleles in 1,367 HAPO study participants15, 16 of European descent whose single nucleotide polymorphism (SNP) genotypes were derived from a GWAS of maternal glycemic and neonatal anthropometric traits17. Study participant characteristics are shown in Table 1C. In these women blood pressure was measured between 24 and 32 weeks of gestation (as close to 28 weeks as possible) using standardised procedures and a calibrated electronic device (Omron 711, Illinois, U.S.A.)18.
Ethical Approval
The Cambridge Baby Growth and Wellbeing Studies were approved by the local ethics committee, Addenbrooke’s Hospital, Cambridge, U.K. In the HAPO Study the protocol was approved by each field centre’s local institutional review board. All procedures followed were in accordance with institutional guidelines. Written informed consent was obtained from the parents in each of the cohorts studied, including consent for inclusion of their infants.
Blood Pressure Categorisation in the Cambridge Cohorts
Routine blood pressure measurements during pregnancy were extracted from hospital notes and categorised into one of four time-points: around 12, 31, 37 and 37~41 weeks of pregnancy. Further details are presented in the Online Supplement.
Gene Selection and Genetics
Genomic DNA from the Cambridge Baby Growth Study blood samples was extracted using an Autopure LS Machine (Qiagen Ltd., Crawley, U.K.). Other genomic DNA, from the Cambridge Baby Growth and Wellbeing Studies, was extracted from mouthswab samples using a standard chloroform-based method. The genes that were studied ((Table S1) DLK1, FAM99A, GNAS, GRB10, H19, IGF2, INS, KCNQ1OT1, MEST, NNAT, PEG3, PEG10, PLAGL1, SGCE, SNRPN, ZIM2) were chosen because they were all imprinted (and paternally-expressed with the exception of H19 whose function includes regulating expression of paternally-expressed IGF2) and are placentally-expressed at some stage of development19. The 155 SNP variants in these 16 genes were haplotype tag SNPs covering the gene and 20kb either side of it, identified by Tagger (r2 > 0.8 and minor allele frequency > 0.2) from the Centre d’Etude du Polymorphisme Humain population (Utah residents with ancestry from Northern and Western Europe) of HapMap Project Build 36 using Haploview20. The one exception to this was IGF2 where 11 of the tagging SNPs were previously identified21. A complete list of the SNPs that were genotyped are listed in Table S1. The DNA samples were genotyped using proprietary Kompetitive Allele Specific PCR assays, which are SNP genotyping assays using Fluorescence Resonance Energy Transfer quencher cassette oligonucleotides (designed and performed by LGC Genomics, Hoddesdon, U.K.). In HAPO the DNA samples were genotyped using the Illumina Human 610 Quad v1 B SNP array (Illumina Inc., San Diego, U.S.A.) and additional SNPs were imputed17. The SNP genotypes that were used in this study were consistent with Hardy Weinberg equilibrium (p > 0.05 using the χ2 test) and had a repeat genotyping discordancy rate of < 1.0%.
Statistical Analyses and Composite Score Formulation
A detailed description of the statistical analyses performed, including the formulation of the composite fetal imprinted gene allele score, the linear regression models used to test associations between maternal mean arterial blood pressures and either individual fetal imprinted gene variants or the allele score, and a power calculation are presented in the Online Supplement. The effects of multiple testing were minimised by considering only the associations between the allelic score and four blood pressure readings primary (all other associations being viewed as secondary). A p-value < 0.0125 was therefore considered statistically significant. Effect sizes are presented as Cohen’s d (where there are two groups being compared) or adjusted r2 values (where there are more than two groups being compared). Unless otherwise stated all other data are presented as means (95% confidence intervals). Statistical analyses were performed using either Stata 13 (StataCorp LP, College Station, Texas, U.S.A.) or R version 3.2.2 (http://cran.r-project.org/bin/windows/base/).
Results
Associations with Maternal Mean Arterial Blood Pressures in the Cambridge Baby Growth and Wellbeing Studies
Table 2 shows the seven strongest associations between fetal imprinted gene SNP alleles and maternal mean arterial blood pressures in univariate linear regression models (the remainder of the statistically significant associations being shown in Table S3). Although two of the SNPs tagged polymorphic variation in DLK1 they were not in linkage disequilibrium in our Cambridge Baby Growth and Wellbeing Studies (r2=0.49, D’=0.89). None of these strongest associations also showed an interaction with fetal sex, although some of the other fetal imprinted gene SNP alleles showed sex-specific associations with maternal blood pressure (Table S4). None of the other p-values, from statistical models involving associations between the 155 fetal SNPs (2 parental transmissions per SNP) and 4 blood pressure readings per subject, crossed the significance threshold.
