

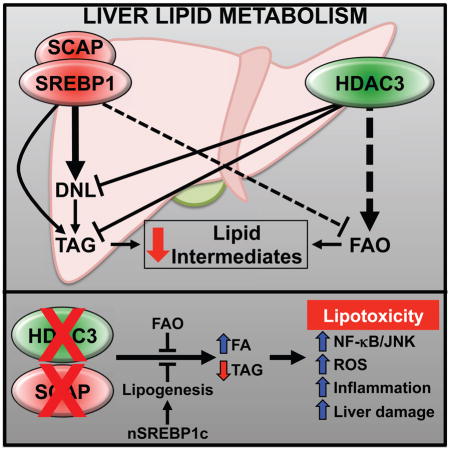
SUMMARY
Liver fat accumulation precedes non-alcoholic steatohepatitis, an increasing cause of end-stage liver disease. Histone Deacetylase 3 (HDAC3) is required for hepatic triglyceride homeostasis and Sterol Regulatory Element Binding Protein (SREBP) regulates the lipogenic response to feeding, but the crosstalk between these pathways is unknown. Here we show that inactivation of SREBP by hepatic deletion of SREBP Cleavage Activating Protein (SCAP) abrogates the increase in lipogenesis caused by loss of HDAC3, but fatty acid oxidation remains defective. This combination leads to accumulation of lipid intermediates and to an energy drain that collectively cause oxidative stress, inflammation, liver damage, and ultimately synthetic lethality. Remarkably, this phenotype is prevented by ectopic expression of nuclear SREBP1c, revealing a surprising benefit of de novo lipogenesis and triglyceride synthesis in preventing lipotoxicity. These results demonstrate that HDAC3 and SCAP control symbiotic pathways of liver lipid metabolism that are critical for suppression of lipotoxicity.
eTOC BLURB
XXX et al show complimentary crosstalk between the lipid metabolism pathways HDAC3 and SCAP/SREBP1c. When both pathways are compromised, toxic accumulation of lipid intermediates in the liver precipitates an aggressively lethal form of non-alcoholic steatohepatitis, which can be prevented by augmenting de novo lipogenesis and triglyceride synthesis to ameliorate lipotoxicity.
INTRODUCTION
Fatty acid metabolism in the liver is fundamentally aligned with the energy status of the organism. Under physiological conditions, the liver orchestrates anabolic and catabolic lipid metabolism to maintain energy homeostasis throughout the day (Gooley and Chua, 2014; Hems et al., 1975). The importance of this integrative function is most evident in metabolic disorders such as obesity, where a surplus of energy leads to an excess of triglycerides (TAG) that escape adipose storage and ectopically accumulate in the liver (hepatosteatosis), skeletal muscle, heart, or kidney (Kang et al., 2014; Snel et al., 2012). While increased hepatic triglycerides per se is well tolerated, it can lead to lipotoxicity through incompletely defined mechanisms that trigger the progression of diseases such as diabetes and non-alcoholic steatohepatitis (NASH) (Browning and Horton, 2004; Listenberger et al., 2003; Monetti et al., 2007; Yamaguchi et al., 2007).
There is increasing evidence that fatty acid intermediates, such as free fatty acids (FFA), acylcarnitines, acyl-CoAs, and ceramides, are mediators of lipotoxicity (Alkhouri et al., 2009; Fon Tacer and Rozman, 2011; Neuschwander-Tetri, 2010a; Williams et al., 2013). Their intracellular levels are tightly regulated by a balance of de novo lipogenesis (DNL) and TAG synthesis under positive energy states, and fatty acid oxidation (FAO) in energy deficient states. Whereas increased DNL is thought to be a major contributor to non-alcoholic fatty liver disease (NAFLD) progression, the role of FAO is less appreciated, but several studies have implicated impaired FAO as a major contributor to fatty liver formation and NASH (Kim et al., 2003; Sahebkar et al., 2014). In fact, PPARα agonists such as fibrates have shown promise in improving liver health by increasing FAO in diabetics and NASH rodent models (Lam and Younossi, 2010; Musso et al., 2016). In this context, both DNL and FAO may be functionally redundant in their ability to eliminate lipotoxic lipid intermediates (Cotter et al., 2014; Fromenty and Pessayre, 1995; Solinas et al., 2015; Yamaguchi et al., 2010).
There is a wealth of data showing that SREBP1a and SREBP1c stimulate hepatic lipogenesis during feeding (Horton et al., 1998a, 2003). A third isoform, SREBP2, is primarily associated with stimulation of cholesterogenic genes. However, all isoforms are functionally redundant to a certain extent especially when overexpressed (Horton et al., 1998b; Shimano et al., 1996, 1997). Full-length precursor SREBP1 (pSREBP1) is bound to the endoplasmic reticulum (ER) and is activated by insulin stimulated proteolytic cleavage of its N-terminus, which translocates to the nucleus (nSREBP1) and functions as a potent lipogenic transcription factor. At the ER, pSREBP is constitutively bound by SCAP, an adapter protein that stabilizes pSREBP structure and also facilitates their cleavage (Matsuda et al., 2001; Moon et al., 2012). To unequivocally assess the function of SREBPs without compensation by their redundant activities, Moon et al. utilized SCAP liver knockout mice to essentially inactivate all three SREBP isoforms. They found that liver-specific loss of SCAP in high-fat diet fed and ob/ob mice inhibited hepatic DNL and prevented hepatosteatosis, demonstrating a singularly important role for SREBPs relative to other nutritionally stimulated lipogenic factors such as Carbohydrate Response Element Binding Protein (ChREBP) (Moon et al., 2012).
HDAC3, a class I histone deacetylase, is a core component of nuclear receptor corepressor complexes whose deletion in liver results in hepatosteatosis due to increased lipogenesis and decreased FAO (Sun et al., 2012). The mechanism involves increased expression of lipogenic genes, many of which are targets of SREBP1. Here we sought to determine the relationship between HDAC3 and SREBPs in the physiological transcriptional regulation of hepatic lipid metabolism. Consistent with complementary mechanisms of action, we found HDAC3 and SREBP1c mainly bound to different sites in the liver genome, and mice lacking hepatic HDAC3 and SCAP exhibited intermediate levels of triglyceride accumulation. Remarkably, in this setting both de novo lipogenesis and fatty acid oxidation were defective, leading to accumulation of toxic lipids and a rapid progression to liver failure and death. Expression of nuclear SREBP1c restored lipogenesis and rescued this lethality despite massive triglyceride accumulation. These results demonstrate that HDAC3 and SCAP control complementary pathways of liver lipid metabolism that are required to prevent lipotoxicity.
