

Abstract
The human c-ErbB2 (HER2) gene is amplified in ~20 % of human breast cancers (BCs), but the protein is overexpressed in ~30% of the cases indicating that multiple different mechanisms contribute to HER2 overexpression in tumors. It has long been used as a molecular marker of BC for sub-categorization for the prediction of prognosis, and determination of therapeutic strategies. In comparison to ER(+) BCs, HER2(+) BCs are more invasive, but the patients respond to monoclonal antibody therapy with trastuzumab or tyrosine kinase inhibitors at least at early stages. To understand the pathophysiology of HER2-driven carcinogenesis and test HER2-targeting therapeutic agents in vivo, numerous mouse models have been created that faithfully reproduce HER2(+) BCs in mice. They include MMTV-neu (active mutant or wild type, rat neu or HER2) models, neu promoter-driven neuNT-transgenic mice, neuNT-knock-in mice at the neu locus, and doxycycline-inducible neuNT-transgenic models. HER2/neu activates the Phosphatidylinositol-3 kinase-AKT-NF-κB pathway to stimulate the mitogenic cyclin D1/Cdk4-Rb-E2F pathway. Of note, overexpression of HER2 also stimulates the cell autonomous Dmp1-Arf-p53 tumor suppressor pathway to quench oncogenic signals to prevent the emergence of cancer cells. Hence tumor development by MMTV-neu mice was dramatically accelerated in mice that lack Dmp1, Arf or p53 with invasion and metastasis. Expressions of neuNT under the endogenous promoter underwent gene amplification, closely recapitulating human HER2(+) BCs. MMTV-HER2 models have been shown to be useful to test humanized monoclonal antibodies to HER2. These mouse models will be useful for the screening of novel therapeutic agents against BCs with HER2 overexpression.
Keywords: HER2/neu, Dmp1, Dmtf1, p53, PI3K, breast cancer, transgenic mice, disease model, target therapy, prevention
Introduction
Breast cancer (BC) is the second most frequent cause of death from cancer and thus is one of the largest public health issues in the United States and the industrialized nations [1, 2]. It is a heterogeneous disorder that can be categorized into 5 different groups - luminal A, luminal B, HER2, basal, and normal types dependent on the expression of markers for hormone-receptors (estrogen receptor [ER] and progesterone receptor [PR]), human ErbB2 [HER2], and Ki67 [3–7]. In a separate thought, intrinsic subtypes of BCs were determined by GeneChip Microarrays of 1800+ genes for categorization [8, 9]. These subtype classifications of BCs have been used to predict patient outcomes and responsiveness to treatments. BCs that are positive for the ER (luminal A/B) are usually responsive to adjuvant hormonal therapy with anti-estrogens and/or aromatase inhibitors, and thus have a favorable prognosis [2]. On the other hand, ER-negative BCs (HER2 and basal-types) are often associated with aggressive disease, including amplification of HER2 or c-Myc oncogenes and mutation of the p53 gene with shorter survival [4–6]. Comprehensive molecular portraits of invasive lobular BC showed that mutations targeting PTEN, TBX3, and FOXA1 were invasive lobular carcinoma-enriched features [10].
The genomic locus for HER2 is amplified in 20 % [11], but the protein is overexpressed in 20–30 % of BCs (21% 3+ and 14% 2+ in our specimens [6]), suggesting additional mechanisms for high HER2 expression exist than gene amplification. HER2/neu overexpression is found in metastatic lesions, and thus is associated with poor prognoses [2, 3, 5, 11]. The use of monoclonal antibody to HER2 (trastuzumab, Herceptin®) has been deployed to treat HER2-positive BC, but the prognosis of such patients is poor since >60% of them experience relapse during the first year due to HER2 modifications, defects in the antibody dependent cellular cytotoxicity or in cell arrest and apoptosis or alterations in HER2 signaling components [12, 13]. The basal-type BC constitutes about 20 % of total BC cases and overlaps with the group of triple-negative BC (TNBC) with worst prognosis since anti-hormonal or monoclonal antibody cannot be performed in these patients [14, 15].
The HER/HER family (HER1-HER4) is made up of four structurally related receptor tyrosine kinases (RTKs) with the EGFR as the founding member of the family [16–18]. Activation of the HER family receptors other than HER2 requires binding of a soluble, growth factor ligand located on the receptor that triggers receptor dimerization, phosphorylation, and activation of downstream pathways to elicit response inside the cell. EGFR (HER1) is activated by growth factor-ligands such as epidermal growth factor (EGF), heparin-binding EGF, amphiregulin, or TGF-α [16, 17]. The product of the human c-ErbB-2 gene (HER2) is a 185-kilodalton glycoprotein with protein-tyrosine kinase activity [19, 20], which is a human version of the rat neu proto-oncogene [21]. However, it is still an orphan receptor to which no specific ligand has been identified [22]. Conversely, HER3 and HER4 are activated by the heregulin or neuregulin family of growth factors (Fig. 1). Each HER molecule, upon activation by growth factor binding, initiates hetero- or homo-dimerization of receptors. This stimulates auto-phosphorylation of the molecule, followed by trans-phosphorylation of the heterodimerization partner. These phosphorylated tyrosine residues recruit adaptor proteins such as Grb2 and the p85 subunit of the PI3K complex, which initiate the activation of several downstream pathways such as protein kinase B (PKB/Akt) and the mitogen activated protein kinase (MAPK) pathways (Fig. 1; refs. 23, 24). Activation of the PI3K and Akt by HER2/neu activates mammalian Target of Rapamycin (mTOR), comprised of two complexes, mTORC1 and mTORC2, which, in turn induce protein synthesis that stimulates cell proliferation, migration, and metabolism [25].
Figure 1. General features of HER2:HER3 signaling involving Ras, Pten, Pi3k, Akt, and NF-κB.
