

Abstract
Acetylation of proteins as a post-translational modification is gaining rapid acceptance as a cellular control mechanism on par with other protein modification mechanisms such as phosphorylation and ubiquitination. Through genetic manipulations and evolving proteomic technologies, identification and consequences of transcription factor acetylation is beginning to emerge. In this review, we summarize the field and discuss newly unfolding mechanisms and consequences of transcription factor acetylation in normal and stressed hearts.
Introduction
Global acetylation of proteins has been observed in many human diseases. [1, 2]. Increased acetylation of key enzymes/proteins and transcription factors have been observed in diabetes associated accelerated atherosclerosis [3], hypertension [4, 5], diabetic cardiomyopathy [6], coronary artery disease (CAD) [7], arrhythmia [8], pulmonary arterial hypertension (PAH) [9, 10], and heart failure [11]. In the sections to follow we will review the mechanisms of transcription factor acetylation and its consequences in the heart.
Overview of Acetylation
With several thousand proteins non-nuclear proteins now identified as being K-acetylated [12, 13], this protein post-translational modification is gaining rapid acceptance as a cellular control mechanism on par with other protein modification mechanisms such as phosphorylation and ubiquitination [14]. Proteomic studies and bioinformatic analysis revealed acetylation of metabolic enzymes and transcription factors in various cell types [12, 15–17]. Acetylation of nucleosomal histone tails provides a critical mechanism for epigenetic control of gene expression. Acetyl groups are transferred to lysine residues by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs). Lysine acetylation also creates binding sites for bromodomain-containing proteins such as bromodomain and extraterminal (BET) proteins. HDAC proteins are grouped into four classes based on function and DNA sequence similarity. Mammalian HDACs are encoded by distinct genes and are classified on the basis of similarity to yeast transcriptional repressors. Class I HDACs (HDACs 1, 2, 3 and 8) are related to yeast RPD3, class II HDACs (HDACs 4, 5, 6, 9 and 10) to yeast HDA1, and class III HDACs (SirT1–7) to yeast Sir2 (Figure 1). Class II HDACs are further divided into two subclasses, IIa (HDACs 4, 5, 7 and 9) and IIb (HDACs 6 and 10). HDAC11 falls into a fourth class [18] (Figure 1). While zinc ion is required for catalysis in classes I, II and IV HDACs, class III HDACs (sirtuins) require nicotinamide adenine dinucleotide (NAD+) as a co-factor for catalytic activity. So far, seven sirtuin family proteins (SIRT1–7) have been identified as mammalian SIR2 orthologs[19]. Figure 1 shows the subcellular compartment for each of the HDACs.
Figure 1.
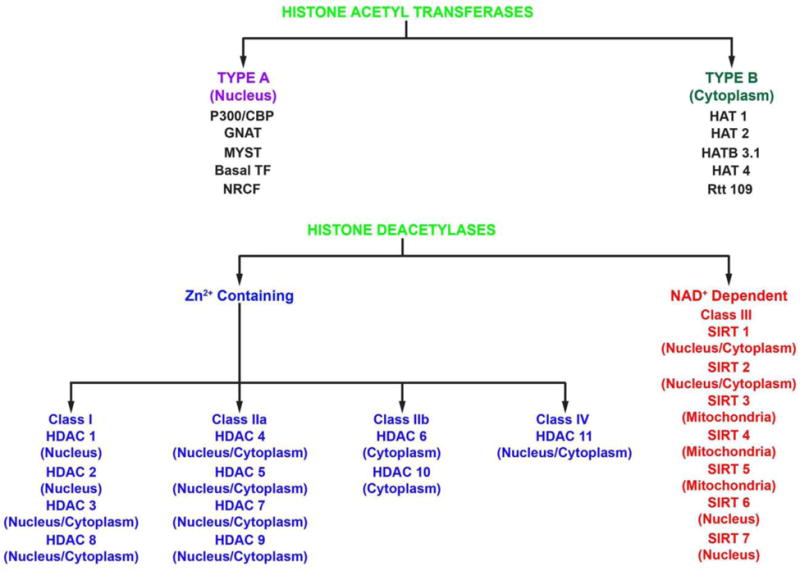
Figure 1 illustrates the various histone acetyltransferases (top) and Zinc dependent and NAD+ dependent histone deacetylases and their subcellular localization in cells (bottom).
Classical studies by Allfrey et al. using radiolabeled acetate identified that histones in nuclei can be acetylated and that these acetylated histones were less inhibitory for RNA polymerase [20]. Several decades later histone HATs and HDACs were cloned and linked to the regulation of gene expression on chromatinized templates [21, 22]. Our current understanding is that there are several acetyl-CoA dependent HATs and HDACs that function to regulate all DNA-templated processes primarily by reversible acetylation of histone lysine residues [23–25] and that site-specific acetylation is sufficient to alter nucleosome dynamics and chromatin folding [26, 27]. In addition, acetylated lysines on histones can function as ‘epitopes’ for the recruitment of acetyl-lysine binding domains (e.g., bromodomains) that are contained within large protein complexes such as histone acetyltransferases, methyltransferases, transcriptional coactivators, and ATP-dependent chromatin-remodelers [28]. The acetylation of p53 and tubulin were early examples that protein acetylation extends beyond histone proteins [29, 30]. The observation that several deacetylases were localized outside the nucleus spurred further interest in exploring protein acetylation as a broader phenomenon [31]. Shortly studies thereafter showed that acetylation renders the acetyl-CoA synthetases inactive, while deacetylation restores full of these acetyl-CoA synthetases [32–34]. It should be noted that studies have shown contradicting outcome on the effect of increased acetylation on fatty acid β oxidation and glycolytic enzymes [35–38]. An accompanying review article by Fukushima and Lopaschuk specifically discusses the impact of acetylation on fatty acid metabolism in the heart [39].
Collectively, these results demonstrated the existence of functionally-relevant non-histone targets, which lead to the use of unbiased acetyl-proteomic discovery methods to identify and characterize other acetylation events. Immunoprecipitation with an antibody against acetyl-lysine followed by liquid chromatography coupled mass spectrometry (LC-MS) was the method of choice. These acetylation studies lead to discovery of high abundant metabolic proteins. With the advances in proteomics methodology, subsequent studies indicated that the acetylation is widespread, including acetylation of transcription factors. In the section to follow, we will briefly provide an overview of HDACs in the heart prior to review acetylation of transcription factors.
Histone acetyltransferases
HATs are divided into two types (Figure 1), nuclear (type A HATs) and cytoplasmic (type B HATs). Type A HATs are localized in the nuclear and are involved in the regulation of gene expression through acetylation of histones. They contain a bromodomain, which helps them recognize and bind to acetylated lysine residues on histone substrates. The nuclear Type A HATs, transcription related, are further subclassified into five families: (1) General Control Nonderepressible (GNAT) related acetyltransferases family represented by GCN5, p300/CREB-binding protein (CBP) associated factor (PCAF), and Elongator complex protein 3 (ELP3); (2) p300/CBP family represented by p300 and CBP; (3) MYST family of histone acetyl transferases (MOZ, YBF2/SAS3, SAS2, and TIP60 protein) family, which consists of MYST1 (HMOF, males absent on the first), MYST2 (HBO1, histone acetyltransferase binding to ORC), MYST3 (MOZ, monocytic leukemia zinc finger), MYST4 (MORF, monocytic leukemia zinc finger protein-related factor), and TIP60 (tat interacting protein 60 kDa); (4) basal TF family; TFIIIC (Transcription Factor IIIC), TAF1, and (5) nuclear receptor cofactors (NRCF) family, steroid receptor coactivator (SRC), ACTR/NCOA3 (nuclear receptor coactivator 3).
Type B HATs, localized in the cytoplasm, are responsible for acetylating newly synthesized histones prior to their assembly into nucleosomes. Type B HATs, lack a bromodomain, recognize newly synthesized unacetylated core histones. HAT1, HAT2 [40], Rtt109 [41], HatB3.1 [42], and HAT4 [43] are the Type B HATs that have been studied so far.