RESULTS
HDAC3 and SREBP1c independently bind near lipogenic genes to regulate their transcription
HDAC3 and SREBP1 target many of the same lipogenic genes (Horton et al., 1998a; Sun et al., 2012). As previously shown, SREBP1 gene expression was not increased in mice lacking HDAC3 (Figure S1A) (Sun et al., 2012), and levels of the processed, nuclear form of SREBP1 were indistinguishable from those of control mice (Figure S1B). Moreover, loss of HDAC3 induced Insig2, a negative regulator of SREBP cleavage, and induced some, but not all of the lipogenic gene targets of SREBP1 including Acly, Acaca, and Srebp1a/c (Figure S1A). Thus, loss of HDAC3 promotes lipogenesis by a mechanism independent of SREBP1 activation.
To further clarify the relationship between hepatic gene regulation by HDAC3 and SREBP, we investigated their cistromes in intact liver. A previous study had uncovered ~400 SREBP1 binding sites (Gilardi et al., 2014), 15–20% of which overlapped with HDAC3 binding sites in mouse liver (Feng et al., 2011). Since HDAC3 and SREBP have opposing effects on lipid metabolism, this raised the question of whether SREBP might interfere with HDAC3 binding. To test this, the active, nuclear form of SREBP1c (nSREBP1c) was expressed with an HA epitope tag in the livers of mice using adeno-associated virus serotype 8 and the hepatocyte-specific thyroxine-binding globulin promoter (AAV8-Tbg) (Figure S1C) (Sun et al., 2012). As expected, HA-nSREBP1c expression led to a striking increase in lipogenic gene expression in mouse livers (Figure S1D).
Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) was used to determine the cistromes of HA-nSREBP1c and endogenous HDAC3 in livers of mice injected with either AAV8:HA-nSREBP1c or AAV8:GFP (control). Using highly stringent cutoffs we identified over 7,700 HA-nSREBP1c binding sites (Figure S1E), almost half of which bound in the same vicinity as HDAC3 (Figure 1A; common sites). HDAC3 and nSREBP1c bound near many lipogenic genes, often at multiple sites both together and separately as at the Scd1 and Gpam loci (Figure S1F). Importantly, expression of HA-nSREBP1c did not interfere with HDAC3 binding indicating that their regulatory elements are adjacent rather than overlapping at common sites (Figure 1A and 1B). De novo motif analysis revealed unique transcription factor (TF) motif signatures demarcating the common, Srebp1c-specifc and HDAC3-specifc groups as well as a core set of lineage-determining TF motifs (HNF4, CEBP and Forkhead) shared by all groups (Figure 1C and Figure S1G). In the common peaks, motif analysis using a smaller search window (30 bp) further unveiled unique TF motifs, reinforcing the conclusion that HDAC3 and nSREBP1c bind to nearby but different sites (Figure S1H). Taken together, these data reveal that HDAC3 and nSREBP1c function in distinct regulatory pathways and bind independently near lipogenic genes in liver.
Figure 1. HDAC3 and SREBP1c independently bind near lipogenic genes to regulate their transcription.

(A) Heat map of ChIP-seq peaks in mouse liver. C57Bl/6 mice (2–3 months old) were tail-vein injected with either AAV8:GFP or AAV8:HA-nSREBP1c and sacrificed 10 days later at ZT10. All ChIPs were performed in parallel. High-confidence peaks were clustered into 3 groups: common peaks where HA-nSREBP1c and HDAC3 peaks overlapped (purple), nSREBP1c-specfic peaks (red), and HDAC3-specific peaks (blue).
(B) Average binding profiles in 2 kb windows. Black and brown lines represent HDAC3 peaks in AAV8:GFP and AAV8:HA-nSREBP1c injected mice, respectively. Green lines represent the HA-nSREBP1c peaks.
(C) De novo motif analysis using 100 bp search windows. The top 6 motifs are shown in each group and ordered according to their percent enrichment (%) in peaks for each assigned group.
See also Figure S1.
HDAC3 partially suppresses fatty liver formation in SCAP depleted livers
SREBP1 controls hepatic DNL and strongly activates TAG synthesis (Horton et al., 2002). As such, its hepatic depletion by SCAP liver knockout is sufficient to prevent hepatosteatosis in genetic and diet-induced models of obesity (Moon et al., 2012). Our finding that HDAC3 and SREBP1 control distinct pathways of hepatic lipid metabolism prompted us to investigate whether HDAC3 suppresses hepatosteatosis in SCAP deficient mice. We crossed Hdac3 floxed (Hdac3f/f) and Scap floxed (Scapf/f) mice to generate double floxed (Hdac3/Scapf/f) mice in the C57Bl/6 background. To delete both alleles conditionally in adult mouse livers, we injected mice with AAV8 expressing either green fluorescent protein (AAV8:GFP; Control) or Cre recombinase (AAV8:Cre; LKO) under the control of the Tbg promoter to obtain hepatocyte-specific conditional knockouts.
The knock out efficiency of AAV8:Cre in Hdac3/Scapf/f mice (DLKO) was comparable to the single knock out controls (HDAC3 LKO and SCAP LKO) (Figure 2A and 2B). Loss of SCAP also leads to reduced expression of SREBP isoforms (Matsuda et al., 2001), which was reproduced in SCAP LKO and DLKO mice (Figure 2B and Figure S2A). All of the cohorts had similar body weights (Figure S2B) while hepatic TAG amounts were increased three-fold in HDAC3 LKO and decreased by 25% in SCAP LKO, as expected (Moon et al., 2012; Sun et al., 2012) (Figure 2C). TAG levels were not significantly increased in DLKO livers compared with controls, but they were nearly double those of SCAP LKO livers (Figure 2C), indicating that HDAC3 depletion supports TAG accumulation even in the absence of SCAP. However, lipogenic genes that were upregulated in HDAC3 LKO and were strongly repressed in SCAP LKO remained repressed in DLKO mice (Figure 2D), demonstrating the effects of SCAP/SREBP on DNL were dominant over those of HDAC3.
Figure 2. HDAC3 partially suppresses fatty liver formation in SCAP depleted livers.