Table 2.
The seven strongest associations between fetal imprinted gene SNP alleles and maternal mean arterial blood pressures in linear regression models that were adjusted for gestational age of the fetus, in the Cambridge Baby Growth and Wellbeing Studies. These fetal variants were used in the construction of the composite fetal imprinted gene allele score. Also shown are the p-values of the associations of maternal mean arterial blood pressures with the equivalent maternal genotypes. The blood pressure readings labelled as being taken at 37~41 weeks were taken in the last two weeks prior to parturition for each pregnancy, the mean (95% confidence interval) of which occurred at 39.3 (39.3, 39.4) weeks.
| Gene | SNP | Parental- Transmission |
Gestation- al Age (weeks) |
Non- Risk Allele (mmHg) |
Risk Allele (mmHg) |
Fetal Allele (p) |
Effe- ct Size (d) |
Fetal Allele Interac- tion With Sex (p) |
Fetal Allele From Other Parent (p) |
Maternal Genotype (p) |
Maternal Untransmitted Allele (p) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FAM99A | rs1489945 | Maternal | 37~41 | 86 (84, 87) (n=170) |
90 (88, 91) (n=168) |
2×10−4 | 0.30 | 0.39 | 0.4 | 0.1 | 1.0 |
| DLK1 | rs10139403 | Maternal | 37~41 | 86 (84, 88) (n=95) |
90 (88, 91) (n=273) |
9×10−4 | 0.39 | 0.33 | 0.01 | 0.04 | 0.7 |
| DLK1 | rs12147008 | Maternal | 37~41 | 85 (82, 87) (n=72) |
89 (88, 90) (n=326) |
1×10−3 | 0.42 | 0.36 | 0.3 | 4.5×10−3 | 0.4 |
| H19 | rs217222 | Paternal | 37~41 | 84 (82, 87) (n=49) |
89 (88, 90) (n=341) |
1×10−3 | 0.43 | 0.10 | 0.05 | 0.5 | 0.1 |
| SNRPN | rs1453556 | Paternal | 37~41 | 87 (85, 88) (n=250) |
90 (89, 92) (n=114) |
1×10−3 | 0.35 | 0.31 | 0.3 | 0.3 | 0.8 |
| IGF2, INS | rs6356 | Paternal | 31 | 81 (80, 82) (n=291) |
84 (83, 86) (n=147) |
1×10−3 | 0.33 | 0.74 | 0.2 | 0.6 | 0.9 |
| NNAT | rs6066671 | Paternal | 31 | 80 (79, 82) (n=167) |
83 (81, 84) (n=288) |
1×10−3 | 0.22 | 0.76 | 0.7 | 0.4 | 0.3 |
Data shown are mean (95 % confidence intervals). Effect size is Cohen’s d.
Confirmation of Univariate Analyses in HAPO and Meta-analysis
Table 3 shows the univariate associations between the seven fetal SNP alleles whose associations with maternal blood pressures in the Cambridge Baby Growth and Wellbeing Studies had the lowest p-values and maternal mean arterial blood pressures around week 28 of pregnancy in HAPO participants. None of these associations reached statistical significance, although, apart from the associations with maternally-transmitted fetal FAM99A rs1489945 and paternally-transmitted fetal rs217222, all the associations were in the same direction as those from the Cambridge Studies. In the meta-analysis that included data from the analysis of the Cambridge Studies one of the associations with maternal mean arterial blood pressure reached genome-wide statistical significance (maternally-transmitted fetal DLK1 rs10139403, p=6.3×10−10). Others associations were also significant (paternally-transmitted fetal IGF2 rs6356, p=6.0×10−6; paternally-transmitted fetal NNAT rs6066671, p=0.006) or borderline significant (maternally-transmitted fetal DLK1 rs12147008, p=0.04; paternally-transmitted fetal SNRPN rs1453556, p=0.02). Only two of the seven fetal SNP alleles showed no sign of any significant association (maternally-transmitted fetal FAM99A rs1489945, p=0.27; paternally-transmitted fetal H19 rs217222, p=0.31).
Table 3.