Among the HATs in muscle, the p300 and the closely related coactivator, CREB-binding protein (CBP), play critical roles in physiological and pathological growth of cardiac myocytes. The p300 knockout mice die between days 9 and 11.5 of gestation. In addition, these mice show reduced expression of muscle structural proteins such as β-myosin heavy chain (MHC) and α-actinin, as well as cardiac structural defects and reduced trabeculation [44]. Further, a gene knock-in study demonstrated the importance of the HAT domain of p300 in heart formation [45]. Emerging studies have linked GCN5 to metabolic changes in hearts from high fat diet fed mice [38] and neonatal mice [46].
Histone deacetylases
HATs and HDACs play an important role in regulating cardiac hypertrophy [47–50]. The activity of NAD+-dependent class III HDACs (Sirtuins) are prominent in the heart and vasculature [49–57]. Sirtuins deacetylate both histones and nonhistone targets in mice and humans [53–57]. Mice deficient in SIRT1 exhibit developmental abnormalities in the heart and only infrequently survive postnatally [57–59]. Sirtuins favor longevity and cell survival and uniquely couple with the cellular metabolic state [57, 59]. Subcellular localization studies initially identified SIRT1 as a nuclear protein [60]. In the heart, the nuclear and cytoplasmic localization of SIRT1 was found to be regulated during development and stress conditions [61, 62]. Impaired nucleocytoplasmic shuttling and increased cytoplasmic SIRT1 during ischemic stress was observed in aged hearts [62]. Studies performed with a cardiac-specific SIRT1 transgenic mouse model showed that SIRT1 exhibits hormesis [63], i.e depending on the magnitude of SIRT1 expression, it can be either beneficial or harmful. Sadoshima and coworkers have shown that moderate SIRT1 overexpression (2.5- to 7.5-fold) protects against the age-dependent increase in cardiac hypertrophy and in I/R [63–65]. SIRT3 inhibits apoptosis by deacetylating Ku70 to sequester Bcl2-associated X protein away from mitochondria [66]. SIRT4, SIRT5, SIRT6, and SIRT7 also exhibit antiapoptotic effects in the heart [67–70]. Hearts from mice deficient in SIRT3 are not protected from ischemia-reperfusion injury [71]. Interestingly, ischemia-reperfsuion injury did not alter the patterns or levels of increased protein acetylation in adult SIRT3−/− compared to adult non-transgenic littermate hearts [71]. Mice with cardiomyocyte specific deletion of SIRT3 exhibited increased vulnerability to hypertrophy when subjected to transaortic constriction [72]. Importantly, SIRT3 deletion lead to increased acetylation of cyclophilin D at lysine 166 and impaired miotochondrial permeability pore opening [72]. Although SIRT5 and SIRT7 protects from inflammatory cardiomyopathy [70], their roles in protecting hearts from ischemia-reperfusion injury remain to be investigated.
Among the class I HDACs, HDAC 2 and 3 are well studied in the heart. Studies have shown that HDAC2 promotes cardiac hypertrophy [73], in part by undergoing acetylation by p300/CBP associated factor [74]. Cardiac-specific deletion of HDAC3 in mice leads to cardiac hypertrophy and excessive myocardial lipid accumulation [75]. The catalytic activity HDAC8, was shown to be elevated in hypertension-induced cardiac hypertrophy [76]. However, the function of HDAC8 in the heart remains unknown. Among Class II HDACs, HDAC6 is well studied in the heart. HDAC6 contributes to structural and functional remodeling of atrial myocytes [77], and its activity is elevated in multiple models of cardiac hypertrophy [78, 79]. HDAC10, as well as the lone class IV HDAC, HDAC11, remain poorly characterized, in part due to the inability to monitor their catalytic activity [80]. In the next section we will review mechanisms and consequences of transcription factor acetylation the heart.
Direct acetylation of transcription factors in heart
Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α)
Peroxisome proliferator-activated receptor γ coactivator 1α(PGC-1α) was originally identified as a transcriptional coactivator of the nuclear receptor PPARγ [81]. Subsequent studies have established that PGC-1α interacts with several other transcription factors, including PPARα, glucocorticoid receptor (GR), hepatic nuclear factor 4α (HNF-4α), estrogen receptor related α (ERRα) and FOXO1 [82–84]. By binding and modulating the activity of these different transcription factors, PGC-1α regulates expression of several genes involved in metabolic pathways, such as fatty acid oxidation, gluconeogenesis, glycolysis and fatty acid synthesis. Several studies have shown that acetylation of PGC-1α represses its ability to function as a transcriptional coactivator [38, 85–87]. Alrob et al [38] demonstrated increased acetylation of PGC-1α along with increased GCN5 acetyltransferase activity in the hearts of obese high fat diet fed mice. They showed that SIRT1, a key activator of PGC-1α, [38], expression was unchanged in hearts from high fat diet fed mice and low fat diet fed mice. Interestingly, they observed significant increases in SIRT6 expression in hearts of high fat diet fed mice. Since SIRT6 can deacetylate and activate GCN5 to increase PGC-1α acetylation [88], they opined that their data in high fat diet fed mice hearts were consistent with GCN5 acetyltransferase as a key regulator of PGC-1α acetylation [89]. Unexpectedly, they showed that increased PGC-1α expression and PGC-1α acetylation was dissociated from the regulation of fatty acid oxidation in the high fat diet fed mice hearts [38].
Recent studies by Fukushima et al showed that acetylation of PGC-1α significantly decreases post-birth and was linked to increases in PPARα but not with the protein levels related to mitochondrial biogenesis [46]. Furthermore, they showed an age-dependent increase in (a) the overall acetylation of cardiac proteins along with an increase in mitochondrial acetyltransferase, GCN5L1, and (b) acetylation of the fatty acid β-oxidative enzymes β-HAD and LCAD along with increased fatty acid oxidation rates. Biphasic changes in SIRT1, SIRT6, and GCN5L1 suggests a complex mechanism regulating PGC-1α acetylation, PPARα, and fatty acid metabolism in neonatal and young hearts. Comprehensive transcriptional and metabolic changes driven PGC-1α acetylation in hearts under various pathological conditions are under active investigation.
Nuclear Factor kappa-light-chain-enhancer of activated B cells (NFκB)
A growing appreciation for NF-κB as a central mediator of various cardiac pathologies has led to the notion that complete inhibition of this pathway will be beneficial in the setting of stress and heart disease [90–93]. In addition to phosphorylation, NF-κB subunits are subject to additional posttranslational modifications. Acetylation has emerged as a significant component of NF-κB signaling linked to the regulation of subunit DNA binding, IκB binding and transactivation potential [94–97]. To date, three NF-κB family members, p50, p65 and p52, have been identified as targets of histone acetylases such as p300/CBP and PCAF [95, 98–103]. Of the three, p65 has been most extensively studied. K122, 123, 218, 218, 221, and 310 have been identified as the key acetylation sites on p65 [104].
Histone lysine acetylation has been associated with induction of gene expression by proinflammatory agents in endothelial cells and vascular smooth muscle cells [105–110]. A complex of NF-kB and cAMP response element-binding protein with HATs including p300/CBP, steroid receptor co-activator-1, and pCAF was required for increased expression of inflammatory genes [105–109, 111]. Increased histone lysine acetylation and increased NF-kB driven gene induction was observed in monocytes obtained from T1D and T2D patients [112, 113].
Resveratrol a known activator of SIRT-1 was found to attenuate cardiac hypertrophy and oxidative stress in Sprague Dawley (SD) rats fed with fructose. These fructose fed rats had increased NOX activity, ROS production, increased activity of NFkB along with acetylation of p65 subunit of NFkB and Histone 3 (H3). Resveratrol improved SIRT-1 actvity, deacetylated NFkB and H3 and decreased ROS, thereby preventing diabetes induced cardiac hypertrophy [114]. Acetylation of NFkB in the lysine 310 position and acetylation of H3 in lysine 9 position in the NFkB binding region has been observed [115, 116]. Functional consequences of NFkB acetylation in hearts are under active investigation.