(A) Quantitative RT-PCR (RT-qPCR) analysis of livers from floxed mice fed a normal chow diet and harvested 10 days post AAV8 injection (Control, AAV8:GFP; LKO, AAV8:Cre). n = 4 to 5 mice per group.
(B) Western blot of HDAC3, SCAP and full-length SREBP1 precursor in total liver lysates. Control mice are random littermate floxed mice injected with AAV8:GFP.
(C) Liver triglyceride (TAG) measurements from normal chow fed mice described in (A).
(D) RT-qPCR analysis of lipogenic genes in livers from normal chow fed mice described in (A).
(E) Hepatic TAG measurements from mice fed a HFD for 8 weeks total (AAV8 injection at 6 weeks). These mice were 8 weeks old at the start of the HFD. n = 4 to 5 mice per group.
(F and G) Representative H&E (F) and ORO (G) staining of livers from HFD fed mice described in (E). Scale bar, 100 μm.
All error bars, s.e.m. Significance was determined by two-tailed Student’s t test (*P < 0.05). See also Figure S2.
The moderate increase in DLKO liver TAG despite low lipogenic gene expression suggested that DLKO livers might become steatotic when the mice are exposed to a high-fat diet (HFD), which provides exogenous fatty acids and reduces the reliance on DNL (Duarte et al., 2014; Leavens et al., 2009; Tripathy et al., 2010). Indeed, while 8 weeks on HFD (AAV8 injections at week 6) had no significant effect on body weight among the cohorts (Figure S2C), DLKO livers were steatotic with more than a five-fold increase in TAG amounts compared to the levels in SCAP LKO (Figure 2E). Histological staining revealed the intermediate accumulation of lipid droplets (Figure 2F) and neutral lipids (Figure 2G) in DLKO liver sections. The gene expression profiles were similar between the normal chow fed and HFD cohorts (Figure S2D), demonstrating that DNL was also downregulated in HFD fed DLKO mice and indicating that they developed fatty liver through alternative lipid metabolic pathways. Notably, Gpam expression was slightly decreased in DLKO, which likely contributed to lowering their levels of TAG accumulation (Figure S2E). Taken together, our results indicate that the net effect of HDAC3 and SCAP depletion results in appreciable accumulation of hepatic TAG via pathways other than DNL.
Decreased fatty acid oxidation and accumulation of lipid intermediates in livers lacking HDAC3 and SCAP
Hepatic TAG levels are also influenced by uptake and oxidation of fatty acids. Fatty acid uptake was unchanged in the DLKO mice (Figure S3A). However, whereas HDAC3 LKO has been shown to block FAO (Sun et al., 2012), we found that SCAP LKO markedly increased FAO (Figure 3A). Importantly, the FAO rate of the DLKO was reduced well below control levels, revealing that the loss of HDAC3 dominantly controlled FAO in DLKO livers. The specificity of the assay for FAO oxidation in mitochondria was confirmed by treatment with CPT1 inhibitor etomoxir, which blocked FAO flux (Figure 3A) and led to an expected increase in intracellular lipids (Figure S3A).
Figure 3. Decreased fatty acid oxidation and accumulation of lipid intermediates in livers lacking HDAC3 and SCAP.

(A) Fatty acid oxidation measured in primary hepatocytes. Control cells are from random littermate floxed mice injected with AAV8:GFP. SCAP LKO and DLKO cells are from SCAPf/f and HDAC3f/f/SCAPf/f mice, respectively, injected with AAV8:Cre. Cells were incubated with 3H-palmitate for 120 min with vehicle (water) or 100 μM etomoxir (an inhibitor of carnitine palmitoyltransferase-1 and fatty acid oxidation). 3H-labelled water (3H2O) was measured by scintillation counting. dpm, disintegrations per minute; 3H-palmitate > 3H2O, 3H-palmitate conversion to 3H2O. n = 6 wells (combined from 3 mice) per group. Significance was determined by one-way ANOVA with Tukey’s correction (*P < 0.05 for comparison between experimental and control cells).
(B and C) Hepatic acylcarnitine (B) and free fatty acid (FFA) (C) content measured by liquid chromatography mass spectrometry. Average ion counts of each metabolite across all conditions are shown. n = 4 mice per group. Significance was determined by two-tailed Student’s t test followed by FDR correction (*P < 0.05). Ion counts are available in Table S1.
(D and E) RT-qPCR analysis of lipid catabolic genes (D) and fatty acid esterification genes (E) in livers. n = 4 to 5 mice per group. Significance was determined by two-tailed Student’s t test (*P < 0.05).
(F) Fatty acid metabolism pathway in DLKO mice. #Acyl-CoA levels were not measured.
All error bars, s.e.m. Control, AAV8:GFP injected; LKO, AAV8:Cre injected. See also Figure S3.
Metabolomics analysis of DLKO livers from mice on normal chow diet revealed a striking increase in acylcarnitines and FFA corroborating the reduced FAO flux (Figure 3B and 3C). Impaired FAO was also apparent from reduced serum β-hydroxybutyrate upon fasting (Figure S3B). The accumulation of FFA in DLKO livers was likely favored by increased expression of lipolytic genes (Figure 3D) coupled with attenuation of genes important for fatty acid incorporation into TAG (Figure 3E and Figure S3C). Lipid intermediates including acylcarnitines were not elevated in single LKO controls likely resulting from increased TAG synthesis in HDAC3 LKO and increased FAO in SCAP LKO mice (Figure S3D and Figure S3E). Thus, total TAG was markedly reduced in DLKO relative to the HDAC3 LKO, with resultant buildup of acylcarnitines and FFA. The logjam in conversion to TAG in DLKO was also reflected by accumulation of other lipid intermediates including lysophosphatidic acid and phosphatidic acid (Figure S3F and Figure S3G). Taken together, loss of HDAC3 and SCAP caused impaired FAO, DNL, and TAG synthesis leading to an accumulation of hepatic lipid intermediates (Figure 3F).