Univariate association between the seven fetal SNP alleles used in the composite fetal imprinted gene allele score and maternal mean arterial blood pressures at 28 weeks gestation in HAPO Study participants with European ancestry.
| Gene | Fetal SNP | Directly Genotyped or Imputed? | Parental Transmission | Risk Allele | n | Standardised β-Coefficient | Standard Error of the β-Coefficient | p-value |
|---|---|---|---|---|---|---|---|---|
| FAM99A | rs1489945 | Genotyped | maternal | C | 999 | −0.99 | 0.50 | 0.046 |
| DLK1 | rs10139403 | Genotyped | maternal | G | 1,067 | 0.11 | 0.60 | 0.860 |
| DLK1 | rs12147008 | Genotyped | maternal | T | 1,099 | 0.52 | 0.81 | 0.520 |
| H19 | rs217222 | Imputed | paternal | G | 1,149 | −0.14 | 0.62 | 0.823 |
| SNRPN | rs1453556 | Genotyped | Paternal | G | 1,016 | 0.01 | 0.53 | 0.988 |
| IGF2, INS | rs6356 | Genotyped | Paternal | A | 1,020 | 0.21 | 0.50 | 0.679 |
| NNAT | rs6066671 | Genotyped | Paternal | A | 986 | 0.25 | 0.50 | 0.613 |
Development of the Composite Fetal Imprinted Gene Allele Score
The effect size for the association between any of the maternal mean arterial blood pressure measurements and the developmental composite fetal imprinted gene allele scores were maximised when the composite score was formulated based on the 7 fetal SNP alleles with the lowest p-values in the univariate analyses: FAM99A rs1489945 transmitted from its mother, DLK1 rs10139403 (mother), DLK1 rs12147008 (mother), H19 rs217222 (father), SNRPN rs1453556 (father), IGF2 rs6356 (father) and NNAT rs6066671 (father). Table 5 shows the associations between the composite fetal imprinted gene allele score and maternal mean arterial blood pressures at different stages of pregnancy. There was no significant association with maternal blood pressures at 12 weeks gestation, but all the readings in the second half of pregnancy were significantly associated with the composite fetal imprinted gene allele score. Fig. 1 shows the composite fetal imprinted gene allele scores plotted against the mean arterial blood pressures at week 12 of pregnancy and in the last two weeks of pregnancy.
Table 5.
The associations between the composite fetal imprinted gene allele score and maternal blood pressure at different stages of pregnancy in the Cambridge Baby Growth and Wellbeing Studies.
| Stage of Pregnancy (Gestational Age (weeks)) |
n | Maternal Mean Arterial Blood Pressure (mmHg) |
Regression Coefficient (β (mmHg)) |
p-value | Effect Size (adjusted r2 (%)) |
|---|---|---|---|---|---|
| 12 | 577 | 80.6 (80.1, 81.2) | 0.2 | 0.5 | 0 |
| 31 | 582 | 81.3 (80.8, 81.9) | 0.6 | 4.1×10−8 | 5.6 |
| 37 | 570 | 85.0 (84.4, 85.6) | 1.4 | 1.1×10−4 | 3.6 |
| 37~41 | 457 | 87.5 (86.7, 88.4) | 2.7 | 1.1×10−10 | 8.7 |
Data are mean (95% confidence interval).
Figure 1.
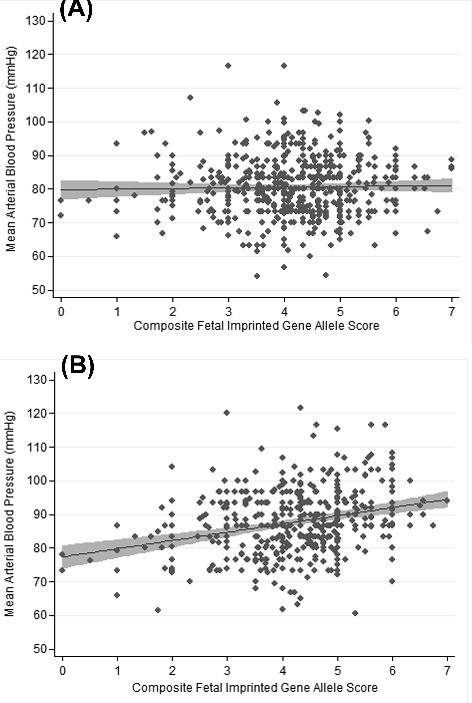
Scatter diagram showing the association between the composite fetal imprinted gene allele score and the mean arterial blood pressure (A) around week 12 of pregnancy and (B) in the final two weeks of pregnancy. The regression lines with their shaded 95% confidence intervals are shown.