Early Growth Response-1 (Egr1)
The hypoxia responsive transcription factor Egr-1 [117], is increased in models of cardiac pathology. Expression of Egr-1 has been previously linked to several aspects of cardiovascular pathology including intimal thickening following acute vascular injury [118], cardiac hypertrophy [119], atherosclerosis [120] and angiogenesis [121]. Doxorubicin induced cardiomyopathy is also mediated by Egr-1 [122] and targeting Egr-1 reduces the pathological effects of acute myocardial infarction in rats [123]. Our studies have reported a novel mechanism linking glucose metabolism to acetylation of Egr-1, leading to proinflammatory and prothrombotic responses in diabetic atherosclerosis [3]. The acetylation was indicated to occur at the consensus KDKK region in the Egr-1 protein [124]. We generated mutants of the Egr-1 in which the “K” was mutated to “A.” In vitro acetylation of the mutant and WT Egr-1 (KDKK) using p300 showed acetylation in the WT Egr-1 and the single mutant (ADKK) but not in the double (ADAK) or triple (ADAA) mutants of Egr-1. We established that glucose flux via polyol pathway reduced NAD+/NADH ratio, leading to reduction in NAD+ dependent SIRT1 activity in endothelial cells. As a consequence of SIRT1 activity reduction, we observed increased acetylation of Egr-1 in endothelial cells from mice models of diabetic atherosclerosis. In cardiomyocytes Egr1 acetylation has been linked to calcium regulation. Kaesneci et al [117] showed that in H9c2 cells calsequestrin (CSQ) expression was reduced in Egr-1-expressing cells. Using in vivo and in vitro chromatin immunoprecipitation they showed Egr-1 binding to the CSQ2 promoter. Inhibition of protein acetylation reduced CSQ expression, thus indicating that protein acetylation is important in CSQ repression [117]. Furthermore, they linked reduction in CSQ protein with abnormal calcium dynamics. Taken together, these studies set the stage for detailed investigations to address in vivo acetylation driven mechanisms of Egr1 acetylation and its cardiovascular consequences in normal and stressed hearts.
Hypoxia-Inducible Factor 1α (HIF 1α)
The transcription factor HIF-1 has an essential role in the maintenance of oxygen homeostasis in mammalian organisms [125–129]. HIF-1 exists as an α/β heterodimer, the activation of which depends on stabilization of an oxygen-dependent degradation domain of the α subunit by the ubiquitin-proteasome pathway [130, 131]. Reduced O2 availability in tissue leads to increased activity of HIF-1, which coordinates adaptive responses through the transcriptional activation of hundreds of target genes [132]. Hif1a−/− mouse embryos, which are homozygous for a knockout allele at the locus encoding HIF-1α, die with cardiac malformations and vascular regression [133].
Lysine acetylation regulates HIF-1 protein stability and function by distinct mechanisms. p300, a component of the HIF-1 transcriptional complex, positively regulates the transactivation of HIF-1. Studies by Geng et al [134] show that p300 increases HIF-1α protein acetylation and stability. They showed that (a) p300 specifically acetylates HIF1α at Lys-709, which increases the protein stability and decreases polyubiquitination in both normoxia and hypoxia, and (b). HDAC1 disrupted HIF1α-p300 interaction. Studies using K709A mutant expressing cancer cells showed that this mutant was transcriptionally more active and less sensitive to hypoxia-induced growth arrest than the cells containing the HIF1α wild-type. These data demonstrated that HIF1α-p300 interaction stabilizes HIF1α via Lys-709 acetylation.
Another mechanism of HIF 1α stabilization was demonstrated by Joo et al [135]. In their studies, it was shown that SIRT1 stabilized HIF-1α via direct binding and deacetylation during hypoxia. SIRT1 depletion or inactivation led to reduced hypoxic HIF-1α accumulation, accompanied by an increase in HIF-1α acetylation. Yoon et al [136] showed that HIF-1α K674 and HIF-2α K741 are acetylated by PCAF and CBP, respectively, but are deacetylated commonly by SIRT1. In this study, SIRT1-mediated accumulation of HIF-1α protein led to increased expression of HIF-1α target genes, including VEGF, GLUT1 and MMP2, and ultimate promotion of cancer cell invasion [135]. These findings imply that hypoxic HIF-1α stabilization requires SIRT1 activation.
While the above two studies in cancer cell showed, contrasting findings on HIF-1α stabilization was published by Lim et al [137]. Authors showed that SIRT1 inactivated HIF-1α by blocking p300 recruitment and consequently repressed HIF-1 target genes. In this study HIF-1α was acetylated at Lys674, SIRT1 binding to HIF-1α lead to deacetylation of this lysine residue. During hypoxia, SIRT1 was downregulated, which which allowed the acetylation and activation of HIF-1α [137]. Transcriptional activity of HIF-1α is largely determined by transactivating domain C-TAD. Studies by Lando et al has shown that C-TAD recruits p300/CBP to facilitate transcription, and that this C-TAD is inhibited by FIH in normoxia [138]. Based on the studies of Lando et al, Lim et al speculated that the acetylation of HIF-1α at Lys674 leads to derepression of C-TAD and increased transcriptional activity both in cells and in vivo.
These contrasting findings have led to comprehensive investigations on the impact of specific lysine reidue (Lys 674, Lys 709) acetylation on transcriptional activation in normoxic and hypoxic cells and tissue including hearts.
Signal Transducer and Activator of Transcription (STATs)
The STAT family contains seven members, STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6 [139, 140], all of which are expressed in the heart. Murine studies showed that STAT3 deletion is embryonically lethal [139, 140]. Hearts from mice with a cardiac-specific knockout of STAT3 have larger infarcts after I/R, increased apoptosis, and increased mortality [141]. Furthrmore, deletion of STAT3 in cardiomyocytes renders hearts more susceptible to inflammatory damage [142]. STAT3 is a pleiotropic transcription factor that is activated by the phosphorylation of tyrosine 705 in response to many cytokines and growth factors. Unphosphorylated STAT3 (U-STAT3) is also a potent transcription factor. Dasgupta et al [143] showed using mass spectroscopic analysis that U-STAT3 is acetylated on Lys-685, and the integrity of Lys-685 is required for the expression of most U-STAT3-dependent genes. They found only a minor role for Lys-685 in gene expression induced in response to tyrosine-phosphorylated STAT3. Importantly, they showed using cellular and molecular approaches that U-STAT3 plays an important role in angiotensin II-induced gene expression and in the consequent development of cardiac hypertrophy. While the role of p300 in acetylating U-STAT3 was demonstrated in this study, specific deacetylases that ensure deacetylation of U-STAT3 remains to be determined.
Forkhead box proteins (FoxOs)
FoxO proteins are a family of transcription factors whose activity can be modulated by phosphorylation, acetylation, ubiquitination, and glycosylation. Acetylation sites of human FoxOs have been characterized and are well described in comprehensive review articles [144, 145]. Briefly, the acetylation sites for each of the human FoxOs are as follows: FoxO1a (K245, K265, K248, K262, K274, K294, K559), FoxO3a (K203, K242, K245, K259, K271, K290, K569), FoxO4 (K186, K189, K215, K237, K407), and FoxO6 (K173, K176, K190, K202, K229). Reversible lysine acetylation is accomplished by the action of histone acetyltransferases and deacetylases: CBP, p300 and p300/CBP-associated factor (PCAF) acetylate FoxOs, using acetyl-CoA as co-substrate, whereas enzymes of the sirtuin family (SIRT1, SIRT2 in some instances) catalyze deacetylation of FoxOs [56, 57, 146–150]. Acetylation sites in human FoxO proteins surround a consensus site for Akt-induced serine phosphorylation within the nuclear localization signal (NLS) motif [145]. This NLS motif is well conserved in all human FoxOs. Acetylation has been shown to result in both stimulation and inhibition of the transcriptional activity of FoxOs, depending on the examined FoxO isoforms and their binding partners such as other transcription factors and transcriptional co-activators, the FoxO target genes and the cell types used in the studies [56, 57, 87, 146–152]. The molecular mechanisms underlying those discrepancies are currently being addressed.