Lack of HDAC3 and SCAP causes hepatic lipotoxicity, oxidative stress, and inflammation
Consistent with reduced FAO, hepatic energy reserves, estimated from ATP/ADP and ATP/AMP levels, were decreased in the DLKO livers (Figure 4A). Moreover, increased peroxidation of the accumulated lipids was observed in DLKO livers (Figure 4B). Lipid peroxidation propagates reactive oxygen species (ROS) (Guéraud et al., 2010) and, consistent with this, RNA-seq analysis revealed that oxidative and inflammatory genes were highly induced in DLKO mice relative to control, HDAC3 LKO or SCAP LKO livers (Figure S4A and S4B). For example, as confirmed by RT-qPCR, hepatic antioxidant genes governing cysteine transport (Slc7a11), glutathione synthesis (Gsta1, Gstm3), and sulfiredoxin (Srxn1) were induced in the DLKO livers (Figure 4C). Hepatic inflammation is a recognized sequela of oxidative stress (Mittal et al., 2014) and, indeed, markers of macrophage/monocytes (Cd68 and Cd14), and targets of NF-κB (Nqo1 and Lcn2), were induced in the DKLO livers (Figure 4D and Figure S4C) with no changes to ER stress genes (Figure S4D). Accordingly, NF-κB was highly activated by phosphorylation in DLKO livers (Figure 4E and Figure S4E). In a subset of DLKO mice the stress levels were such that activation of pro-apoptotic factor JNK was also greatly induced (Figure 4E). Flow cytometric analysis confirmed the infiltration of inflammatory cells including neutrophils, eosinophils and macrophages into the livers of DLKO mice as compared with control mice, while T and B cell numbers were similar between these groups, indicating the development of an innate inflammatory response in the absence of HDAC3 and SCAP (Figure 4F and S4F).
Figure 4. Lack of HDAC3 and SCAP causes hepatic lipotoxicity, oxidative stress, and inflammation.

(A) Relative ATP/ADP and ATP/AMP ratios in DLKO livers as measured by liquid chromatography mass spectrometry. n = 4 mice per group.
(B) Liver thiobarbituric acid reactive substances (TBARS) assay showing levels of malondialdehyde (MDA) formation in 24 h fasted mice. n = 7 to 9 mice per group.
(C and D) RT-qPCR analysis of antioxidant genes (C) and inflammation genes (D) identified by RNA-seq. n = 4 to 5 mice per group.
(E) Western blot of total liver lysates. # denotes the subset of DLKO mice in which the pro-apoptotic factor JNK was activated by phosphorylation.
(F) Number of immune cells from control (n = 4) or DLKO (n = 3) mice, as determined by flow cytometry and total cell number. Control and DLKO mice were sacrificed 15 days post AAV8 injections.
All error bars, s.e.m. Control, AAV8:GFP injected; LKO, AAV8:Cre injected. Significance was determined by two-tailed Student’s t test (*P < 0.05). See also Figure S4.
Loss of hepatic HDAC3 and SCAP is synthetic lethal
The above findings in DLKO livers were observed within 15 days of AAV8-Cre driven depletion of HDAC3 and SCAP. Remarkably, all DLKO mice died within 3 weeks of the AAV8 injection (Figure 5A). By contrast, the SCAP LKO remained viable for months while HDAC3 LKO mice eventually developed jaundice and died 7–11 weeks post AAV8 injection with liver tumors that were absent in DLKO livers (data not shown).
Figure 5. Loss of hepatic HDAC3 and SCAP is synthetic lethal.

(A) Kaplan-Meier survival curve of indicated LKO mice. n = 4 mice per group. Day 10 cohort (d10), mice were harvested 10 days post AAV8 injection. n = 4 to 5 mice per group. Moribund cohort (Pre-†), DLKO mice were monitored daily and were sacrificed when they presented with weight loss, inactivity, and non-responsiveness. Livers from control floxed mice in this cohort were harvested ~15 days post AAV8:GFP injection. n = 3 to 6 mice per group.
(B) Body mass of mice from d10 and Pre-† mice.
(C) Mass of epididymal white adipose tissue of Pre-† mice.
(D) Levels of serum non-esterified fatty acids (NEFA) in d10 and Pre-† mice.
(E) Levels of serum alanine transaminase (ALT) in d10 and Pre-† mice.
(F) H&E staining of representative livers from d10 and Pre-† mice. Green arrowheads indicate immune cell infiltrates. Yellow arrowheads indicate damaged tissue. Scale bar, 100 μm.
All error bars, s.e.m. Control, AAV8:GFP injected; LKO, AAV8:Cre injected. Significance was determined by two-tailed Student’s t test (*P < 0.05). See also Figure S5.
The rapid onset of death in DLKO mice prompted us to systematically compare the moribund mice (Figure 5A, cohort “Pre-†”) with a cohort sacrificed at day 10 post AAV8 injection (cohort “d10”) to distinguish the primary and secondary phenotypes associated with DLKO. Moribund DLKO mice had generally unresponsive behavior (Figure S5A) and marked weight loss (Figure 5B). In line with these results, moribund DLKO mice exhibited a loss of adipose tissue (Figure 5C), and a dramatic increase in serum non-esterified fatty acids (Figure 5D), suggesting an exaggerated fasting physiology. Indeed, moribund DLKO mice abruptly ceased feeding activity (Figure S5B), and this was reflected by biomarkers of starvation including elevated hepatic Fgf21 gene expression (Figure S5C) as well as marked decreases in blood glucose, serum insulin and liver glycogen (Figure S5D, S5E and S5F).
Because the AAV8-Tbg Cre delivery system is highly specific to hepatocytes (Sun et al., 2012), we reasoned that liver failure was the likely cause of death. The near-comatose state of moribund DLKO mice fulfilled the criteria for hepatic encephalopathy (Figure S5A) (Hori et al., 2011), and serum ALT levels skyrocketed, consistent with massive liver damage (Figure 5E). Increased severity of hepatitis just before death was manifest by increased expression of the macrophage marker Cd68 (Figure S5G), as well as the marked inflammatory infiltrates seen by H&E staining, which also revealed damaged hepatocytes (Figure 5F). Of note, there was no evidence of cholestasis, ruling out a major role for abnormal bile acid or sterol metabolism in this phenotype. Thus, the severe liver damage that results in the demise of DLKO mice develops from lipotoxicity-induced inflammation and hepatocyte cell death stemming from molecular changes that were already apparent in d10 mice.