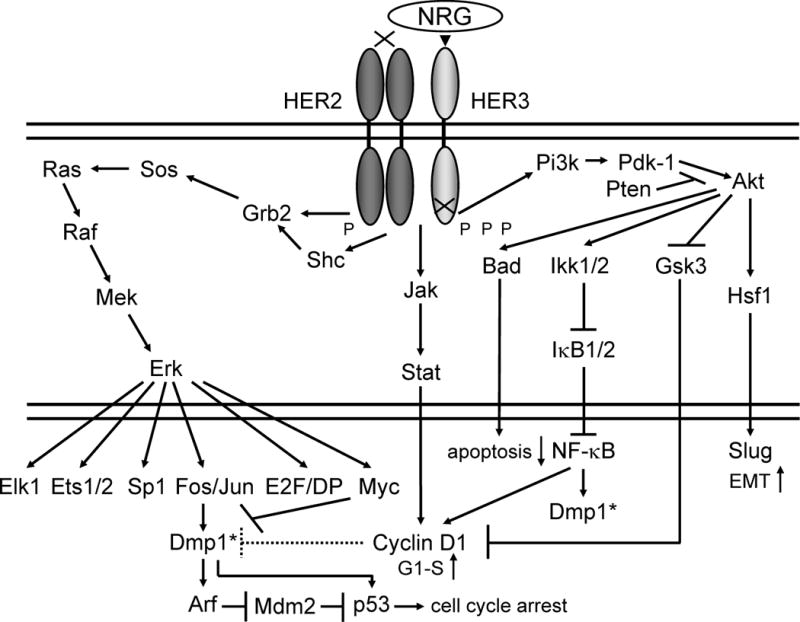
The genomic locus for HER2 is amplified in ~20 % of human BCs, and is associated with aggressive disease with shorter overall and disease-free survival [11]. The protein is overexpressed in ~30 % of BCs in our samples [6] suggesting additional mechanisms for HER2 overexpression exist. The signaling pathways stimulated by the ligand NRG activates ErbB2:ErbB3 (HER2:HER3) heterodimers [17, 23]. Following ligand engagement, HER3 engages and allosterically activates its kinase partner, in this case HER2. Although HER2 molecules make both homo-and hetero-dimers, HER3 do not form homodimers, and does not have protein-tyrosine kinase activity. Phosphorylation of its C-terminal tail leads to recruitment of adapter proteins leading to activation of Pi3k and Ras pathways [23]. Activation of Pi3k leads to phosphorylation of membrane phosphoinositides producing Pip3, which in turn docks the PH domain-containing proteins Pdk1 and Akt [87]. Membrane - bound Akt is phosphorylated and activated by Pdk1. Activated Akt proceeds to phosphorylate a plethora of cellular substrates involved in diverse biological processes. These processes include accelerated G1-S progression as demonstrated by increased cyclin D1 and decreased p27Kip1 levels, and enhanced cell survival through increased phosphorylation of Bad and increased NF-κB levels [17, 23]. Indeed aberrant overexpression of cyclin D1 is frequently observed in human cancers, caused by different mechanisms [88, 89]. The Jak-Stat pathway activation by HER2:HER3 also leads to cell proliferation through cyclin D1 induction. The signaling cascades also stimulate epithelial-mesenchymal transition through stimulation of Hsf1-Slug signaling to cause invasion/metastasis [87]. Thus deregulation of HER2/HER3 can lead to tumorigenesis. Aberrant overexpression of HER2 activates the Dmp1 promoter to stimulate the Arf-Mdm2-p53 self-autonomous tumor surveillance pathway through Pi3k-Akt-NF-κB and Ras-Raf-Mek-Erk-Jun cascades to eliminate incipient cancer cells by cell cycle arrest or apoptosis [37, 42]. D-type cyclins inhibit the transcriptional activity of Dmp1α in a Cdk-independent fashion on promoters without E2F sites [38, 89], but cyclin D1 collaborates with Dmp1α in Ink4a and Arf transactivation [42, 48]. Tumor surveillance by the DMP1-p53 pathway is mediated by DMP1α, but not DMP1β, since the latter is an oncogenic splice variant that blocks the tumor-suppressive activity of DMP1α [90]; it also has p53-independent functions for cell proliferation, and is a novel marker for BC [91, 92]. Dmp1*: these are identical molecules.
The PI3K-Akt signaling has also been linked to the induction of NF-κB (Fig. 1; refs. 23, 26). Two serine threonine IκB kinases (IKK1/2) are responsible for phosphorylating and targeting IκB molecules for degradation. Importantly, both human BC cell lines and primary specimens often show constitutive activation of NF-κB [27], which results in decreased apoptosis and induction of cyclin D1 (Fig. 1), suggesting oncogenic roles of NF-κB in BC development [28].
The transforming potential of HER2/neu has been demonstrated in a number of ways. Both the transforming neu oncogene (neuNT) and the proto-oncogene neu were isolated in the Weinberg’s lab through NIH 3T3 transformation assays [21]. The level of expression was shown to be critical for transformation by HER2 [29]. Long terminal repeat - based expression vectors with neu were proven to achieve transformation in NIH 3T3 cells [30]. The effect of overexpression in murine mammary epithelium was first analyzed through the creation of MMTV-neuNT transgenic mice [FVB/N-Tg(MMTVneu)202Mul/J; ref. 31]. Then a different model was created to overexpress the proto oncogenic form of neu under the control of the MMTV promoter [32]. Subsequently a mouse model for BC induced by amplification and overexpression of the activated neu gene driven by the neu promoter was generated [33]. A knock-in model for neuNT was also created at the mouse neu locus [34]. More recently, a doxycycline (dox)-inducible model for neuNT overexpression has been established [35]. Finally, MMTV-driven overexpression of human ErbB2 (HER2) was generated to test the therapeutic effects of monoclonal antibodies to HER2 since the rat neu protein is not recognized by trastuzumab [36]. The details for these different HER2/neu-transgenic models are discussed in this review. These mouse models will be useful to screen drugs that could be effective for novel therapy for human cancers overexpressing HER2, and thus are extremely important tools for future target therapies.
Dmp1, a cyclin D-binding myb-like protein 1, directly binds and activates the Arf promoter, thereby stimulates p53 to quench oncogenic Ras signaling [37–48; see 49–51 for Arf reviews; 52–54 for Dmp1 reviews]. To demonstrate the tumor suppressive role for Dmp1 in oncogenic HER2/neu signaling, we analyzed the responsiveness of Dmp1 and Arf promoters to HER2/neu [37]. We also crossed MMTV-neuNT mice with Dmp1-knockout mice in FVB/N to demonstrate the role of Dmp1 in HER2/neu-driven carcinogenesis [37]. Then we established MMTV-FlagDmp1α mice and crossed them with MMTV-neuNT to directly demonstrate the tumor suppressive potential of Dmp1α in vivo [55]. The results from these studies are also reviewed here.
Oncogenic mouse models for HER2/neu
MMTV-mutant neu (neuNT) and MMTV-neu (wild type) models
The cDNA clones for the neu oncogene, which is frequently activated in neuro- and glioblastomas of BDIX rats, possessed a valine to glutamic acid substitution in its transmembrane domain of the epidermal growth factor receptor EGFR [56, 57]. This mutation resulted in the constitutive activation of the receptor in the absence of ligand [58, 59]. c-ErbB2 plays critical roles in ductal morphogenesis of the mammary glands as demonstrated by the results of gene-knockout studies [60, 61]. Although the transmembrane point mutation has not been detected in primary human BCs overexpressing HER2, recent studies have detected an aberrant alternative splice isoform in human BCs lacking 16 amino acids at exon 20 [62, 63; reviewed in 18]. The first transgenic mouse expressing oncogenic rat neu (neuNT) was reported [31]. Unlike the stochastic occurrence of solitary mammary tumors in transgenic mice bearing the MMTV-c-myc or v-Ha-ras oncogenes, transgenic mice uniformly expressing the MMTV-neuNT gene developed multi-focal mammary tumors at the median age of 197 days in FVB strain (non-parous) with frequent potential to metastasize to the lung [37]. Because these tumors arise synchronously and are polyclonal in origin, expression of the activated c-neu oncogene appears to be sufficient to induce malignant transformation in this tissue in a single step (Table 1).