Hariharan et al [153] investigated the role of FoxOs and its posttranslational modification in mediating starvation-induced autophagy in heart. Starvation in cardiomyocytes was induced by glucose deprivation They showed that starvation increased autophagic flux and upregulation of SIRT1 and FoxO1 in cardiomyocytes and that both SIRT1 and FoxO1 were required for starvation induced autophagy. They showed that starvation increased deacetylation of FoxO1, and SIRT1 was essential for deacetylation of FoxO1. Delving into mechanisms they overexpressed FoxO1(3A/LXXAA), a mutant FoxO1 which cannot interact with SIRT1or p300, and observed increased acetylation of FoxO1 and inhibition of starvation induced autophagy. Furthermore, their studies revealed that FoxO1 driven expression of Rab7, is a key component mediating FoxO1-induced increases in autophagic flux. In vivo studies in mice with cardiac-specific overexpression of FoxO1(3A/LXXAA) and those with cardiac-specific homozygous deletion of FoxO1 (c-FoxO1(−/−))showed that cardiac function was reduced and autophagy was inhibited in these hearts [153]. This study linked SIRT1-mediated deacetylation of FoxO1 and upregulation of Rab7 in hearts to autophagic flux.
Sin et al [154] investigated the effects of long-term treatment of SIRT1 activator resveratrol on cardiac function and FoxO1-associated pro-apoptotic signaling in senescent mice. They observed that aging significantly reduced the deacetylase activity and that this reduction was accompanied by increased acetylation of the FoxO1 transcription factor and transactivation of its target, pro-apoptotic Bim. Furthermore, their data showed thatresveratrol restored SIRT1 activity and suppressed elevations of Foxo1 acetylation, Bim and pro-apoptotic signaling in the aged heart. This study linked SIRT1 activity and FoxO1 acetylation to pro-apoptotic signaling in the aged heart.
FoxO1 nuclear localization has been shown to essential for regulating pyruvate dehydrogenase kinase-4 (PDK4) in the rat cardiomyocytes [155]. Puthanveetil et al showed that dexamethasone increased nuclear FoxO1, as well as SIRT1-FoxO1 interaction, decreased FoxO1 acetylation with consequent increases in PDK4 expression in cardiomyocytes. Importantly, this study linked FoxO1 acetylation changes to increased glucose oxidation in cardiomyocytes.
Taken together these studies indicate acetylation of FoxOs and consequent regulation of autophagy, glucose metabolism, and apoptosis in hearts.
GATA binding protein 4 (GATA4)
GATA4 is a critical transcription factor for proper mammalian cardiac development and essential for survival of the embryo. Bnip3 is a hypoxia-regulated member of the Bcl-2 family of proteins that is implicated in apoptosis, programmed necrosis, autophagy and mitophagy. Bnip3 is induced in the heart by ischemia and pressure-overload, and may contribute to cardiomyopathy and heart failure. Studies by Thompson et al [156], showed novel activity of Bnip3 that is independent of mitochondrial function and programmed cell death. Using cultured cardiomyocytes and transgenic mice overexpressing Bnip3 in the heart, they demonstrated novel activity of Bnip3 that involves binding and activation of p300-acetyltransferase with downstream effects on transcription and histone acetylation. Importantly, cell culture date revealed that Bnip3 bound and activated the acetyltransferase p300 driving increased acetylation of histones and the transcription factor GATA4. Morphological changes conferred by Bnip3 overexpression were significantly blocked by curcumin treatment or knockdown of GATA4. In their studies, mice overexpressing Bnip3 mice exhibited age-dependent ventricular dilation and dilated cardiomyopathy. Inhibition of p300 with curcumin partially prevented cardiac dysfunction in the Bnip3 transgenic mice. The results suggest that GATA4 acetylation by Bnip3 regulates cardiac gene expression and function.
Nuclear factor erythroid 2-related factor (Nrf2)
The transcription factor Nrf2, an oxidative stress response modifier, induces transcription of a variety of genes via binding to the antioxidant response element (ARE) in target gene promoters [157–159]. Nrf2-dependent induction of cytoprotective proteins enable cells to combat oxidative stress [157–160] Nrf2 possesses six highly conserved domains called Nrf2-ECH homology (Neh) domains. Acetylation of Neh domains in Nrf2 was thought to regulate its transcriptional activity. Mutational analysis of the Neh1 domain led Sun et al [161] to conclude that acetylation-dependent modulation of the transcriptional activity of Nrf2 is, in part, due to the lysyl residues in the Neh1 domain. Mutating all the 18 lysine residues in Neh1 domain led to a 40–45% decrease in Nrf2-induced transcription [162], suggesting that other Neh domains may harbor acetylation sites that may also contribute to reductions in Nrf2 activity. In this regard, studies by Kawai et al [163] showed that acetylation/deacetylation plays a crucial role in nucleocytoplasmic shuttling of Nrf2 and that acetylatable lysine residues in its Neh3 domain participate in modulating its transcriptional activity. Their studies in K562 cells and HepG2 cells revealed that the Neh3 domain modulates the transcriptional activity of Nrf2 through acetylation-dependent regulation that can be ascribed to Lys591 and Lys588 located in that domain. Using molecular and pharmacological strategies they showed that SIRT1 deacetylated Nrf2 and blocked Nrf2 driven gene transcription. In mice models of stroke, HDAC inhibition using trichostatin A activated Nrf2 and upregulated proteins downstream of Nrf2, including HO1, NAD(P)H:quinone oxidoreductase 1, and glutamate-cysteine ligase catalytic subunit [164]. Since trichostatin A does not inhibit NAD+ dependent SIRT activity, it appears that regulation of Nrf2 deacetylation in brain may be driven by non-NAD+ dependent HDACs. Mechanisms and consequences of Nrf2 acetylation in normal and stressed hearts are under active investigation.
In the next section we will review how indirect actions of acetylation can impact transcription factors in the heart.
Indirect actions of acetylation on transcription factors
Transforming growth factor β (TGF-β)
Tissue fibrosis is a major cause of organ dysfunction during chronic diseases and aging. A critical step in this process is TGF-β1-mediated transformation of fibroblasts into myofibroblasts, a cell capable of synthesizing extracellular matrix. Studies by Sunderesan et al [165] demonstrate that SIRT3 controls transformation of fibroblasts into myofibroblasts by suppressing the pro-fibrotic TGF-β1 signaling. They showed SIRT3 deficiency caused induction of TGFβ1 expression and hyper-acetylation of GSK3β at K15 residue. This hyperacetylation negatively regulated GSK3β activity to phosphorylate the substrates, Smad3 and β-catenin. Consequently, reduced phosphorylation led to stabilization and activation of these transcription factors regulating profibrotic genes. Data from rescue studies utilizing overexpression of SIRT3 reveled deacetylation and activation of GSK3β, and consequent attenuation of TGFβ1 signaling and tissue fibrosis.
Myocardin
Myocardin belongs to the SAF-A/B, Acinus, PIAS (SAP) domain family of transcription factors and is specifically expressed in cardiac and smooth muscle. Myocardin functions as a transcriptional coactivator of SRF and is sufficient and necessary for smooth muscle gene expression. Myocardin has been shown to induce the acetylation of nucleosomal histones surrounding SRF-binding sites in the control regions of cardiac and smooth muscle genes through recruiting chromatin-modifying enzyme p300 [166]. Cao et al [167] showed that myocardin is a direct target for p300-mediated acetylation and that p300 acetylates lysine residues at the N terminus myocardin. Importantly, they showed that interaction between p300 and myocardin, mediated by the C terminus of myocardin, is required for the acetylation. Myocardin acetylation by p300 (a) enhanced the association of myocardin and SRF as well as the formation of the myocardin-SRF-CArG box ternary complex, (b) decreased the binding of histone deacetylase 5 (HDAC5) to myocardin, and (c) activated smooth muscle gene expression [167].