Glucose treatment only briefly extends viability of DLKO mice
Given the hypoglycemia of DLKO mice, which likely resulted from a combination of their liver failure and starvation, we tested whether supplementation of their drinking water with 10% glucose would extend their life. Glucose treatment did prolong the viability of DLKO mice, but only by ~5.5 days before all mice in the cohort (n = 4) succumbed (Figure S6A), with a similar gross liver morphology as untreated DLKO mice. Although it was possible that glucose stimulation of ChREBP-induced lipogenesis contributed to the increased viability (Benhamed et al., 2012), we did not detect an increase in the expression of Fasn or other DNL factors in this cohort (Figure S6B). Thus, the partial rescue with glucose suggested that energy deficiency contributed to the progression of the disease, but was not the main driver of the pathophysiology of DLKO mice.
Restoration of DNL and TAG synthesis prevents lipotoxicity-induced liver damage
We next considered whether lipotoxicity is the major contributor to liver damage and death in DLKO mice. To address this issue, we utilized the AAV8:HA-nSREBP1c gene delivery system to ectopically express HA-nSREBP1c in DLKO mice. Western blot analysis showed efficient expression of HA-nSREBP1c protein in DLKO livers (Figure S6C). Consistent with the ChIP-seq results, expression of HA-nSREBP1c in DLKO mice led to a robust increase in expression of genes involved in DNL and TAG synthesis with a modest increase in cholesterogenic gene expression (Figure 6A), resulting in a more than 3-fold increase in hepatic TAG content (Figure 6B). Hematoxylin & eosin staining revealed an increase in steatotic hepatocytes and, remarkably, a loss of immune cell infiltrates in DLKO+nSREBP1c mice (Figure 6C). In addition, the gene expression profile of DLKO+nSREBP1c mice showed a marked improvement in the inflammatory and antioxidant stress response pathways (Figure 6D), although we did not see enrichment of nSREBP1c binding at most of these inflammatory and antioxidant genes, suggesting that nSREBP1c indirectly improves these deleterious pathways in DLKO mice. Most impressively, nSREBP1c expression completely prevented death in DLKO mice (Figure 6E).
Figure 6. Restoration of DNL and TAG synthesis prevents lipotoxicity-induced liver damage.

(A) RT-qPCR analysis of liver gene expression from Control (HDAC3f/f/SCAPf/f injected with AAV8:GFP + AAV8:Null), DLKO (HDAC3f/f/SCAPf/f injected with AAV8:Cre + AAV8:Null) and DLKO+nSREBP1c (HDAC3f/f/SCAPf/f injected with AAV8:Cre + AAV8:HA-nSREBP1c) mice sacrificed 10 days post AAV8 injection. n = 5 mice per group.
(B) Hepatic TAG measurements in mice described in (A).
(C) H&E staining of representative livers from Control, DLKO, and DLKO+nSREBP1c mice from (A). Green arrowhead points to a density of immune cell infiltrates that are lacking in Control and DLKO+nSREBP1c livers. Scale bar, 100 μm.
(D) RT-qPCR analysis of liver antioxidant and inflammatory gene expression from the mice described in (A).
(E) Kaplan-Meier survival curve of DLKO and DLKO+nSREBP1c mice. n = 4 mice per group.
(F) Hepatic DNL rate in vivo measured by tracing hepatic 2H-palmitate synthesis from deuterated water during a 6-hour interval. n = 4 mice per group.
(G and H) Hepatic levels of acylcarnitines (G) and other lipids (H) as measured by liquid chromatography mass spectrometry. Average ion counts of each metabolite across all conditions are shown in (G). Biological replicates are depicted in each row and each column represents a specific lipid metabolite in (H). The heat map highlights relative changes for each lipid metabolite with respect to its average ion counts in all three conditions. FFA, free fatty acids. MAG, monoacylglycerols. DAG, diacylglycerol. TAG, triacylglycerol. n = 4 to 5 mice per group. These mice were sacrificed 12 days after AAV8 injections. Ion counts and lipid identifications are available in Table S4.
(I) Summary of lipid metabolism in HDAC3 LKO, SCAP LKO, and DLKO livers.
All error bars, s.e.m. Significance was determined by one-way ANOVA followed by Holm-Sidak correction (A,B) or Tukey’s correction (B,F) (*P < 0.05 for comparison between control and experimental mice). (G and H) Significance of lipidomics data was determined by one-way ANOVA followed by FDR correction (*P < 0.05). See also Figure S6.
We next addressed the mechanism by which nSREBP1c expression prevented lipotoxicity in mice lacking SCAP and HDAC3. As expected from the gene expression data, hepatic DNL was upregulated in DLKO+nSREBP1c mice (Figure 6F) while, despite the increased fatty acid uptake, FAO remained attenuated in their hepatocytes (Figure S6D). Lipidomics analysis of a second cohort with no changes in body mass (Figure S6E) and similar gene expression profile (Figure S6F and S6G) revealed a dramatic reduction in lipotoxic acylcarnitines (Figure 6G) and ceramides (Figure S6H). Furthermore, HA-nSREBP1c expression reduced monoacylglycerol (MAG) levels in DLKO mice, consistent with decreased lipolysis, increased FA esterification, and elevated TAG levels (Figure 6H). FFA levels, generally increased in DLKO mice, remained elevated in DLKO+nSREBP1c mice with an increase in monounsaturated FFA, which are products of DNL and favorable substrates for TAG synthesis (Figure S6I). Together, these data show that SREBP1c-mediated induction of DNL and FA esterification prevents hepatic lipotoxicity in DLKO mice.
DISCUSSION
Our work has uncovered complementary and critical roles for HDAC3 and SCAP in the control of hepatic fatty acid metabolism. Their combined depletion led to a loss of lipogenesis and impaired FAO, resulting in an accumulation of extrahepatic-derived lipid intermediates (Figure 6I). The acute onset of liver damage and death in DLKO mice underscores the inherent toxicity of intermediate lipid species, and our data suggest that their elimination by FAO or sequestration by TAG synthesis is cytoprotective.
Lipotoxicity is increasingly recognized as a major pathophysiological driver of liver injury because it can interfere with hormonal signaling, disrupt cellular bioenergetics, and cause inflammation (Neuschwander-Tetri, 2010b). The mechanisms that transform inert hepatosteatosis into a lipotoxic state are largely unknown. A popular explanation invokes a successive progression, whereby a backlog of toxic lipid intermediates triggers oxidative stress and inflammation (Day and James, 1998). The source of oxidative stress in lipotoxic states remains controversial although mitochondrial dysfunction leading to lipid peroxidation has been suggested (Koliaki et al., 2015; Sunny et al., 2011; Zhang et al., 2007). Most likely multiple hits accumulate over the course of years, and still only a portion of NAFLD patients progress to NASH (Arrese et al., 2016).