Table 1. Mouse models for human breast cancer overexpressing neu, neuNT, HER2 or Dmp1α.
The name of the transgene in each mouse model, mean latency of mammary tumor development, pathological findings, potential clinical uses, and references are shown.
| Transgene | Mouse model | Mean latency of tumor development | Pathology/genetics | Use | References |
|---|---|---|---|---|---|
| neu/ErbB2 | MMTV-neuNT (mutant) | Dmp1+/+: 197 days (NP), 100% | Invasive adca | S, PP, TKI | 31 |
| MMTV-neuNT; Dmp1KO | Dmp1+/−: 162 days (NP) | Aggressive adca, multi-focal, larger tumor, highly invasive/metastatic | 37 | ||
| Dmp1−/−: 154 days (NP) | Aggressive adca, multi-focal, larger tumor, highly invasive/metastatic | ||||
| MMTV-neu (wild type) | 205 (#202) – 367 (#510) days (MP) | Focal Adca, lung metastasis after long latency (#1) | S, PP, TKI | 32, 65 | |
| MMTV-neu; Ink4a/Arf+/− | 240 days in neu TG, 259 days in neu; Ink4a/Arf+/− (nm) | Increased S Phase, higher cyclin D1 and Ki67; increased apoptosis | 76 | ||
| MMTV-neu; WAP-p53-172H | 240 days in neu TG, 154 days in neu;p53-172H (nm) | Higher grade adca, anaplastic and aneuploid; increased apoptosis | 72 | ||
| neu promoter - neuNT | 14.5 months (MP) | Solid nests of anaplastic cells, micropapillary adca Hyperplastic mammary glands producing lactoferrin and lipid droplets |
S, PP, TKI | 33 | |
| neuNT knock-in at the neu locus | 8 (TM1) - 17 (TM7/8) months (nm) | Accelerated lobuloalveolar development and formation of focal mammary tumors after a long latency (#2) | S, PP, TKI | 34, 67, 68 | |
| TetO- neuNT (dox-inducible) | 7 weeks with doxycycline (nm) | Multiple invasive mammary adca with metastasis that regresses to a clinically undetectable state following transgene de-induction | S, PP, TKI | 35 | |
| MMTV-HER2* (wild type) | 28 weeks (nm) | Adca with areas of solid, tubular, and papillary growth cellular polymorphism with mitosis and metastasis (#3) |
S, PP, TKI, MoAb | 71 | |
| MMTV-HER2Δ16* | 17 weeks (nm) | Tubular adca consisting of three zones: an outer zone (pale cells), an intermediate zone (darker fusiform cells), and an inner zone (pinkish cytoplasm); ER(−) PR(−) PCNA(+) E-CAD(+) | S, PP, TKI, MoAb | 66 | |
| Dmp1α | MMTV-FLAG Dmp1α | No tumor development in 24 months (NP) | No tumor | S, PP | 55 |
| MMTV-FLAG Dmp1α;neuNT | Delayed mammary tumor development (NP) | Adca; smaller tumors; Flag Dmp1α(+) cells undergo apoptosis, Ki67(−) | |||
|
| |||||
| neuNT: mutant rat neu HER2*: human ErbB2 NP: Nulliparous MP: Multiparous nm: parous status, not mentioned |
Adca: Adenocarcinoma #1: in-frame deletions in the extracellular region of neu close to the transmembrane domain #2: gene amplification and protein overexpression of neu #3: in-frame deletion in the juxtamembrane region of HER2 |
S: signaling pathways PP: pathophysiology TKI: tyrosine kinase inhibitor screening MoAb: monoclonal antibody screening |
64 for a review of MMTV models | ||
Mice strains bearing the wild type ErbB2 allele under the control of the MMTV promoter have also been established (MMTV-neu in Table 1) [32; reviewed in 64]. In contrast to the rapid tumor progression observed in several transgenic strains carrying the activated neu transgene, expression of unactivated neu in the mammary epithelium resulted in the development of focal mammary tumors with longer latency than activated neu (7–12 months dependent on the strain, Table 1). Overexpression of neu in these mammary tumors was associated with elevated intrinsic tyrosine kinase activity of neu and the de novo tyrosine phosphorylation of several cellular proteins. Interestingly, many of the tumor-bearing transgenic mice developed secondary metastatic lesions in the lung. These observations suggest that overexpression of the unactivated neu protein induces metastatic disease after long latency [32]. The same group later reported that that activation of neu in these transgenic mice occurred through somatic mutations located within the transgene itself [65]. Sequence analyses of these mutations revealed that the tumors contain in-frame deletions of 7 to 12 amino acids in the extracellular region proximal to the transmembrane domain [65]. Since transmembrane deletion of HER2 is frequently found in human BCs [18, 66], these mice are naturally-occurring models for human BC with HER2 overexpression.
Wild type neu and neuNT expression under the control of the neu promoter - a more physiological model
Although most human BCs are not associated with pregnancy, the promoters that have been used in mouse models of mammary tumors (MMTV and whey acidic protein [WAP] promoters) require the induction of pregnancy to reach high levels of transcriptional activity [64]. Of note, HER2/neu is expressed in the virgin, pregnant, lactating and regressing mammary gland of the mouse [33], making it difficult for the transgenic promoters, such as those of MMTV or WAP, to accurately model the consequence of amplification of the HER2 gene in human BC throughout the course of murine development. To overcome this problem, transgenic mice containing the 1.2 kb upstream sequence of the mouse neu gene fused to either the normal or transforming (Val664Glu) rat neu gene has been established [33]. With only moderate overexpression of wild type neu, the virgin mammary tree underwent lobulo-alveolar hyperplasia throughout the gland [33]. NeuNT-transgenic mice developed solid nests of anaplastic cells, micropapillary adenocarcinoma, or hyperplastic mammary glands producing lactoferrin and lipid droplets (Table 1). The proliferative phenotype was enhanced once the mouse had undergone multiple rounds of pregnancy (mean tumor latency of 14 months in MPs; Table 1) and regression. Rather than reverting to a near virgin state, the gland continued to produce milk proteins and lipids long after weaning. This may be due to the fact that neu, under the direction of the transgenic neu promoter, is expressed evenly throughout the gland, as can be seen by its proliferative effects in a mammary gland whole mount [33]. In summary, this is an improved mouse model for human BC where HER2 is amplified and overexpressed since the neu gene is driven by the neu promoter.