Krüppel-like factor (KLF)
Krüppel-like factor KLF5, a member of the Sp/KLF family of zinc finger factors and a key regulator of cardiovascular remodeling. KLF5, is regulated positively by the acetylase p300 and negatively by the oncogenic regulator SET through coupled interaction and regulation of acetylation, [168]. Nagai and his researchers [169] have shown that the deacetylase HDAC1 can negatively regulate KLF5 through direct interaction. Using gThey showed that KLF5 interaction with HDAC1 inhibits the DNA binding activity of KLF5 and suppresses KLF5-dependent promoter activation. Overexpression of HDAC1 suppressed KLF5-dependent activation of its endogenous downstream gene, platelet-derived growth factor-A chain gene. They also showed that HDAC1 binding to the first zinc finger of KLF5, inhibits binding of p300 to KLF5. Other studies have demonstrated in non-cancerous epithelial cells, KLF5 converts from pro-proliferative to anti-proliferative activity upon acetylation [170–172]. KLF5 acetylation alters its transcriptional complex and the expression of genes such as p15 and MYC [Ref A–C]. Li et al showed that, in prostate cancer cell lines, KLF5 inhibited the proliferation, and the inhibition was dependent on KLF5 acetylation [173]. KLF5 suppressed tumor growth in an acetylation-dependent manner in Nude mice. Using RNA-Seq studies they determined that multiple molecules, including RELA, p53, CREB1, MYC, JUN, ER, AR and SP1, mediate the opposing functions of acetylated KLF5 and unacetylated KLF5. Their results indicated a novel acetylation driven mechanism that controls how KLF5 can be both pro- and anti-tumorigenic. Taken together, these studies indicate multiple mechanisms by transcriptional activity of KLF5 cn be altered in multiple cell types.
While all these studies highlight indirect mechanisms by which acetylation influences transcription factors in the heart. In the next section we will review novel and emerging mechanisms impacting HDACs in the heart.
Novel mechanisms impacting HDAC function
HDAC3 degradation
HATs and HDACs form large activator and repressor complexes, resulting in transcriptional gene regulation [174, 175]. HDAC3 is recruited to the SMRT (silencing mediator of retinoid and thyroid receptor) complex, where it interacts with the DAD (deacetylase activation domain) domain of either NCOR1 (nuclear corepressor 1) or SMRT (nuclear corepressor 2; [176]). HDAC3 synthesis is an ongoing process, and this protein is degraded if unbound to the corepressors. The HDAC3-corepressor complex represses several nuclear receptors including thyroid receptor (TR), retinoic acid receptor (RAR), vitamin D receptor (VDR), androgen receptor, glucocorticoid, receptor (GR), and others [177–180].
In our recent study [181], we showed that the interaction of polyol pathway enzyme aldose reductase (AR) with the nuclear corepressors, NCOR1 and SMRT, could lead to HDAC3 degradration (Figure 2). HDAC3, a class I enzyme exists as a multiprotein complex including HDAC3, nuclear corepressors and cofactors. HDAC3 binds to the deacetylation domain (DAD) of either silencing mediator of retinoic and thyroid receptor (SMRT) or nuclear corepressor 1 (NCOR1) [176, 182]. Molecular, cellular, and in vivo studies revealed that AR interaction with the DAD domain of nuclear corepressors drives HDAC3 degradation, consequently leading to the expression of nuclear corepressor’s cofactors including Gps2, Tblr1, PPARγ activation and lipid accumulation in the hearts [181]. Our studies demonstrated that regulating the levels of nuclear corepressors or its cofactors could also result in changes of HDACs and consequently transcription.
Figure 2.
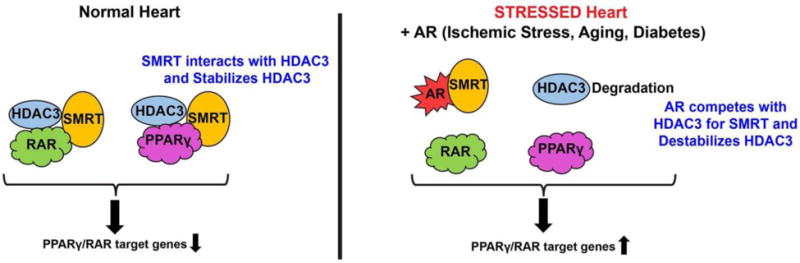
Figure 2 depicts an emerging mechanism of HDAC3 regulation in cells. This mechanism scheme adapted from [181] shows that polyol pathway enzyme aldose reductase (AR) competes with HDAC3 for corepressor complex binding leading to HDAC3 degradation and derepression of the PPARg and RAR pathways.
HDAC3-NCOR complexes take place mostly in the cytoplasm [183]. The HDAC3-corepressor complex represses several nuclear receptors like TR, RXR, RAR, VDR, GR, PXR (pregnane X receptor), and LXR [177–179, 184]. In our study, though we have shown low HDAC3-corepressor complex formation, derepression is specific to PPARg and RARB, altering lipid and retinoid metabolism. Our study revealed a key role for AR as a dissociation factor of the HDAC3-corepressor complex via interaction with the DAD domain, thereby derepressing target gene expression, mimicking the liganded PPARg and RAR state.
Interclass cross talk between HDACs
Histone deacetylases (HDACs) are closely involved in cardiac reprogramming. Although the functional roles of class I and class IIa HDACs are well established, the significance of interclass crosstalk in the development of cardiac hypertrophy is evolving. Recently, Eom et al [74] reported a novel post-translational activation mechanism of HDAC2 that involves acetylation of HDAC2 mediated by p300/CBP-associated factor/HDAC5. They showed that the enzymatic activity of Hdac2 was positively correlated with its acetylation status. p300/CBP-associated factor bound to Hdac2 and induced acetylation. The HDAC2 K75 residue was responsible for hypertrophic stress-induced acetylation. The acetylation-resistant Hdac2 K75R showed a significant decrease in phosphorylation on S394, which led to the loss of intrinsic activity (Figure 3). Hdac5, one of class IIa HDACs, directly deacetylated Hdac2 (Figure 3). Acetylation of Hdac2 was increased in Hdac5-null mice. When an acetylation-mimicking mutant of Hdac2 was infected into cardiomyocytes, the antihypertrophic effect of either nuclear tethering of Hdac5 with leptomycin B or Hdac5 overexpression was reduced. Taken together, these data indicated a novel mechanism by which the balance of HDAC2 acetylation is regulated by p300/CBP-associated factor and HDAC5 in the development of cardiac hypertrophy.
Figure 3.
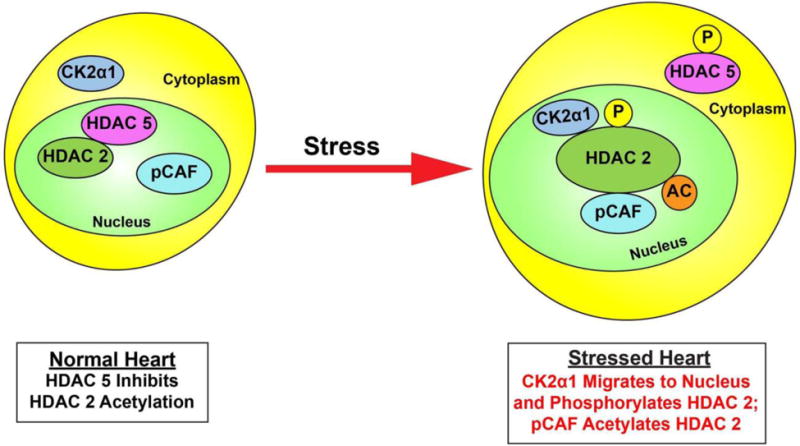
Figure 3 adapted from [74] depicts post-translational activation mechanism of HDAC2 that involves acetylation of HDAC2 mediated by p300/CBP-associated factor/HDAC5. Cardiac stress induces phosphorylation of HDAC5 and subsequent export to the cytoplasm. Casein kinase 2α1 (CK2α1) phosphorylates HDAC2, and p300/CBP-associated factor (pCAF) binds to HDAC2 and induces acetylation. “P’ indicates phosphorylation, “Ac” indicates acetylation.