Our DLKO model shares many of the same hallmarks of NASH development with a notable distinction that DLKO mice died reproducibly and acutely within 3 weeks of the conditional knockout. Despite the abbreviated disease progression, DLKO livers contained increased levels of oxidative stress and intermediate fatty acids in combination with impaired mitochondrial FAO. In line with human studies, lesions in DLKO led to NF-κB and JNK activation, which mediate lipotoxicity-induced inflammation and cell death (Cazanave et al., 2009). The innate inflammation in DLKO livers parallels that of human NASH (Arrese et al., 2016) and our data suggest that altered hepatic lipid profiles may be an early and potent driver of inflammation in NAFLD. Furthermore, DLKO mice developed severe liver damage with increased serum ALT levels, another hallmark of advanced NAFLD. It is important to note the acute onset of death in DLKO mice likely thwarted the development of overt hepatic fibrosis, which is a product of chronic liver injury in NAFLD.
Within this context, our work supports the notion that lipogenesis can have opposing roles depending on the severity of hepatosteatosis (Solinas et al., 2015). Early in the course of hepatosteatosis, lipogenesis may actually prevent lipotoxicity by sequestering deleterious lipid intermediates into TAG. However, as the disease progresses, the capacity to store TAG becomes saturated leading to accumulation of toxic lipids (Moore, 2012). Importantly, FAO flux can synergize with lipogenesis to remove fatty acid intermediates. Thus, although mitochondrial FAO may itself contribute to oxidative stress in NAFLD (Sunny et al., 2011), we show here that impaired FAO is also deleterious to the health of the liver, consistent with observations in human NASH (Koliaki et al., 2015). Furthermore, in addition to lipogenesis, abnormal fatty acid uptake, oxidation and secretion can also contribute to fatty liver formation (Donnelly et al., 2005; Lambert et al., 2014; Sunny et al., 2011). Our finding that HDAC3 physiologically suppresses hepatosteatosis in SCAP LKO mice highlights this point and suggests that loss of HDAC3 activity may worsen lipotoxicity not only by promoting lipogenesis, but also by impairing FAO.
The DLKO mice showed that SREBP is dominant over HDAC3 with respect to the expression of genes involved in DNL, while HDAC3 plays a greater role in FAO regulation. From a physiological standpoint, HDAC3 and SCAP are mutually reinforcing, where HDAC3 likely supports SREBP by suppressing lipogenesis and stimulating FAO during inactive periods. During the active phase when mice are eating, HDAC3 is less bound to chromatin (Feng et al., 2011; Sun et al., 2012), supporting an increase in lipogenic gene expression and reducing FAO. Together, our research identifies HDAC3/SCAP as partners in suppressing lipotoxicity.
EXPERIMENTAL PROCEDURES
Mice
Hdac3 floxed mice on a C57Bl/6 background described previously (Feng et al., 2011) were backcrossed to the C57Bl/6J genetic background for at least 3–4 generations. Scap floxed mice previously generated (Matsuda et al., 2001) were backcrossed to the C57Bl/6J genetic background for at least 7–8 generations (T.F. Osborne). Analysis at Jackson Labs reported that the C57Bl/6 Scap and Hdac3 floxed mice were 100% and 84–91% 6J, respectively. Hdac3 and Scap floxed mice were crossed to generate homozygous double floxed Hdac3/Scap (Hdac3f/f/Scapf/f) mice. Analysis at Jackson Labs indicated that the Hdac3f/f/Scapf/f mice were 88% 6J. All mice were housed under a 12 h light and 12 h dark cycle (lights on at 7 a.m. [ZT0] and lights off at 7 p.m. [ZT12]). Unless otherwise stated, we used adult male mice (8 weeks old) with ad lib access to normal chow that were sacrificed 10 days post AAV8 injections at ZT10. AAV8 expressing either GFP (control) or Cre were tail-vein injected at 1.5×1011 genome copies (GC) per mouse. For HA-nSREBP1c expression, C57Bl/6 mice were injected with 1.5×1011 GC AAV8:GFP (Control) or 1.5×1011 GC AAV8:HA-nSREBP1c. For HA-nSREBP1c expression in DLKO, mice were co-injected with either 1.5×1011 GC AAV8:Cre and 1.0×1011 GC AAV8:HA-nSREBP1c, or 1.5×1011 GC AAV8:GFP + 1.0×1011 GC AAV8:Null for control. All genes delivered by AAV8 in this study were under the control of the thyroxine-binding globulin (Tbg) promoter. For fasting studies, mice were restricted access to food starting at ZT10 and sacrificed 24 h later. HFD containing 60% kcal% from fat was purchased from Research Diets Inc. (D12492). All animal care and use procedures followed the guidelines of the Institutional Animal Care and Use Committee of the University of Pennsylvania in accordance with the guidelines of the US National Institutes of Health.
Western blot and gene expression analysis
Tissues were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with phosphatase and protease inhibitors using a tissuelyser and steel beads (Qiagen), and were resolved in 4–12% Bis-Tris NUPAGE gradient gels with MOPS running buffer (ThermoFisher Scientific). Proteins were transferred onto polyvinylidene difluoride membranes and blotted with the indicated antibodies. Nuclear fractionation to determine nSREBP1 levels in HDAC3 LKO livers were performed as previously described (Wan et al., 2011). For RT-qPCR, all data were normalized to 36b4 expression. Additional details on western blots and gene expression procedures are found in Supplemental Experimental Procedures.
DNA constructs and adeno-associated virus
The nSREBP1c transgene gene expressing amino acids 2–427 of mouse SREBP1c was directly cloned from total mouse liver RNA using primers designed for Gibson cloning technology (New England Biolabs). These primers also included the sequence for an N-terminal HA/Flag tag. HA-nSREBP1c[2–427] cDNA was cloned into the pENN-AAV8-Tbg vector (provided by the UPenn Viral Vector Core) and confirmed by sequencing. The Viral Vector Core at Penn Diabetes Research Center generated the AAV8:Tbg-GFP, AAV8:Tbg-Cre, AAV8:Tbg-Null, and AAV8:Tbg-HA-nSREBP1c viruses.