The neuNT knock-in mouse model
To test whether expression of activated neu under the control of the endogenous promoter in the mammary gland contributes to tumor development, knock-in mice for activated neu were developed [34]. They showed accelerated lobulo-alveolar development and formation of focal mammary tumors after a long latency period (mean latency, 8–17 months, Table 1). Thus, the expression of neuNT under the endogenous neu promoter was not sufficient for to overt mammary carcinogenesis. In comparison to mammary tumors from MMTV-neuNT mice, the neuNT knock-in mice showed a significantly low metastasis rate [31, 34]. Of note, all tumors isolated from these mice had amplified copies (2–22 copies) of the neuNT allele and expressed highly elevated levels of the neu protein. In summary, like human HER2-positive BCs, mammary tumorigenesis in this mouse model requires gene amplification and overexpression of neu [34, 67, 68].
Doxycycline (dox)-inducible mouse mammary tumor models for neuNT
To determine the impact of tumor progression on the reversibility of neu-induced tumorigenesis, a tetracycline-regulatory system that conditionally expresses activated neu in the mammary epithelium of transgenic mice has been developed (Table 1; ref. 35). When induced with dox, bitransgenic MMTV-rtTA;TetO-neuNT mice developed multiple invasive mammary carcinomas, which regressed to a clinically undetectable state following transgene de-induction, indicating that that neuNT-initiated tumorigenesis was reversible. Strikingly, extensive lung metastases arising from neuNT-induced mammary tumors also rapidly and fully regressed following the disappearance of neu expression. However, despite the near universal dependence of both primary and metastatic tumors on neuNT-transgene expression, most animals bearing fully regressed tumors ultimately experienced recurrent disease that progressed to a neu-independent state [35]. These findings highlight the importance of elucidation of the mechanisms by which tumor cells escape from their dependence on the initial oncogenic pathways for the treatment of relapsed cancer.
The AMP-activated protein kinase-related molecule Hunk is overexpressed in aggressive subsets of human breast, ovarian, and colon cancers, and is required for metastasis of c-Myc-induced mammary tumors [69]. By crossing Hunk−/− mice with MMTV-rtTA;TetO-neuNT mice, Hunk was shown to be essential for tumor development driven by HER2/neu [69]. Most recently, the same group demonstrated the role of Notch signaling in tumor recurrence from dormant residual cells, which are ideal drug targets in BC [70]. Thus this dox-inducible/de-inducible neuNT-transgenic model will be very useful in the analysis of oncogenic signaling cascades as well as for the screening of drugs targeting HER2/neu.
MMTV-driven transgenic mouse models employing human ErbB2 (HER2)
Transgenic mouse models offer the advantage of having immune system components that may be important in the action of antibodies, but are lacking in immunodeficient hosts. Although numerous transgenic mice have been developed using rat neu, they cannot be used to test the efficacy of Herceptin® (trastuzumab), a monoclonal antibody to HER2 since the rat neu protein is not recognized by the antibody [36]. To overcome this problem, three different groups made MMTV-driven transgenic mice using the human ErbB2 (HER2) gene [36, 66, 71]. In a MMTV-human ErbB2 model, 76 % of female transgenics developed asynchronous mammary tumors with an average latency of 28 weeks (Table 1; ref. 71). At 24 weeks of age, the founder mouse had a primary mammary tumor of 1 mm3 that ultimately grew to span two adjacent mammary glands. Later primary mammary tumors (rapidly growing adenocarcinomas) developed in a stochastic manner in the transgenic progeny. Pulmonary metastases were found in 23 % of the female transgenics with high HER2 expression. Sequencing of HER2 in mammary tumors revealed an in-frame 15-bp deletion in the HER2 juxtamembranous region [71]. This deletion resulted in the repositioning of two cysteines (634 and 641) to within two amino acids of each other, potentially affecting cysteine-mediated dimerization. These findings are consistent with the published data from the MMTV-neu-transgenic model (wild type; ref. 32) that showed the presence of in-frame deletions confined to the juxta-membranous region of the neu extracellular domain in mammary tumors via activation of intrinsic tyrosine kinase activity by the neu protein [65].
A splice isoform of the HER2 receptor that lacks exon 20 (Δ16HER2) is expressed in many HER2-positive BCs [18], but the impact of Δ16HER2 on tumor pathobiology and its therapeutic response remains uncertain. Therefore MMTV-ErBB2 (Δ16HER2) mice have been created to provide genetic evidence that expression of Δ16HER2 was sufficient to accelerate mammary tumorigenesis [66]. A comparative analysis of spontaneous mammary tumor development assay showed that the tumor-free survival (TFS) was shortened from 45 weeks (wild type HER2) to 17 weeks (Δ16HER2). Clinically, HER2-positive BCs from patients who received trastuzumab exhibited a positive correlation in Δ16HER2 and pSRC abundance, consistent with the mouse genetic results [66]. Moreover, patients expressing high pSRC or an activated Δ16HER2 metagene were found to receive the greatest benefit from trastuzumab treatment suggesting that the Δ16HER2 signaling axis is a signature for decreased risk of relapse after trastuzumab treatment [66].
Compound HER2/neu-transgenic mice with inactivation of the Dmp1-Arf-p53 tumor suppressor pathway
MMTV-neu;WAP-p53-172H bitransgenic mice
As many as 37 % of tumors arising in MMTV-neu (wild type) mice have missense mutations in p53 [72]. To directly test the cooperativity between wild type neu and mutant p53 in mammary tumorigenesis, they crossed MMTV-neu (line N#202; ref. 30) and WAP-p53-172H [70]. The tumor latency was shortened from 240 days (MMTV-neu) to 154 days in bitransgenic mice, indicating strong cooperativity (Table 1) [72]. Tumors arising in WAP-p53-172H; MMTV-neu bitransgenic mice were anaplastic, aneuploid and exhibited increased apoptosis, in contrast to tumors arising in p53-null mice. They recapitulated the two common genetic lesions that occur in human BC, and have shown that p53 mutation is an important cooperating event in HER2/neu-mediated oncogenesis [72].
MMTV-neu; Ink4a/Arf+/− mice
p16INK4a is deleted in 40–60 % of human BC cell lines, and p16INK4a inactivation by DNA methylation occurs in 20–30 % of human BCs [73, 74]. To test the effects of Ink4a/Arf inactivation in ErbB2-induced breast carcinogenesis, MMTV-neu (wild type) mice [32] were crossed Ink4a/Arf-null mice [75]. Compared with MMTV-neu; Ink4a/Arf+/− mice, MMTV-neu; Ink4a/Arfwt mammary tumors showed increased p16Ink4a, reduced Ki67 expression, reduced cyclin D1 protein, and decreased apoptosis with no significant change in the risk of developing mammary tumors. Their study demonstrated the contribution of Ink4a/Arf-heterozygosity in mammary tumorigenesis [76].