Associations with Maternal Gestational Hypertension in the Cambridge Baby Growth and Wellbeing Studies
The composite fetal imprinted gene allele score was significantly associated with the development of gestational hypertension in the Cambridge Baby Growth and Wellbeing Studies (odds ratio 1.54 (1.14, 2.09) per allele; p = 0.005; 45 cases and 549 controls).
Discussion
This study has shown novel fetal genetic variants that are robustly associated with maternal blood pressure in the second half of pregnancy in a non sex-specific manner. This adds to previous reports of genetic associations found between autosomal fetal but not maternal variants and gestational hypertension in small, unreplicated studies22,23 and variable associations between maternal blood pressure and interactions between fetal sex and maternal variants24–26. Our results are consistent with the suggestion that the fetal genome is able to influence maternal blood pressure in pregnancy7,8, although they are associations rather than demonstrating causality. When the alleles from these variants were amalgamated as a composite score there were significant associations demonstrated with each of three maternal blood pressure readings taken across the second half of pregnancy in our Cambridge cohorts.
Interestingly there were only weak associations with the maternal blood pressure reading taken in the first trimester of pregnancy. Any hypertension detected at that stage of pregnancy would be considered by definition chronic hypertension rather than gestational hypertension. Fetal genes cannot contribute to chronic hypertension per se in pregnancy (as opposed to gestational hypertension superimposed upon chronic hypertension) since their effects would not be present outside of pregnancy. Therefore the finding that the composite fetal imprinted gene allele score was associated with the readings in the second half of pregnancy but not with that in the first is not unexpected. The association between the gene score and gestational hypertension itself is consistent with this. It may be that the genetic regulation of maternal blood pressure is dynamic throughout pregnancy as the effects of fetal genes slowly increase27, since our regression coefficients increase as pregnancy progresses. As a major role of imprinted genes is in fetoplacental growth and development10,19, it may also not be surprising that the effect sizes increase when fetal growth is maximal. Indeed our largest effect size came just prior to delivery, when fetal growth is likely to have just peaked28. At this stage 8.7% of the variance in the maternal blood pressure could be explained by associations with the score, consistent with the Swedish Birth and Multi-Generation Registries where 20% of the variability in liability of proteinuric gestational hypertension (preeclampsia) could be attributed to fetal genetic effects9. More of the variance in maternal blood pressure might be attributable to additional, as yet untested fetal variants or sex-specific associations24.
The list of fetal variants with the strongest non sex-specific associations with maternal blood pressure were from a range of imprinted genes and different parental transmissions (other associations were present in a sex-specific manner suggesting different causal mechanisms). Consistent with imprinting effects, these associations were all parental-transmission-dependent. The parental transmission with the significant association was not always as would be expected (e.g. maternally-transmitted DLK1 variants and a paternally-transmitted H19 variant). However the variants with the significant associations were genotyped as tags rather than potential functional variants and no assertions of causality can be made. Their loci tend to be in clusters of imprinted genes, some of which are maternally-expressed and some paternally. Taking fetal DLK1 rs10139403 as an example, its association with maternal blood pressure is with the maternally-transmitted allele despite DLK1 being paternally-expressed. However this SNP appears to be an expression quantitative trait locus (eQTL) for neighbouring MEG3 which is imprinted and maternally-expressed29.
Most of the associations with variants that were included in the composite fetal imprinted gene allele score showed some degree of replication in the meta-analysis that included results from HAPO, even if it were only of borderline significance for some of them. Differences between the HAPO Study populations and the Cambridge populations are suggested by the different direction of the association between maternally-transmitted fetal FAM99A rs1489945 and maternal mean arterial blood pressures in the three cohorts. Its link with maternal blood pressure in pregnancy is biologically plausible as it appears to be an eQTL for BRSK226 whose placental expression is raised 8.6 fold in severe preeclampsia30. The other association of note in the meta-analysis is that between maternally-expressed fetal DLK1 rs10139403 and maternal blood pressure since it reached genome wide significance. This is the first association between a fetal SNP allele and maternal blood pressure in pregnancy that crosses this threshold, with its implied validity across populations. As stated above this SNP, positioned in intron 1 of DLK1, appears to be a stronger eQTL for MEG3 than DLK129, a gene whose reduced placental expression is linked to preeclampsia and for whom in vitro data suggest a potential role in uterine spiral artery remodelling failure due to the aberrant conditioning of placental trophoblast cells31. Reduced placental MEG3 expression is also associated with intrauterine growth restriction32, another feature of preeclampsia. The other SNP tagging the DLK1 region that contributed to the composite fetal imprinted gene allele score in our study, rs12147008, probably relates to being a strong eQTL for Genbank ascension number AL117190.6, which is located in a putative intergenic differentially-methylated region which may regulate both DLK1 and MEG3 expression33. Of the other fetal SNPs used in the composite score rs6356 appears to be an eQTL for TH29, itself associated with blood pressure variability34. Fetal SNRPN rs1453556 could affect SNRPN expression through altering a transcription factor binding site35, a defect in which can cause Prader Willi syndrome36 which itself is associated with hypertension. Fetal rs217222 is a tag for genetic variation in the H19 gene region, where alterations in placental imprinting have been implicated in the development of preeclampsia37. Fetal rs6066671 is a tag for rs6066727 which is also predicted to alter a transcription factor binding site35 and for NNAT whose placental expression is raised 2.3 fold in severe preeclampsia30. All the SNPs used to construct the fetal imprinted gene allele score therefore have plausible links with gestational hypertension.