ChIP sequencing and analysis
All ChIPs were performed in parallel as described before (Feng et al., 2011) with a few minor changes. Briefly, mouse livers were dounce homogenized in 1% formaldehyde/PBS solution and incubated for 20 min while rotating at room temperature. Crosslinking was quenched with 1/20 volume of 2.5 M glycine solution for 5 min and the pellets were washed with PBS twice. Pellets were resuspended in ChIP buffer (50 mM HEPES pH 7.5, 155 mM NaCl, 1.1% triton x-100, 0.11% Sodium Deoxylcholate, 0.1% SDS, 1 mM EDTA and protease inhibitors) and sonicated. Around 30 mg liver were used per ChIP with either 10 μg anti-HDAC3 antibody (ab7030, Abcam) or 25 μL bead volume of anti-HA conjugated agarose (A2095, Sigma-Aldrich). Proteins were immunoprecipitated in ChIP buffer, and crosslinks were reversed overnight at 65°C in SDS buffer (50 mM Tri s-HCl pH8, 10 mM EDTA, 1% SDS). DNA was isolated using phenol/chloroform/isoamyl alcohol extractions. Additional details are found in Supplemental Experimental Procedures.
Metabolic profiling, histology and flow cytometry
For triglyceride measurement, livers were homogenized in lysis buffer (140 mM NaCl, 50 mM Tris pH 7.4 and 1% Triton-X100) using a tissuelyser with steal beads (Qiagen). Triglyceride concentration was then measured using the LiquiColor Triglyceride kit following the manufacturer’s protocol (StanBio). Liver TBARS (Cayman Chemical) and glycogen (Biovision Inc.), and serum NEFA (Wako Chemicals USA, Inc.), ALT (Biovision Inc.), insulin (Crystal Chem), and BHB (Stanbio) were measured using commercial kits following manufacturer’s protocol. Blood glucose concentrations were measured with a glucometer (OneTouch) by sampling from the tail. For H&E staining, tissues were harvested and immediately fixed overnight in 10% buffered formalin, dehydrated with 70% ethanol, and embedded. For Oil Red O staining, fixed tissues were snap frozen in HistoPrep(TM) media (Fisher Scientific) and sectioned. The sections were stained with 0.5% Oil Red O in propylene glycerol overnight and then with hematoxylin for 5 sec. Histological procedures were performed by the Penn Digestive Disease Center Morphology Core. A blinded board certified Veterinary Pathologist examined the H&E slides. Details for flow cytometry are found in Supplemental Experimental Procedures.
Metabolomics
Liquid chromatography mass spectrometry (LC-MS) analysis of water-soluble metabolites and lipids from mice liver samples. Frozen liver samples were divided into two portions, for the analysis of water-soluble metabolites and lipids, respectively. About 30 mg of frozen tissue samples were weighed and then pulverized in a CryoMill machine (Retsch, Germany) with stainless ball at liquid nitrogen temperature. Additional details are found in Supplemental Experimental Procedures.
Fatty acid oxidation and uptake in hepatocytes
Primary hepatocytes were isolated as before (Titchenell et al., 2015) and plated in M199 media (ThermoFisher Scientific) supplemented with 10% FBS, 0.1% BSA, 100 nM thyroid hormone (T3), 500 nM dexamethasone, 1 nM insulin, and 1.1 g sodium bicarbonate/500 mL. After the cells attached, the media was replaced with complete DMEM + 100 nM dexamethasone and incubated overnight. The cells were washed 2X with DMEM and serum starved in DMEM for 2 h with or without 100 μM etomoxir. Then the cells were incubated with 125 μM 3H-palmitate conjugated on BSA and 1 mM carnitine for an additional 2 h with or without 100 μM etomoxir. The media was delipidated and 3H20 was measured by scintillation counting. For uptake measurements, the attached cells were washed 2X with PBS and the lipids were extracted using the Folch method and measured in a scintillation counter.
Measurement of de novo lipogenesis in vivo
Levels of newly made palmitate in liver was determined as previously described (Titchenell et al., 2016). Briefly, ad lib fed mice were injected i.p. with deuterated water (20 μL per gram of body weight) and sacrificed 6 hours later at ZT10. Lipids were extracted from the liver and analyzed using gas chromatography-mass spectrometry.
Statistical Analysis
We used an unpaired, two-tailed Student’s t test when two conditions were compared. For three conditions, we used a one-way ANOVA followed by the indicated corrections. Statistical tests were performed using R, MS Excel, or Graphpad Prism 7.0 software. Error bars represent SEM in all figures. A P value < 0.05 was considered significant in all cases.
Supplementary Material
HIGHLIGHTS.
HDAC3 and SCAP/SREBP1 control distinct pathways of lipid metabolism in the liver
Their depletion paralyzes liver lipid metabolism, leading to lipotoxicity and NASH
Lipotoxicity is prevented by activation of lipogenesis and triglyceride synthesis
Lipid storage as triglyceride can be beneficial in liver inflammation and injury
Acknowledgments
We thank T.F. Osborne (Sanford, Burnham, Prebys Institute) for the SCAP floxed mice backcrossed to the C57Bl/6J genetic background. We thank the Viral Vector Core, Functional Genomics Core and Princeton Regional Metabolomics Core of the Penn Diabetes Research Center (P30 DK19525), and the Penn Digestives Disease Center Morphology Core (P30 DK050306). Deuterium-labeled palmitate levels were measured by the IDOM Metabolic Tracer Resource (J. Millar). We thank Q. Chu, Y.H. Foong, W. Teng, A. Angueira and other members of the Lazar Lab for technical support and S. Hackett (Princeton U) for assistance with statistical analysis of lipidomics data. This work was supported by US National Institutes of Health grants R37 DK43806 (M.A.L.), F32 DK108555 (R.P.), R00 DK099443 (Z.S.), F32 DK101175 (P.M.T.), T32 GM008216 (Y.H.K) and the JPB Foundation. M.A.L is on the scientific advisory boards for Pfizer Inc. and Eli Lilly and Company, and M.W. is currently employed at Agios Pharmaceuticals.
Footnotes
ACCESSION NUMBERS
The GEO accession number for all sequencing data is GSE85929.