MMTV-neuNT; Dmp1 knockout mice
HER2 overexpression stimulates cell growth in p53-mutated cells while it inhibits cell proliferation in those with wild type p53, but the molecular mechanism remained poorly understood. We conducted a study to demonstrate the roles of Dmp1 and Arf in HER2/neu signaling and breast carcinogenesis [37]. The Dmp1 promoter was activated by HER2/neu through the PI3K-Akt-NF-κB pathway, which in turn stimulated Arf transcription. Both Dmp1 and p53 were induced in pre-malignant lesions from MMTV-neu mice, suggesting that the Dmp1-p53 tumor suppressor pathway plays a crucial role in quenching aberrant HER2 signaling in vivo. To study the cooperation between Dmp1-loss and HER2/neu overexpression, we crossed MMTV-neuNT mice [31] with Dmp1-deficient mice [40] in FVB/N (Table 1; ref. 37). HER2/neu-induced mammary tumor development was significantly accelerated from 197 days to 162 days in Dmp1+/− and 154 days in Dmp1−/− mice (p < 0.0001), with no statistically significant differences between Dmp1+/− and Dmp1−/− (p = 0.23; Fig. 2A; ref. 37). The tumors from Dmp1+/− mice expressed the Dmp1 protein, showing haploid insufficiency (37, 77). Molecular analyses of tumors revealed that overexpression of the Ink4a/Arf repressor Tbx2/Pokemon [52] was found in >50 % of wild type MMTV-neuNT mammary tumors while the deficiency of Arf or overexpression of Mdm2 was rare. Tumors from Dmp1+/− or Dmp1−/−; neuNT mice showed significant downregulation of p53 and p21Cip1/WAF1, showing p53 inactivity, with more aggressive phenotypes (i.e. increased invasion and metastasis) than those without Dmp1 deletion. Notably, endogenous hDMP1 mRNA decreased when HER2 was depleted in human BC cells, suggesting the existence of HER2-DMP1 axis in human cells as well [37]. Interestingly, the mouse Dmp1 gene was hemizygously deleted in ~50 % of neu mammary tumors from Dmp1+/+;neuNT mice with significant downregulation of the Dmp1 protein [37]. neuNT-induced mammary tumors are different from human BCs in that the Ink4a/Arf, Mdm2, or p53 locus is not frequently involved. Conversely, we found frequent overexpression of Ink4a/Arf repressors in neuNT tumors [37]. Further studies will be required to reveal how these Ink4a/Arf repressors collaborate with Dmp1-loss in breast (or mammary) carcinoma development.
Figure 2. Tumor development in MMTV-neuNT mice.
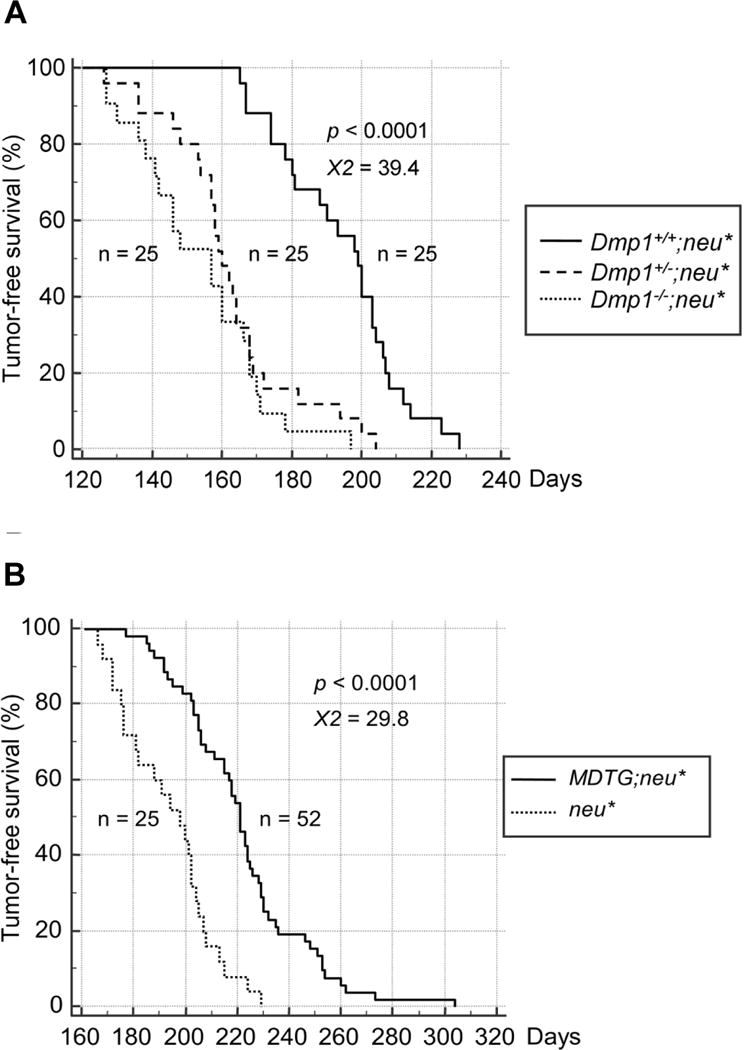
A. TFS curves for nulliparous Dmp1+/+ (straight line), Dmp1+/− (discontinuous line), and Dmp1−/− (dotted line); MMTV-neuNT compound transgenic mice are shown [37]. The median TFS was significantly shortened from 197 days in Dmp1+/+ mice to 154 and 162 days in Dmp1−/− and Dmp1+/− mice, respectively (both p < 0.0001), with little difference between the two cohorts (p = 0.23).
B. TFS curves of MMTV-FLAG-Dmp1α (MDTG);MMTV-neuNT and single MMTV-neuNT mice [55]. TFS curves for nulliparous MDTG;neuNT (straight line, n = 52) and MMTV-neuNT (dotted line, n = 25) females are shown. The median TFS was significantly prolonged (p < 0.0001) from 197 to 221 days with decreased burden of mammary tumors in MDTG;neuNT mice in comparison to neuNT mice.
Tumor prevention model with MMTV-FLAG Dmp1α
The study shown above suggests a pivotal role of Dmp1α in quenching hyperproliferative signals from HER2 to the Arf-p53 pathway as a safety mechanism to prevent breast carcinogenesis [37]. To directly demonstrate the role of Dmp1α in preventing HER2/neu-driven mammary tumorigenesis, we established Flag-Dmp1α transgenic mice under the control of the MMTV promoter (MMTV-FlagDmp1α transgenic: MDTG) [55]. All mice were viable, but exhibited poorly developed mammary glands with markedly reduced milk production; hence > 50% of the parous females were unable to support the lives of new-born pups. The mammary glands of the MDTG mice had very low Ki67 expression, but high levels of Arf, Ink4a, p53, and p21Cip1, markers of senescence [55]. None of the transgenic females, regardless of their parous status, exhibited any malignant transformation within two-year observation period (Table 1). In all three strains of MDTG;neuNT mice, TFS was significantly prolonged (from 197 to 221 days by median) with decreased weight of mammary tumors [55]. The extent of TFS elongation was dependent on the copy number of the Flag-Dmp1α transgene - i.e. the higher, the more protection of tumors [55].