Although the associations between maternal blood pressures in the second half of pregnancy and the composite fetal imprinted gene allele score appear to be strong the study does have limitations including the fact that the composite score associations were only tested in the Cambridge cohorts and therefore could be less evident in other populations. Some of the limitations such as the relatively modest size of the Cambridge cohorts and the lack of paternity testing have been addressed previously14. Of the remaining limitations one is that none of the strongest associations in the Cambridge cohorts could be replicated independently in HAPO (just indirectly by meta-analysis). This could be for a number of possible reasons including: (1) the only recorded blood pressure measurement in HAPO was at a stage of pregnancy slightly earlier than when the strongest associations were found in the Cambridge cohorts and the Cambridge data suggest that generally these fetal associations are more evident as pregnancy progresses, (2) the exclusion of women with explicit diabetes from HAPO may have resulted in a depletion of hypertension risk genes given the link between what would previously have been considered gestational diabetes and gestational hypertension38, and (3) in some studies related to hyperglycemia such as HAPO there is a requirement for hyperglycemia to be present for certain genetic associations to become evident39. Another limitation is that the analysis used to construct the composite fetal imprinted gene allele score could be considered post hoc in nature although we constructed it this way due to the lack of fully established fetal imprinted gene allele associations with maternal blood pressures in pregnancy. A further limitation is that in our meta-analysis converting standardised β-coefficients into r-values sometimes had to use values which were outside the validated range of coefficients (−0.5 < β < 0.5, too few values being outside this range in Peterson and Browne’s original study to validate them)40. Since this only applied to rs1489945 and rs12147008, one of which was not significant and the other of which was only of borderline significance in the meta-analysis, it did not appear to influence the major associations observed. A final limitation is that our study has only shown associations with tag SNPs and therefore causality cannot be inferred.
Perspectives
This study has shown novel genetic associations between fetal imprinted gene variant alleles and maternal blood pressures in the second half of pregnancy, the time point over which gestational hypertension may emerge. One of these, maternally-transmitted fetal DLK1 rs10139403 reached genome-wide significance in the meta-analysis of its association with maternal blood pressure. An unweighted composite fetal imprinted gene allele score, constructed from the variants showing the strongest associations, was robustly associated with maternal blood pressures across the second half of pregnancy and with gestational hypertension in our Cambridge cohorts. These data support the concept that polymorphic variation in certain fetal genes are related to the development of gestational hypertension.
Supplementary Material
Table 4.
Results of the meta-analysis of the associations between maternal mean arterial blood pressures and fetal imprinted gene SNP alleles that were used in the composite score in the Cambridge Baby Growth Study, the Cambridge Wellbeing Study and HAPO Study participants with European ancestry.