Therapeutic Opportunities to modulate acetylation in hearts
Sirtuins are attractive targets for therapeutic interventions in several diseases, including cancer, inflammation, metabolic disorder, and cardiovascular disease. Chemical compounds such as flavones, stilbenes, and anthocyanidins, can directly stimulate SIRT1 activity by an allosteric mechanism [185]. Natural product resveratrol (3,5,4=-trihydroxystilbene), is a well-known potent activator of SIRT1 [185]. Recently, several clinical trials on SIRT1 activators have been reported. Longevinex, a modified form of resveratrol, has been shown to improve endothelial function in patients with metabolic syndrome [186]. SRT2104, a SIRT1 activator, was demonstrated to improve lipid profiles in otherwise healthy cigarette smokers [187]. SRT2104 was also associated with improvement in lipid profiles in a phase-II, randomized, placebo-controlled, double-blind, multidose study of subjects with type 2 diabetes [188].
Preclinical studies using HDAC inhibitors demonstrated protection in cardiac hypertrophy [189, 190]. Recently, a study demonstrated that SAHA (vorinostat), an FDA-approved pan-HDAC inhibitor for cancer, was efficacious in a rabbit model of cardiac ischemia-reperfusion injury [191]. This proof-of-concept study has set the stage for a clinical trial in humans to assess effects of HDAC inhibitor SAHA on pathological cardiac remodeling post-myocardial infarction. Entinostat, a novel and selective Class I HDAC inhibitor, has been shown to be efficacious in the rat model of ischemia reperfusion injury [192]. Further characterization of these molecules and determining the optimal dose and timing for application to humans will help in advancing clinical use of HDAC inhibitors and sirtuin activators as therapeutic interventions for cardiovascular diseases. Taken together, these considerations highlight examples of the diverse efforts to target acetylation impacting HDACs for protection of the hearts.
Challenges and limitations in determining acetylation stoichiometry and its functional implications
In a recent review Baeza et al [193] discussed in depth the challenges in quantitation and establishing stoichiometry of acetylation in proteins. In the case of transcription factors, low abundance and complexities involved in isolation makes mass spectrometric studies challenging. Nevertheless, as discussed in the earlier sections, studies have identified specific acetylation sites for some of the transcription factors. Most challenging aspect is identifying number of acetylation sites and it impact on transcription factor activity. Acetylation studies for high abundant proteins/enzymes have shown that (a) single acetylation site can directly control enzymatic activity (an example is acetyl-CoA synthetase 2 (AceCS2) [32, 33] and (b) acetylation in multiple sites affect enzymatic activity (examples include ACAT1, HMGCS2 and SOD2) [194–198]. Advances in mass spectrometric techniques and the use of Knockin strategies to generate cells expressing single and multiple site acetylation resistant mutant transcription factors will help address the impact of site specific acetylation on transcription factor activity.
Conclusions
Lysine acetylation has emerged as a major posttranslational modification in the regulation of transcription in cardiovascular cells. Given the wide range of regulatory mechanisms it impacts, acetylation regulation of transcription factors seems poised to only grow in significance as we continue to discover its functions and mechanisms. Comprehensive proteomics, cellular, molecular, and in vivo (including generation of knockin mice) strategies will help delineate mechanisms and consequences of transcription factor acetylation in mediating cardiovascular disease. Intensive research efforts are underway to uncover the potential of inhibiting acetylation as a therapeutic target in cardiovascular diseases.
Highlights.
Transcription factor acetylation in hearts
HDACs and HATs in transcription factor acetylation
Novel mechanisms impacting HDAC function
Acknowledgments
Authors were supported by grants HL60901, AG026467, HL102022, and HL61783 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J, Cai L. Dysregulation of histone acetyltransferases and deacetylases in cardiovascular diseases. Oxid Med Cell Longev. 2014;2014:641979. doi: 10.1155/2014/641979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vedantham S, Thiagarajan D, Ananthakrishnan R, Wang L, Rosario R, Zou YS, Goldberg I, Yan SF, Schmidt AM, Ramasamy R. Aldose reductase drives hyperacetylation of Egr-1 in hyperglycemia and consequent upregulation of proinflammatory and prothrombotic signals. Diabetes. 2014;63:761–774. doi: 10.2337/db13-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee HA, Lee DY, Cho HM, Kim SY, Iwasaki Y, Kim IK. Histone deacetylase inhibition attenuates transcriptional activity of mineralocorticoid receptor through its acetylation and prevents development of hypertension. Circ Res. 2013;112:1004–1012. doi: 10.1161/CIRCRESAHA.113.301071. [DOI] [PubMed] [Google Scholar]
- 5.Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, Fujita T. Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17:573–580. doi: 10.1038/nm.2337. [DOI] [PubMed] [Google Scholar]
- 6.Vadvalkar SS, Baily CN, Matsuzaki S, West M, Tesiram YA, Humphries KM. Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of type 1 diabetes. Biochem J. 2013;449:253–261. doi: 10.1042/BJ20121038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thal MA, Krishnamurthy P, Mackie AR, Hoxha E, Lambers E, Verma S, Ramirez V, Qin G, Losordo DW, Kishore R. Enhanced angiogenic and cardiomyocyte differentiation capacity of epigenetically reprogrammed mouse and human endothelial progenitor cells augments their efficacy for ischemic myocardial repair. Circ Res. 2012;111:180–190. doi: 10.1161/CIRCRESAHA.112.270462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Q, Lin X, Andrews L, Patel D, Lampe PD, Veenstra RD. Histone deacetylase inhibition reduces cardiac connexin43 expression and gap junction communication. Front Pharmacol. 2013;4:44. doi: 10.3389/fphar.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soubrier F, Chung WK, Machado R, Grunig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, Humbert M. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D13–21. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 10.Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU. Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1001–1010. doi: 10.1152/ajplung.00092.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie M, Hill JA. HDAC-dependent ventricular remodeling. Trends Cardiovasc Med. 2013;23:229–235. doi: 10.1016/j.tcm.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong Y, Guan KL. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol. 2012;198:155–164. doi: 10.1083/jcb.201202056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, Lombard DB. Calorie restriction alters mitochondrial protein acetylation. Aging Cell. 2009;8:604–606. doi: 10.1111/j.1474-9726.2009.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 22.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 23.Guarente L, Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 2011;364:2235–2244. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- 24.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 26.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 28.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987;104:289–302. doi: 10.1083/jcb.104.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 31.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 32.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 35.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsusaka T, Guo T, Yagura T, Inoue T, Yokode M, Inagaki N, Kondoh H. Deacetylation of phosphoglycerate mutase in its distinct central region by SIRT2 down-regulates its enzymatic activity. Genes Cells. 2014;19:766–777. doi: 10.1111/gtc.12176. [DOI] [PubMed] [Google Scholar]
- 37.Xu Y, Li F, Lv L, Li T, Zhou X, Deng CX, Guan KL, Lei QY, Xiong Y. Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res. 2014;74:3630–3642. doi: 10.1158/0008-5472.CAN-13-3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res. 2014;103:485–497. doi: 10.1093/cvr/cvu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukushima A, Lopaschuk GD. Acetylation control of cardiac fatty acid beta-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim Biophys Acta. 2016;16 doi: 10.1016/j.bbadis.2016.07.020. pii:S0925–4439 30188–30180. [DOI] [PubMed] [Google Scholar]
- 40.Parthun MR, Widom J, Gottschling DE. The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell. 1996;87:85–94. doi: 10.1016/s0092-8674(00)81325-2. [DOI] [PubMed] [Google Scholar]