Notwithstanding the tumor-suppressive properties of Dmp1α, mammary tumors arose later in MDTG;neuNT mice, irrespective of their transgenic strain. Immunohistochemical studies showed that Flag-Dmp1α and Ki67 protein expressions were mutually exclusive [55] indicating that Flag-Dmp1α-positive areas were not proliferating, and thereby preventing tumor development in vivo. Moreover, we saw significant overlap between Flag-Dmp1α-positive areas and cleaved caspase 3 expressing areas in MDTG;neuNT tumors [55] indicating that Flag-Dmp1α induces apoptotic cell death as well. In summary, our research provides direct evidence that Dmp1α has a tumor suppressive activity in vivo [55]. Future studies should focus on the molecular mechanisms of how the Dmp1α protein is downregulated in the mammary glands of MDTG;neuNT mice. Molecules that specifically activate the Dmp1 promoter or stabilize the Dmp1α protein may thus be effective to induce regression of tumor growth in vivo.
Discussion and conclusive remarks
We have reviewed different mouse models of human BC overexpressing wild type or mutant HER2/neu driven by MMTV, neu, or dox-inducible promoters. These mice have been used to analyze the biological behavior of mammary tumors overexpressing HER2/neu (tumor growth, angiogenesis/metastasis), oncogenic signaling pathways, oncogene - tumor suppressor gene interactions [78, 79], and to test therapeutic efficacies of HER(2) tyrosine kinase inhibitors to treat the disease. Accumulating data suggests that the mutant neu is more oncogenic than the wild type, and usage of the neu promoter for neu expression provides more physiological models than MMTV-driven ones. MMTV-HER2 (wild type, Δ16) models have been created since trastuzumab does not recognize the rat neu protein. These mouse models will be useful for the screening of novel humanized monoclonal antibodies that specifically target HER2.
Since human BCs have been categorized into 5 different groups to predict the prognosis and determine therapeutic modalities, it is reasonable to compare gene expression in human BCs and that in gene-engineered mouse models (GEMs). Surprisingly, it has been reported that the MMTV-neu model represents the luminal subtypes more than HER2-enriched tumors [80]. Conserved with humans, murine NeuEx tumors highly expressed several tyrosine kinase pathway-related gene-signatures, namely EGFR and HER2, which would be expected from the nature of the neu/ERBB2 transgene [81, 82]. Hence MMTV-neu tumors regressed with the dual tyrosine kinase inhibitor lapatinib [83]. It has been reported that mice with conditional expression of neuNT under the endogenous promoter undergo gene amplification [34], closely recapitulating human HER2+BC [67]. Consistent with the non-invasive nature of mammary tumors induced by expression of neuNT under the endogenous promoter, these tumors expressed markers of a highly differentiated state, which are closely linked to HER2 and often co-amplified in non-invasive ductal BCs [68, 84], indicating that it is a faithful model of human BC with HER2 amplification.
In addition to GEMs employing HER2/neu, patient-derived xenografts (PDXs) generated from fresh specimens recapitulate the diversity of BC and reflect the histopathology, the tumor behavior, and metastatic properties of the original tumor [85, 86]. There are several advantages of PDX models in examination of therapeutic responses to drugs. They are i) one can use human tumor tissue retaining the complexity of genetic/epigenetic abnormalities, which can be applied in the development of individualized molecular therapeutics and ii) stroma from the human tumor microenvironment can be included in the xenograft to mimic the tumor microenvironment. The disadvantages are i) PDX tumor models are expensive and technically challenging, and ii) it is necessary to use immune-deficient mice in which the lymphocyte-mediated response to the tumor is lost; thus we cannot precisely evaluate the tumor growth in immune-competent individuals.
Conversely, the advantages of the GEMs are i) specific genetic abnormalities present in human tumors can be reproduced in an inducible/de-inducible manner in specific tissue, ii) the mice are immune-competent, such that the tumor microenvironment can be reproduced, and iii) multiple genetic interactions can be tested just by crossing GEMs with different genetic backgrounds. The major disadvantage of GEM is that the tumors are mouse tumors, but not human versions where more than four genetic alterations are present; hence it requires multiple crossbreedings to reproduce human conditions. Since both mouse models have advantages and disadvantages, they will complement each other to predict BC patients’ responses to novel therapies.
Acknowledgments
We thank all other members of Dr. Inoue’s lab for sharing unpublished research data.
Financial Support: K. Inoue was supported by NIH/NCI 2R01CA106314, ACS RSG-07-207-01-MGO, and KG080179.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Literature Cited
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Ross JS, Hortobagyi GN. Molecular Oncology of Breast Cancer. Publishers; Sudbury, Massachusetts: 2005. [Google Scholar]
- 3.Taneja P, Maglic D, Kai F, et al. Classical and novel prognostic markers for breast cancer and their clinical significance. Clin Med Oncol. 2010;4:15–34. doi: 10.4137/cmo.s4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kittaneh M, Montero AJ, Glück S. Molecular profiling for breast cancer: a comprehensive review. Biomark Cancer. 2013;5:61–70. doi: 10.4137/BIC.S9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patani N, Martin LA, Dowsett M. Biomarkers for the clinical management of breast cancer: international perspective. Int J Cancer. 2013;133:1–13. doi: 10.1002/ijc.27997. [DOI] [PubMed] [Google Scholar]
- 6.Maglic D, Zhu S, Fry EA, et al. Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast cancer. Oncogene. 2013;32:4120–9. doi: 10.1038/onc.2012.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inwald EC, Koller M, Klinkhammer-Schalke M, et al. 4-IHC classification of breast cancer subtypes in a large cohort of a clinical cancer registry: use in clinical routine for therapeutic decisions and its effect on survival. Breast Cancer Res Treat. 2015;153:647–58. doi: 10.1007/s10549-015-3572-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 9.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciriello G, Gatza ML, Beck AH, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163:506–19. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 12.Patel TA, Dave B, Rodriguez AA, et al. Dual HER2 blockade: preclinical and clinical data. Breast Cancer Res. 2014;16:419. doi: 10.1186/s13058-014-0419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madrid-Paredes A, Cañadas-Garre M, Sánchez-Pozo A, et al. Non-HER2 signaling pathways activated in resistance to anti-HER2 therapy in breast cancer. Breast Cancer Res Treat. 2015;153:493–505. doi: 10.1007/s10549-015-3578-x. [DOI] [PubMed] [Google Scholar]
- 14.Guestini F, McNamara KM, Ishida T, et al. Triple negative breast cancer chemosensitivity and chemoresistance: current advances in biomarkers identification. Expert Opin Ther Tar. 2016;20:705–20. doi: 10.1517/14728222.2016.1125469. [DOI] [PubMed] [Google Scholar]