| Gene | Fetal SNP | Parental Transmission | Study | n | r | Meta-analysis r |
Meta-analysis p |
|---|---|---|---|---|---|---|---|
| FAM99A | rs1489945 | maternal | CBGS | 277 | 0.102 | −0.498 | 0.271 |
| Wellbeing | 61 | 0.345 | |||||
| HAPO | 999 | −0.970 | |||||
| DLK1 | rs10139403 | maternal | CBGS | 301 | 0.191 | 0.161 | 6.277×10−10 |
| Wellbeing | 47 | 0.017 | |||||
| HAPO | 1,067 | 0.158 | |||||
| DLK1 | rs12147008 | maternal | CBGS | 306 | 0.228 | 0.299 | 0.044 |
| Wellbeing | 65 | 0.017 | |||||
| HAPO | 1,099 | 0.560 | |||||
| H19 | rs217222 | paternal | CBGS | 325 | 0.114 | 0.056 | 0.314 |
| Wellbeing | 65 | 0.251 | |||||
| HAPO | 1,149 | −0.137 | |||||
| SNRPN | rs1453556 | paternal | CBGS | 300 | 0.216 | 0.121 | 0.024 |
| Wellbeing | 64 | 0.079 | |||||
| HAPO | 1,016 | 0.060 | |||||
| IGF2, INS | rs6356 | paternal | CBGS | 387 | 0.139 | 0.214 | 5.965×10−6 |
| Wellbeing | 51 | 0.263 | |||||
| HAPO | 1,020 | 0.259 | |||||
| NNAT | rs6066671 | paternal | CBGS | 395 | 0.103 | 0.199 | 0.006 |
| Wellbeing | 60 | 0.171 | |||||
| HAPO | 986 | 0.295 |
CBGS = Cambridge Baby Growth Study, Wellbeing = Cambridge Wellbeing Study, HAPO = Hyperglycemia and Adverse Pregnancy Outcome Study. For the HAPO study the r-values were estimated from the standardised β-values using the method of Peterson & Brown23.
Novelty and Significance.
What is new
We have shown, for the first time, a group of fetal imprinted gene variants that are robustly associated with maternal blood pressure in pregnancy. The association with one of them, DLK1 rs10139403, reached genome-wide significance.
What is relevant
A composite fetal imprinted gene allele score was strongly associated with both maternal blood pressure across the second half of pregnancy (explaining up to 8.7% of the variance in maternal mean arterial blood pressure), and gestational hypertension.
Summary
This study implies a potential role for fetal imprinted genes in the development of gestational hypertension.
Acknowledgments
The authors acknowledge the excellent technical assistance for this project that was provided by Niamh Campbell, Dianne Wingate, Rachel Seear, and Katrin Mooslehner. The authors would like to thank all the families that took part in the Cambridge Baby Growth Study, and acknowledge the crucial role played by the research nurses especially Suzanne Smith, Ann-Marie Wardell, Karen Forbes and Catherine Fullah, staff at the Addenbrooke’s Wellcome Trust Clinical Research Facility, and midwives at the Rosie Maternity Hospital.
Sources of Funding
The genotyping part of the Cambridge Baby Growth Study was funded by the Evelyn Trust (EW9035322), Diabetes U.K. (11/0004241) and the Wellbeing of Women (the Royal College of Obstetricians and Gynaecologists, U.K.) (RG1644). Other core funding has come from the Medical Research Council (7500001180); European Union Framework 5 (QLK4-1999-01422); the Mothercare Charitable Foundation (RG54608); Newlife Foundation for Disabled Children (07/20) and the World Cancer Research Fund International (2004/03). In addition there has been support from National Institute for Health Research Cambridge Biomedical Research Centre. The HAPO Study was supported by NIH grants HD-34242, HD-34243, HG-004415, and CA-141688, Institutes of Health Research–INMD (Funding Reference Number 110791), and by the American Diabetes Association.
Footnotes
Conflicts of Interest/Disclosures
None.
References
- 1.Salonen Ros H, Lichtenstein P, Lipworth L, Cnattingius S. Genetic effects on the liability of developing pre-eclampsia and gestational hypertension. Am J Med Genet. 2000;91:256–260. [PubMed] [Google Scholar]
- 2.Williams PJ, Broughton Pipkin F. The genetics of pre-eclampsia and other hypertensive disorders of pregnancy. Best Pract Res Clin Obstet Gynaecol. 2011;25:405–417. doi: 10.1016/j.bpobgyn.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basson J, Simino J, Rao DC. Between candidate genes and whole genomes: time for alternative approaches in blood pressure genetics. Curr Hypertens Rep. 2012;14:46–61. doi: 10.1007/s11906-011-0241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laivuori H. Pitfalls in setting up genetic studies on preeclampsia. Pregnancy Hypertens. 2013;3:60. doi: 10.1016/j.preghy.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 5.Padmanabhan S, Caulfield M, Dominiczak AF. Genetic and molecular aspects of hypertension. Circ Res. 2015;116:937–959. doi: 10.1161/CIRCRESAHA.116.303647. [DOI] [PubMed] [Google Scholar]