- 41.Burgess RJ, Zhou H, Han J, Zhang Z. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell. 2010;37:469–480. doi: 10.1016/j.molcel.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sklenar AR, Parthun MR. Characterization of yeast histone H3-specific type B histone acetyltransferases identifies an ADA2-independent Gcn5p activity. BMC Biochem. 2004;5:11. doi: 10.1186/1471-2091-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang X, Yu W, Shi L, Sun L, Liang J, Yi X, Li Q, Zhang Y, Yang F, Han X, Zhang D, Yang J, Yao Z, Shang Y. HAT4, a Golgi apparatus-anchored B-type histone acetyltransferase, acetylates free histone H4 and facilitates chromatin assembly. Mol Cell. 2011;44:39–50. doi: 10.1016/j.molcel.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 44.Yao TP, Oh SP, Fuchs M, Zhou ND, Ch’ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 45.Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth JF, Marino S, Wittwer J, Scheidweiler A, Eckner R. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 2003;22:5175–5185. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukushima A, Alrob OA, Zhang L, Wagg CS, Altamimi T, Rawat S, Rebeyka IM, Kantor PF, Lopaschuk GD. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am J Physiol Heart Circ Physiol. 2016;311:H347–363. doi: 10.1152/ajpheart.00900.2015. [DOI] [PubMed] [Google Scholar]
- 47.Hamamori Y, Schneider MD. HATs off to Hop: recruitment of a class I histone deacetylase incriminates a novel transcriptional pathway that opposes cardiac hypertrophy. J Clin Invest. 2003;112:824–826. doi: 10.1172/JCI19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 49.Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem. 2003;278:28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denu JM. The Sir 2 family of protein deacetylases. Curr Opin Chem Biol. 2005;9:431–440. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 52.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 53.Smith J. Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol. 2002;12:404–406. doi: 10.1016/s0962-8924(02)02342-5. [DOI] [PubMed] [Google Scholar]
- 54.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 55.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 56.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 57.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 58.Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 59.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282:6823–6832. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- 62.Tong C, Morrison A, Mattison S, Qian S, Bryniarski M, Rankin B, Wang J, Thomas DP, Li J. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. FASEB J. 2013;27:4332–4342. doi: 10.1096/fj.12-216473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 64.Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95:971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 65.Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S, Sadoshima J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170–2182. doi: 10.1161/CIRCULATIONAHA.110.958033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28:6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu B, Che W, Xue J, Zheng C, Tang K, Zhang J, Wen J, Xu Y. SIRT4 prevents hypoxia-induced apoptosis in H9c2 cardiomyoblast cells. Cell Physiol Biochem. 2013;32:655–662. doi: 10.1159/000354469. [DOI] [PubMed] [Google Scholar]
- 68.Liu B, Che W, Zheng C, Liu W, Wen J, Fu H, Tang K, Zhang J, Xu Y. SIRT5: a safeguard against oxidative stress-induced apoptosis in cardiomyocytes. Cell Physiol Biochem. 2013;32:1050–1059. doi: 10.1159/000354505. [DOI] [PubMed] [Google Scholar]
- 69.Maksin-Matveev A, Kanfi Y, Hochhauser E, Isak A, Cohen HY, Shainberg A. Sirtuin 6 protects the heart from hypoxic damage. Exp Cell Res. 2015;330:81–90. doi: 10.1016/j.yexcr.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 70.Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 71.Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol. 2014;306:H1602–1609. doi: 10.1152/ajpheart.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eom GH, Kook H. Role of histone deacetylase 2 and its posttranslational modifications in cardiac hypertrophy. BMB Rep. 2015;48:131–138. doi: 10.5483/BMBRep.2015.48.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eom GH, Nam YS, Oh JG, Choe N, Min HK, Yoo EK, Kang G, Nguyen VH, Min JJ, Kim JK, Lee IK, Bassel-Duby R, Olson EN, Park WJ, Kook H. Regulation of acetylation of histone deacetylase 2 by p300/CBP-associated factor/histone deacetylase 5 in the development of cardiac hypertrophy. Circ Res. 2014;114:1133–1143. doi: 10.1161/CIRCRESAHA.114.303429. [DOI] [PubMed] [Google Scholar]
- 75.Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kee HJ, Bae EH, Park S, Lee KE, Suh SH, Kim SW, Jeong MH. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive rats via regulation of HDAC6/HDAC8 enzyme activity. Kidney Blood Press Res. 2013;37:229–239. doi: 10.1159/000350148. [DOI] [PubMed] [Google Scholar]
- 77.Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, Nattel S, Henning RH, Brundel BJ. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129:346–358. doi: 10.1161/CIRCULATIONAHA.113.005300. [DOI] [PubMed] [Google Scholar]
- 78.Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, Schuetze KB, Horn TR, Chen B, Ferrara C, Scellini B, Piroddi N, Tesi C, Poggesi C, Jeong MY, McKinsey TA. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol. 2014;307:H252–258. doi: 10.1152/ajpheart.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, Irwin DC, McCune SA, Chung E, Leinwand LA, McKinsey TA. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol. 2011;51:41–50. doi: 10.1016/j.yjmcc.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 82.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 83.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 85.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 86.Sack MN, Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a013102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 88.Dominy JE, Jr, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R, Denu JM, Clish CB, Yang X, Shulman GI, Gygi SP, Puigserver P. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell. 2012;48:900–913. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, Baar K, Schenk S. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J Biol Chem. 2011;286:30561–30570. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dawn B, Xuan YT, Marian M, Flaherty MP, Murphree SS, Smith TL, Bolli R, Jones WK. Cardiac-specific abrogation of NF-kappa B activation in mice by transdominant expression of a mutant I kappa B alpha. J Mol Cell Cardiol. 2001;33:161–173. doi: 10.1006/jmcc.2000.1291. [DOI] [PubMed] [Google Scholar]
- 91.Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- 92.Sakaguchi T, Sawa Y, Fukushima N, Nishimura M, Ichikawa H, Kaneda Y, Matsuda H. A novel strategy of decoy transfection against nuclear factor-kappaB in myocardial preservation. Ann Thorac Surg. 2001;71:624–629. doi: 10.1016/s0003-4975(00)01906-8. discussion 629–630. [DOI] [PubMed] [Google Scholar]
- 93.Trescher K, Bernecker O, Fellner B, Gyongyosi M, Krieger S, Demartin R, Wolner E, Podesser BK. Adenovirus-mediated overexpression of inhibitor kappa B-alpha attenuates postinfarct remodeling in the rat heart. Eur J Cardiothorac Surg. 2004;26:960–967. doi: 10.1016/j.ejcts.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 94.Chen LF, Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J Mol Med (Berl) 2003;81:549–557. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 95.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The Transcriptional Activity of NF-κB Is Regulated by the IκB-Associated PKAc Subunit through a Cyclic AMP– Independent Mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 97.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 98.Deng WG, Wu KK. Regulation of inducible nitric oxide synthase expression by p300 and p50 acetylation. J Immunol. 2003;171:6581–6588. doi: 10.4049/jimmunol.171.12.6581. [DOI] [PubMed] [Google Scholar]
- 99.Deng WG, Wu KK. Augmentation of NF-kappa B mediated gene expression by a positive p300 andp50 regulatory loop. FASEB J. 2003;17:A173. [Google Scholar]
- 100.Deng W-G, Zhu Y, Wu KK. Up-regulation of p300 Binding and p50 Acetylation in Tumor Necrosis Factor-α-induced Cyclooxygenase-2 Promoter Activation. Journal of Biological Chemistry. 2003;278:4770–4777. doi: 10.1074/jbc.M209286200. [DOI] [PubMed] [Google Scholar]
- 101.Furia B, Deng L, Wu K, Baylor S, Kehn K, Li H, Donnelly R, Coleman T, Kashanchi F. Enhancement of nuclear factor-kappa B acetylation by coactivator p300 and HIV-1 Tat proteins. J Biol Chem. 2002;277:4973–4980. doi: 10.1074/jbc.M107848200. [DOI] [PubMed] [Google Scholar]
- 102.Hu J, Colburn NH. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res. 2005;3:100–109. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 103.Vermeulen L, De Wilde G, Notebaert S, Vanden Berghe W, Haegeman G. Regulation of the transcriptional activity of the nuclear factor-kappaB p65 subunit. Biochem Pharmacol. 2002;64:963–970. doi: 10.1016/s0006-2952(02)01161-9. [DOI] [PubMed] [Google Scholar]