- 15.Palma G, Frasci G, Chirico A, et al. Triple negative breast cancer: looking for the missing link between biology and treatments. Oncotarget. 2015;6:26560–74. doi: 10.18632/oncotarget.5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mujoo K, Choi B-K, Huang Z, et al. Regulation of ERBB3/HER3 signaling in cancer. Oncotarget. 2014;5:10222–36. doi: 10.18632/oncotarget.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roskoski R., Jr The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Inoue K, Fry EA. Aberrant Splicing of Estrogen Receptor, HER2, and CD44 genes in breast cancer. Genet Epigenet. 2015;7:19–32. doi: 10.4137/GEG.S35500. eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto T, Ikawa S, Akiyama T, et al. Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature. 1986;319:230–4. doi: 10.1038/319230a0. [DOI] [PubMed] [Google Scholar]
- 20.Akiyama T, Sudo C, Ogawara H, et al. The product of the human c-erbB-2 gene: a 185-kilodalton glycoprotein with tyrosine kinase activity. Science. 1986;232:1644–6. doi: 10.1126/science.3012781. [DOI] [PubMed] [Google Scholar]
- 21.Hung MC, Schechter AL, Chavray PY, et al. Molecular cloning of the neu gene: absence of gross structural alteration in oncogenic alleles. Proc Natl Acad Sci USA. 1986;83:261–4. doi: 10.1073/pnas.83.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moasser MM. The oncogene HER2; Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469–87. doi: 10.1038/sj.onc.1210477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Citri A, Skaria KB, Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 24.Wilks ST. Potential of overcoming resistance to HER2-targeted therapies through the PI3K/Akt/mTOR pathway. Breast. 2015;24:548–55. doi: 10.1016/j.breast.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Xu K, Liu P, Wei W. mTOR signaling in tumorigenesis. Biochim Biophys Acta. 2014;1846:638–54. doi: 10.1016/j.bbcan.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biswas DK, Iglehart JD. Linkage between EGFR family receptors and nuclear factor kappaB (NF-kappaB) signaling in breast cancer. J Cell Physiol. 2006;209:645–52. doi: 10.1002/jcp.20785. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Nag SA, Zhang R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr Med Chem. 2015;22:264–89. doi: 10.2174/0929867321666141106124315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hudziak RM, Schlessinger J, Ullrich A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci USA. 1987;84:7159–63. doi: 10.1073/pnas.84.20.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Marco E, Pierce JH, Knicley CL, et al. Transformation of NIH 3T3 cells by overexpression of the normal coding sequence of the rat neu gene. Mol Cell Biol. 1990;10:3247–52. doi: 10.1128/mcb.10.6.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller WJ, Sinn E, Pattengale PK, et al. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–15. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 32.Guy CT, Webster MA, Schaller M, et al. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–82. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinstein EJ, Kitsberg DI, Leder P. A mouse model for breast cancer induced by amplification and overexpression of the neu promoter and transgene. Mol Med. 2000;6:4–16. [PMC free article] [PubMed] [Google Scholar]
- 34.Andrechek ER, Hardy WR, Siegel PM, et al. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci USA. 2000;97:3444–9. doi: 10.1073/pnas.050408497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moody SE, Sarkisian CJ, Hahn KT, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451–61. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- 36.Schwall RH, Hollingshead P, Erickson S, et al. Herceptin-sensitivity of HER2-transgenic mouse mammary tumors. Breast Cancer Res. 2003;5(Suppl 1):14. [Google Scholar]
- 37.Taneja P, Maglic D, Kai F, et al. Critical roles of DMP1 in human epidermal growth factor receptor 2/neu-Arf-p53 signaling and breast cancer development. Cancer Res. 2010;70:9084–94. doi: 10.1158/0008-5472.CAN-10-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol. 1998;18:1590–600. doi: 10.1128/mcb.18.3.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inoue K, Roussel MF, Sherr CJ. Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci USA. 1999;96:3993–8. doi: 10.1073/pnas.96.7.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue K, Wen R, Rehg JE, et al. Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev. 2000;14:1797–809. [PMC free article] [PubMed] [Google Scholar]
- 41.Inoue K, Zindy F, Randle DH, et al. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001;15:2934–9. doi: 10.1101/gad.929901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sreeramaneni R, Chaudhry A, McMahon M, et al. Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol. 2005;25:220–32. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mallakin A, Taneja P, Matise LA, et al. Expression of Dmp1 in specific differentiated, nonproliferating cells and its repression by E2Fs. Oncogene. 2006;25:7703–13. doi: 10.1038/sj.onc.1209750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taneja P, Mallakin A, Matise LA, et al. Repression of Dmp1 and Arf promoter by anthracyclins: critical roles of the NF-κB subunit p65. Oncogene. 2007;26:7457–66. doi: 10.1038/sj.onc.1210568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mallakin A, Sugiyama T, Taneja P, et al. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell. 2007;12:381–94. doi: 10.1016/j.ccr.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mallakin A, Sugiyama T, Kai F, et al. The Arf-inducing transcription factor Dmp1 encodes transcriptional activator of amphiregulin, thrombospondin-1, JunB and Egr1. Int J Cancer. 2010;126:1403–16. doi: 10.1002/ijc.24938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frazier DP, Kendig RD, Kai F, et al. Dmp1 physically interacts with p53 and positively regulates p53′s stabilization, nuclear localization, and function. Cancer Res. 2012;72:1740–50. doi: 10.1158/0008-5472.CAN-11-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu S, Mott RT, Fry EA, et al. Cooperation between cyclin D1 expression and Dmp1-loss in breast cancer. Am J Pathol. 2013;183:1339–50. doi: 10.1016/j.ajpath.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozenne P, Eymin B, Brambilla E, et al. The ARF tumor suppressor: structure, functions and status in cancer. Int J Cancer. 2010;127:2239–47. doi: 10.1002/ijc.25511. [DOI] [PubMed] [Google Scholar]
- 50.Maggi LB, Jr, Winkeler CL, Miceli AP, et al. ARF tumor suppression in the nucleolus. Biochim Biophys Acta. 2014;1842:831–9. doi: 10.1016/j.bbadis.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- 52.Inoue K, Mallakin A, Frazier DP. Dmp1 and tumor suppression. Oncogene. 2007;26:4329–35. doi: 10.1038/sj.onc.1210226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inoue K, Fry EA, Frazier DP. Transcription factors that interact with p53 and Mdm2. Int J Cancer. 2016;138:1577–85. doi: 10.1002/ijc.29663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inoue K, Fry EA. Aberrant splicing of the DMP1-INK4a/ARF-MDM2-p53 pathway in cancer. Int J Cancer. 2016;139:33–41. doi: 10.1002/ijc.30003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fry EA, Taneja P, Maglic D, et al. Dmp1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS One. 2013;8:e77870. doi: 10.1371/journal.pone.0077870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45:649–57. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- 57.Bargmann CI, Hung MC, Weinberg RA. The neu oncogene encodes an epidermal growth factor receptor-related protein. Nature. 1986;319:226–30. doi: 10.1038/319226a0. [DOI] [PubMed] [Google Scholar]