- 6.Haig D. Placental hormones, genomic imprinting, and maternal–foetal communication. J Evol Biol. 1996;9:357–380. [Google Scholar]
- 7.Petry CJ, Ong KK, Dunger DB. Does the fetal genotype affect maternal physiology during pregnancy? Trends Mol Med. 2007;13:414–421. doi: 10.1016/j.molmed.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Petry CJ, Beardsall K, Dunger DB. The potential impact of the fetal genotype on maternal blood pressure during pregnancy. J Hypertens. 2014;32:1553–1561. doi: 10.1097/HJH.0000000000000212. [DOI] [PubMed] [Google Scholar]
- 9.Cnattingius S, Reilly M, Pawitan Y, Lichtenstein P. Maternal and fetal genetic factors account for most of familial aggregation of preeclampsia: a population-based Swedish cohort study. Am J Med Genet A. 2004;130A:365–371. doi: 10.1002/ajmg.a.30257. [DOI] [PubMed] [Google Scholar]
- 10.Moore GE, Ishida M, Demetriou C, et al. The role and interaction of imprinted genes in human fetal growth. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140074. doi: 10.1098/rstb.2014.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haig D. Genetic conflicts in human pregnancy. Q Rev Biol. 1993;68:495–532. doi: 10.1086/418300. [DOI] [PubMed] [Google Scholar]
- 12.Reik W, Constância M, Fowden A, Anderson N, Dean W, Ferguson-Smith A, Tycko B, Sibley C. Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol. 2003;547:35–44. doi: 10.1113/jphysiol.2002.033274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wangler MF, Chang AS, Moley KH, Feinberg AP, Debaun MR. Factors associated with preterm delivery in mothers of children with Beckwith-Wiedemann syndrome: a case cohort study from the BWS registry. Am J Med Genet A. 2005;134A:187–191. doi: 10.1002/ajmg.a.30595. [DOI] [PubMed] [Google Scholar]
- 14.Petry CJ, Seear RV, Wingate DL, Manico L, Acerini CL, Ong KK, Hughes IA, Dunger DB. Associations between paternally transmitted fetal IGF2 variants and maternal circulating glucose concentrations in pregnancy. Diabetes. 2011;60:3090–3096. doi: 10.2337/db11-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.HAPO Study Cooperative Research Group. Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, Coustan DR, Hadden DR, McCance DR, Hod M, McIntyre HD, Oats JJ, Persson B, Rogers MS, Sacks DA. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358:1991–2002. doi: 10.1056/NEJMoa0707943. [DOI] [PubMed] [Google Scholar]
- 16.HAPO Study Cooperative Research Group. The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. Int J Gynaecol Obstet. 2002;78:69–77. doi: 10.1016/s0020-7292(02)00092-9. [DOI] [PubMed] [Google Scholar]
- 17.Hayes MG, Urbanek M, Hivert MF, et al. Identification of HKDC1 and BACE2 as genes influencing glycemic traits during pregnancy through genome-wide association studies. Diabetes. 2013;62:3282–3291. doi: 10.2337/db12-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study Cooperative Research Group. Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study: preeclampsia. Am J Obstet Gynecol. 2010;202:255.e1–7. doi: 10.1016/j.ajog.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frost JM, Moore GE. The importance of imprinting in the human placenta. PLOS Genet. 2010;6:e1001015. doi: 10.1371/journal.pgen.1001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 21.Rodríguez S, Gaunt TR, O’Dell SD, Chen XH, Gu D, Hawe E, Miller GJ, Humphries SE, Day IN. Haplotypic analyses of the IGF2-INS-TH gene cluster in relation to cardiovascular risk traits. Hum Mol Genet. 2004;13:715–725. doi: 10.1093/hmg/ddh070. [DOI] [PubMed] [Google Scholar]
- 22.Xiang P, Li Z, Di H, Nie S, Yan W. The associations between maternal and fetal angiotensinogen M235T polymorphism and pregnancy-induced hypertension in Chinese women. Reprod Sci. 2011;18:640–644. doi: 10.1177/1933719110395405. [DOI] [PubMed] [Google Scholar]
- 23.Wilson ML, Brueggmann D, Desmond DH, Mandeville JE, Goodwin TM, Ingles SA. A fetal variant in the GCM1 gene is associated with pregnancy induced hypertension in a predominantly Hispanic population. Int J Mol Epidemiol Genet. 2011;2:196–206. [PMC free article] [PubMed] [Google Scholar]