AUTHOR CONTRIBUTIONS
R.P. and M.A.L. conceived of the hypothesis and designed the experiments. R.P., Y.H.K., P.M.T., D.A.H., M.W., Y.Z., W.L., and E.B. performed the experiments. M.D. provided bioinformatic analysis of ChIP-seq data. R.P., Y.H.K., P.M.T., D.A.H., Z.S., M.W., W.L., J.D.R., and M.A.L. analyzed and interpreted the data. R.P. and M.A.L. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445–451. doi: 10.1586/egh.09.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61:1294–1303. doi: 10.1007/s10620-016-4049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, Ratziu V, Serfaty L, Housset C, Capeau J, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122:2176–2194. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–52. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, Kahraman A, Garrison SP, Zambetti GP, Charlton MR, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooley JJ, Chua EC. Diurnal regulation of lipid metabolism and applications of circadian lipidomics. J Genet Genomics. 2014;41:231–250. doi: 10.1016/j.jgg.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Cotter DG, Ercal B, Huang X, Leid JM, D’Avignon DA, Graham MJ, Dietzen DJ, Brunt EM, Patti GJ, Crawford PA. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J Clin Invest. 2014;124:5175–5190. doi: 10.1172/JCI76388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, James OFW. Steatohepatitis: A tale of two “Hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JAG, Carvalho F, Pearson M, Horton JD, Browning JD, Jones JG, Burgess SC. A high-fat diet suppresses de novo lipogenesis and desaturation but not elongation and triglyceride synthesis in mice. J Lipid Res. 2014;55:2541–2553. doi: 10.1194/jlr.M052308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, Liu XS, Lazar MA. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–1319. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon Tacer K, Rozman D. Nonalcoholic Fatty liver disease: focus on lipoprotein and lipid deregulation. J Lipids. 2011;2011:783976. doi: 10.1155/2011/783976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- Gilardi F, Migliavacca E, Naldi A, Baruchet M, Canella D, Le Martelot G, Guex N, Desvergne B, Delorenzi M, Deplancke B, et al. Genome-Wide Analysis of SREBP1 Activity around the Clock Reveals Its Combined Dependency on Nutrient and Circadian Signals. PLoS Genet. 2014;10:e1004155. doi: 10.1371/journal.pgen.1004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guéraud F, Atalay M, Bresgen N, Cipak A, Eckl PM, Huc L, Jouanin I, Siems W, Uchida K. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res. 2010;44:1098–1124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- Hems DA, Rath EA, Verrinder TR. Fatty acid synthesis in liver and adipose tissue of normal and genetically obese (ob/ob) mice during the 24-hour cycle. Biochem J. 1975;150:167–173. doi: 10.1042/bj1500167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Chen F, Baine AMT, Gardner LB, Nguyen JH. Fulminant liver failure model with hepatic encephalopathy in the mouse. Ann Gastroenterol. 2011;24:294–306. [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Bashmakov Y, Shimomura I, Shimano H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci U S A. 1998a;95:5987–5992. doi: 10.1073/pnas.95.11.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998b;101:2331–2339. doi: 10.1172/JCI2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park ASD, Tao J, Sharma K, Pullman J, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2014;21:37–46. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Haluzik M, Asghar Z, Yau D, Joseph JW, Fernandez AM, Reitman ML, Yakar S, Stannard B, Heron-Milhavet L, et al. Peroxisome proliferator-activated receptor-α agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes. 2003;52:1770–1778. doi: 10.2337/diabetes.52.7.1770. [DOI] [PubMed] [Google Scholar]
- Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel, Rudo WT, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Lam B, Younossi ZM. Treatment options for nonalcoholic fatty liver disease. Therap Adv Gastroenterol. 2010;3:121–137. doi: 10.1177/1756283X09359964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 Is Required for Hepatic Lipid Accumulation in Models of Insulin Resistance. Cell Metab. 2009;10:405–418. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M, Korn BS, Hammer RE, Moon YA, Komuro R, Horton JD, Goldstein JL, Brown MS, Shimomura I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 2001;15:1206–1216. doi: 10.1101/gad.891301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, Stevens RD, Bain JR, Newgard CB, Farese RV, et al. Dissociation of Hepatic Steatosis and Insulin Resistance in Mice Overexpressing DGAT in the Liver. Cell Metab. 2007;6:69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Moon YA, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Brown MS, Goldstein JL, Horton JD. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012;15:240–246. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DD. Nuclear receptors reverse McGarry’s vicious cycle to insulin resistance. Cell Metab. 2012;15:615–622. doi: 10.1016/j.cmet.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016;15:249–274. doi: 10.1038/nrd.2015.3. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA. Nontriglyceride hepatic lipotoxicity: The new paradigm for the pathogenesis of NASH. Curr Gastroenterol Rep. 2010a;12:49–56. doi: 10.1007/s11894-009-0083-6. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology. 2010b;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- Sahebkar A, Chew GT, Watts GF. New peroxisome proliferator-activated receptor agonists: potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin Pharmacother. 2014;15:493–503. doi: 10.1517/14656566.2014.876992. [DOI] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel M, Jonker JT, Schoones J, Lamb H, De Roos A, Pijl H, Smit JWA, Meinders AE, Jazet IM. Ectopic fat and insulin resistance: Pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. 2012;2012 doi: 10.1155/2012/983814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas G, Boren J, Dulloo AG. De novo lipogenesis in metabolic homeostasis: More friend than foe? Mol Metab. 2015;4:367–377. doi: 10.1016/j.molmet.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, Zhang D, Graham MJ, Unterman TG, Shulman GI, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. 2012;18:934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD, et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016;23:1154–1166. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy S, Torres-Gonzalez M, Jump DB. Elevated hepatic fatty acid elongase-5 activity corrects dietary fat-induced hyperglycemia in obese C57BL/6J mice. J Lipid Res. 2010;51:2642–2654. doi: 10.1194/jlr.M006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Leavens KF, Saleh D, Easton RM, Guertin Da, Peterson TR, Kaestner KH, Sabatini DM, Birnbaum MJ. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab. 2011;14:516–527. doi: 10.1016/j.cmet.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Argus JP, Zhu Y, Wilks MQ, Marbois BN, York AG, Kidani Y, Pourzia AL, Akhavan D, Lisiero DN, et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013;73:2850–2862. doi: 10.1158/0008-5472.CAN-13-0382-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Yang L, McCall S, Huang J, Xing XY, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H, Okanoue T, Yoshikawa T. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab Invest. 2010;90:1169–1178. doi: 10.1038/labinvest.2010.75. [DOI] [PubMed] [Google Scholar]
- Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood Pa, Shulman GI. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.