- 58.Stern DF, Heffernan PA, Weinberg RA. p185, a product of the neu proto-oncogene, is a receptor like protein associated with tyrosine kinase activity. Mol Cell Biol. 1986;6:1729–40. doi: 10.1128/mcb.6.5.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie Y, Li K, Hung MC. Tyrosine phosphorylation of Shc proteins and formation of Shc/Grb2 complex correlate to the transformation of NIH3T3 cells mediated by the point-mutation activated neu. Oncogene. 1995;10:2409–13. [PubMed] [Google Scholar]
- 60.Jackson-Fisher AJ, Bellinger G, Ramabhadran R, et al. ErbB2 is required for ductal morphogenesis of the mammary gland. Proc Natl Acad Sci USA. 2004;101:17138–43. doi: 10.1073/pnas.0407057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andrechek ER, White D, Muller WJ. Targeted disruption of ErbB2/Neu in the mammary epithelium results in impaired ductal outgrowth. Oncogene. 2005 Jan 27;24:932–7. doi: 10.1038/sj.onc.1208230. [DOI] [PubMed] [Google Scholar]
- 62.Kwong KY, Hung MC. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog. 1998;23:62–8. doi: 10.1002/(sici)1098-2744(199810)23:2<62::aid-mc2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 63.Siegel PM, Ryan ED, Cardiff RD, et al. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999;18:2149–64. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taneja P, Frazier DP, Kendig RD, et al. MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert Rev Mol Diagn. 2008;9:423–40. doi: 10.1586/ERM.09.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siegel PM, Dankort DL, Hardy WR, et al. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol Cell Biol. 1994;14:7068–77. doi: 10.1128/mcb.14.11.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Castagnoli L, Iezzi M, Ghedini GC, et al. Activated d16HER2 homodimers and SRC kinase mediate optimal efficacy for trastuzumab. Cancer Res. 2014;74:6248–59. doi: 10.1158/0008-5472.CAN-14-0983. [DOI] [PubMed] [Google Scholar]
- 67.Montagna C, Andrechek ER, Padilla-Nash H, et al. Centrosome abnormalities, recurring deletions of chromosome 4 and genomic amplification of HER2/neu define mouse mammary gland adenocarcinomas induced by mutant HER2/neu. Oncogene. 2002;21:890–8. doi: 10.1038/sj.onc.1205146. [DOI] [PubMed] [Google Scholar]
- 68.Andrechek ER, Laing MA, Girgis-Gabardo AA, et al. Gene expression profiling of neu-induced mammary tumors from transgenic mice reveals genetic and morphological similarities to ErbB2-expressing human breast cancers. Cancer Res. 2003;63:4920–6. [PubMed] [Google Scholar]
- 69.Yeh ES, Yang TW, Jung JJ, et al. Hunk is required for HER2/neu-induced mammary tumorigenesis. J Clin Invest. 2011;121:866–79. doi: 10.1172/JCI42928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abravanel DL, Belka GK, Pan TC, et al. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J Clin Invest. 2015;125:2484–96. doi: 10.1172/JCI74883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Finkle D, Quan ZR, Asghari V, et al. HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin Cancer Res. 2004;10:2499–511. doi: 10.1158/1078-0432.ccr-03-0448. [DOI] [PubMed] [Google Scholar]
- 72.Li B, Rosen JM, McMenamin-Balano J, et al. neu/ERBB2 cooperates with p53-172H during mammary tumorigenesis in transgenic mice. Mol Cell Biol. 1997;17:3155–63. doi: 10.1128/mcb.17.6.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–77. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 74.Silva J, Silva JM, Domínguez G, et al. Concomitant expression of p16INK4a and p14ARF in primary breast cancer and analysis of inactivation mechanisms. J Pathol. 2003;199:289–97. doi: 10.1002/path.1297. [DOI] [PubMed] [Google Scholar]
- 75.Serrano M, Lee H, Chin L, et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 76.D’Amico M, Wu K, Di Vizio D, et al. The role of Ink4a/Arf in ErbB2 mammary gland tumorigenesis. Cancer Res. 2003;63:3395–402. [PubMed] [Google Scholar]
- 77.Inoue K, Fry EA. A book chapter in ‘Advances in Genetics Research’. Nova Science Publishers, Inc; 2016. Haplo-insufficient tumor suppressor genes. [Google Scholar]
- 78.Taneja P, Zhu S, Maglic D, et al. Transgenic and knockout mice models to reveal the functions of tumor suppressor genes. Clin Med Oncol. 2011;5:235–7. doi: 10.4137/CMO.S7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Inoue K, Fry EA, Taneja P. Recent progress in mouse models for tumor suppressor genes and its implications in human cancer. Clin Med Oncol. 2013;7:103–22. doi: 10.4137/CMO.S10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Landis MD, Seachrist DD, Montañez-Wiscovich ME, et al. Gene expression profiling of cancer progression reveals intrinsic regulation of transforming growth factor-beta signaling in ErbB2/Neu-induced tumors from transgenic mice. Oncogene. 2005;24:5173–90. doi: 10.1038/sj.onc.1208712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pfefferle AD, Herschkowitz JI, Usary J, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013;14:R125. doi: 10.1186/gb-2013-14-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roberts PJ, Usary JE, Darr DB, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18:5290–03. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hollern DP, Andrechek ER. A genomic analysis of mouse models of breast cancer reveals molecular features of mouse models and relationships to human breast cancer. Breast Cancer Res. 2014;16:R59. doi: 10.1186/bcr3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whittle JR, Lewis MT, Lindeman GJ, et al. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. doi: 10.1186/s13058-015-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Richmond A, Su Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Model Mech. 2008;1:78–82. doi: 10.1242/dmm.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dey N, Williams C, Leyland-Jones B, et al. A critical role for HER3 in HER2-amplified and non-amplified breast cancers: function of a kinase-dead RTK. Am J Transl Res. 2015;7:733–50. [PMC free article] [PubMed] [Google Scholar]
- 88.Aggarwal P, Vaites LP, Kim JK, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010;18:329–40. doi: 10.1016/j.ccr.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Inoue K, Fry EA. Aberrant expression of Cyclin D1 in cancer. Signal Transduction Insights. 2015;4:1–13. doi: 10.4137/STI.S30306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tschan MP, Federzoni EA, Haimovici A, et al. Human DMTF1β antagonizes DMTF1α regulation of the p14ARF tumor suppressor and promotes cellular proliferation. Biochim Biophys Acta. 2015;1849:1198–208. doi: 10.1016/j.bbagrm.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maglic D, Stovall DB, Cline JM, et al. DMP1β, a splice isoform of the tumor suppressor DMP1 locus, induces proliferation and progression of breast cancer. J Pathol. 2015;236:90–102. doi: 10.1002/path.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Inoue K, Fry EA. Novel biomarkers for breast cancer. Biomark Cancer. 2016;8:1–18. doi: 10.4137/BIC.S38394. [DOI] [PMC free article] [PubMed] [Google Scholar]