- 24.Hocher B, Chen YP, Schlemm L, Burdack A, Li J, Halle H, Pfab T, Kalk P, Lang F, Godes M. Fetal sex determines the impact of maternal PROGINS progesterone receptor polymorphism on maternal physiology during pregnancy. Pharmacogenet Genomics. 2009;19:710–718. doi: 10.1097/FPC.0b013e328330bc7a. [DOI] [PubMed] [Google Scholar]
- 25.Hocher B, Schlemm L, Haumann H, Poralla C, Chen YP, Li J, Guthmann F, Bamberg C, Kalache KD, Pfab T. Interaction of maternal peroxisome proliferator-activated receptor gamma2 Pro12Ala polymorphism with fetal sex affects maternal glycemic control during pregnancy. Pharmacogenet Genomics. 2010;20:139–142. doi: 10.1097/FPC.0b013e3283357337. [DOI] [PubMed] [Google Scholar]
- 26.Hocher B, Schlemm L, Haumann H, Jian Li, Rahnenführer J, Guthmann F, Bamberg C, Kalk P, Pfab T, Chen YP. Offspring sex determines the impact of the maternal ACE I/D polymorphism on maternal glycaemic control during the last weeks of pregnancy. J Renin Angiotensin Aldosterone Syst. 2011;12:254–261. doi: 10.1177/1470320310387843. [DOI] [PubMed] [Google Scholar]
- 27.Kovanci E, Gregg AR. Blood pressure regulation across pregnancy: evidence of a paradigm shift in gene expression. Hypertens Pregnancy. 2010;29:236–247. doi: 10.3109/10641950903452394. [DOI] [PubMed] [Google Scholar]
- 28.Guihard-Costa AM, Droullé P, Thiebaugeorges O, Hascoet JM. A longitudinal study of fetal growth variability. Biol Neonate. 2000;78:8–12. doi: 10.1159/000014239. [DOI] [PubMed] [Google Scholar]
- 29.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877–881. doi: 10.1093/nar/gkv1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sitras V, Paulssen RH, Grønaas H, Leirvik J, Hanssen TA, Vårtun A, Acharya G. Differential placental gene expression in severe preeclampsia. Placenta. 2009;30:424–433. doi: 10.1016/j.placenta.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Zou Y, Wang W, Zuo Q, Jiang Z, Sun M, De W, Sun L. Down-regulated long non-coding RNA MEG3 and its effect on promoting apoptosis and suppressing migration of trophoblast cells. J Cell Biochem. 2015;116:542–550. doi: 10.1002/jcb.25004. [DOI] [PubMed] [Google Scholar]
- 32.McMinn J, Wei M, Schupf N, Cusmai J, Johnson EB, Smith AC, Weksberg R, Thaker HM, Tycko B. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta. 2006;27:540–549. doi: 10.1016/j.placenta.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Khoury H, Suarez-Saiz F, Wu S, Minden MD. An upstream insulator regulates DLK1 imprinting in AML. Blood. 2010;115:2260–2263. doi: 10.1182/blood-2009-03-212746. [DOI] [PubMed] [Google Scholar]
- 34.Rao F, Zhang L, Wessel J, et al. Tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis: discovery of common human genetic variants governing transcription, autonomic activity, and blood pressure in vivo. Circulation. 2007;116:993–1006. doi: 10.1161/CIRCULATIONAHA.106.682302. [DOI] [PubMed] [Google Scholar]
- 35.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37:W600–W605. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carrel AL, Huber S, Allen DB, Voelkerding KV. Assessment of SNRPN expression as a molecular tool in the diagnosis of Prader-Willi syndrome. Mol Diagn. 1999;4:5–10. doi: 10.1016/s1084-8592(99)80044-7. [DOI] [PubMed] [Google Scholar]
- 37.Yu L, Chen M, Zhao D, Yi P, Lu L, Han J, Zheng X, Zhou Y, Li L. The H19 gene imprinting in normal pregnancy and pre-eclampsia. Placenta. 2009;30:443–447. doi: 10.1016/j.placenta.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Ros HS, Cnattingius S, Lipworth L. Comparison of risk factors for preeclampsia and gestational hypertension in a population-based cohort study. Am J Epidemiol. 1998;147:1062–1070. doi: 10.1093/oxfordjournals.aje.a009400. [DOI] [PubMed] [Google Scholar]
- 39.Teumer A, Tin A, Sorice R, et al. Genome-wide association studies identify genetic loci associated with albuminuria in Diabetes. Diabetes. 2016;65:803–817. doi: 10.2337/db15-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peterson RA, Brown SP. On the use of beta coefficients in meta-analysis. J Appl Psych. 2005;90:175–181. doi: 10.1037/0021-9010.90.1.175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.