- 104.Schmitz ML, Mattioli I, Buss H, Kracht M. NF-kappaB: a multifaceted transcription factor regulated at several levels. Chembiochem. 2004;5:1348–1358. doi: 10.1002/cbic.200400144. [DOI] [PubMed] [Google Scholar]
- 105.Edelstein LC, Pan A, Collins T. Chromatin modification and the endothelial-specific activation of the E-selectin gene. J Biol Chem. 2005;280:11192–11202. doi: 10.1074/jbc.M412997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D’Abreo C, Marsden PA. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem. 2005;280:24824–24838. doi: 10.1074/jbc.M502115200. [DOI] [PubMed] [Google Scholar]
- 107.Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res. 2011;90:421–429. doi: 10.1093/cvr/cvr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reddy MA, Sahar S, Villeneuve LM, Lanting L, Natarajan R. Role of Src tyrosine kinase in the atherogenic effects of the 12/15-lipoxygenase pathway in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2009;29:387–393. doi: 10.1161/ATVBAHA.108.179150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sahar S, Reddy MA, Wong C, Meng L, Wang M, Natarajan R. Cooperation of SRC-1 and p300 with NF-kappaB and CREB in angiotensin II-induced IL-6 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:1528–1534. doi: 10.1161/ATVBAHA.107.145862. [DOI] [PubMed] [Google Scholar]
- 110.Vanden Berghe W, De Bosscher K, Boone E, Plaisance S, Haegeman G. The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem. 1999;274:32091–32098. doi: 10.1074/jbc.274.45.32091. [DOI] [PubMed] [Google Scholar]
- 111.Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, Forstermann U, Zeiher AM, Dimmeler S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837–844. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- 112.Miao F, Gonzalo IG, Lanting L, Natarajan R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem. 2004;279:18091–18097. doi: 10.1074/jbc.M311786200. [DOI] [PubMed] [Google Scholar]
- 113.Miao F, Li S, Chavez V, Lanting L, Natarajan R. Coactivator-associated arginine methyltransferase-1 enhances nuclear factor-kappaB-mediated gene transcription through methylation of histone H3 at arginine 17. Mol Endocrinol. 2006;20:1562–1573. doi: 10.1210/me.2005-0365. [DOI] [PubMed] [Google Scholar]
- 114.Bagul PK, Deepthi N, Sultana R, Banerjee SK. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J Nutr Biochem. 2015;26:1298–1307. doi: 10.1016/j.jnutbio.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 115.Hwang YJ, Lee EW, Song J, Kim HR, Jun YC, Hwang KA. MafK positively regulates NF-kappaB activity by enhancing CBP-mediated p65 acetylation. Sci Rep. 2013;3:3242. doi: 10.1038/srep03242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kasneci A, Kemeny-Suss NM, Komarova SV, Chalifour LE. Egr-1 negatively regulates calsequestrin expression and calcium dynamics in ventricular cells. Cardiovasc Res. 2009;81:695–702. doi: 10.1093/cvr/cvn357. [DOI] [PubMed] [Google Scholar]
- 118.Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circ Res. 2006;98:186–191. doi: 10.1161/01.RES.0000200177.53882.c3. [DOI] [PubMed] [Google Scholar]
- 119.Rayner BS, Figtree GA, Sabaretnam T, Shang P, Mazhar J, Weaver JC, Lay WN, Witting PK, Hunyor SN, Grieve SM, Khachigian LM, Bhindi R. Selective inhibition of the master regulator transcription factor Egr-1 with catalytic oligonucleotides reduces myocardial injury and improves left ventricular systolic function in a preclinical model of myocardial infarction. J Am Heart Assoc. 2013;2:e000023. doi: 10.1161/JAHA.113.000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harja E, Bucciarelli LG, Lu Y, Stern DM, Zou YS, Schmidt AM, Yan SF. Early growth response-1 promotes atherogenesis: mice deficient in early growth response-1 and apolipoprotein E display decreased atherosclerosis and vascular inflammation. Circ Res. 2004;94:333–339. doi: 10.1161/01.RES.0000112405.61577.95. [DOI] [PubMed] [Google Scholar]
- 121.Fahmy RG, Dass CR, Sun LQ, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat Med. 2003;9:1026–1032. doi: 10.1038/nm905. [DOI] [PubMed] [Google Scholar]
- 122.Arai M, Yoguchi A, Takizawa T, Yokoyama T, Kanda T, Kurabayashi M, Nagai R. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca(2+)-ATPase gene transcription. Circ Res. 2000;86:8–14. doi: 10.1161/01.res.86.1.8. [DOI] [PubMed] [Google Scholar]
- 123.Bhindi R, Khachigian LM, Lowe HC. DNAzymes targeting the transcription factor Egr-1 reduce myocardial infarct size following ischemia-reperfusion in rats. J Thromb Haemost. 2006;4:1479–1483. doi: 10.1111/j.1538-7836.2006.02022.x. [DOI] [PubMed] [Google Scholar]
- 124.Dje N’Guessan P, Riediger F, Vardarova K, Scharf S, Eitel J, Opitz B, Slevogt H, Weichert W, Hocke AC, Schmeck B, Suttorp N, Hippenstiel S. Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:380–386. doi: 10.1161/ATVBAHA.108.178319. [DOI] [PubMed] [Google Scholar]
- 125.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 126.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of Intracellular Oxygen in Hypoxia by Nitric Oxide: Effect on HIF1α. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 127.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Poellinger L, Johnson RS. HIF-1 and hypoxic response: the plot thickens. Curr Opin Genet Dev. 2004;14:81–85. doi: 10.1016/j.gde.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 129.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 130.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117:862–865. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Geng H, Liu Q, Xue C, David LL, Beer TM, Thomas GV, Dai MS, Qian DZ. HIF1alpha protein stability is increased by acetylation at lysine 709. J Biol Chem. 2012;287:35496–35505. doi: 10.1074/jbc.M112.400697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Joo HY, Yun M, Jeong J, Park ER, Shin HJ, Woo SR, Jung JK, Kim YM, Park JJ, Kim J, Lee KH. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1alpha (HIF-1alpha) via direct interactions during hypoxia. Biochem Biophys Res Commun. 2015;462:294–300. doi: 10.1016/j.bbrc.2015.04.119. [DOI] [PubMed] [Google Scholar]
- 136.Yoon H, Shin SH, Shin DH, Chun YS, Park JW. Differential roles of Sirt1 in HIF-1alpha and HIF-2alpha mediated hypoxic responses. Biochem Biophys Res Commun. 2014;444:36–43. doi: 10.1016/j.bbrc.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 137.Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010;38:864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 138.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Knight RA, Scarabelli TM, Stephanou A. STAT transcription in the ischemic heart. JAKSTAT. 2012;1:111–117. doi: 10.4161/jkst.20078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Santos CI, Costa-Pereira AP. Signal transducers and activators of transcription-from cytokine signalling to cancer biology. Biochim Biophys Acta. 2011;1816:38–49. doi: 10.1016/j.bbcan.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 141.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 142.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dasgupta M, Unal H, Willard B, Yang J, Karnik SS, Stark GR. Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. J Biol Chem. 2014;289:30763–30771. doi: 10.1074/jbc.M114.603894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 145.Obsil T, Obsilova V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene. 2008;27:2263–2275. doi: 10.1038/onc.2008.20. [DOI] [PubMed] [Google Scholar]
- 146.Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T, Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- 148.Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6:105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nakae J, Oki M, Cao Y. The FoxO transcription factors and metabolic regulation. FEBS Lett. 2008;582:54–67. doi: 10.1016/j.febslet.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 150.Perrot V, Rechler MM. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1-induced transcription. Mol Endocrinol. 2005;19:2283–2298. doi: 10.1210/me.2004-0292. [DOI] [PubMed] [Google Scholar]
- 151.Dansen TB, Smits LM, van Triest MH, de Keizer PL, van Leenen D, Koerkamp MG, Szypowska A, Meppelink A, Brenkman AB, Yodoi J, Holstege FC, Burgering BM. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat Chem Biol. 2009;5:664–672. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- 152.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sin TK, Yu AP, Yung BY, Yip SP, Chan LW, Wong CS, Ying M, Rudd JA, Siu PM. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J Physiol. 2014;592:2535–2548. doi: 10.1113/jphysiol.2014.271387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Puthanveetil P, Wang Y, Wang F, Kim MS, Abrahani A, Rodrigues B. The increase in cardiac pyruvate dehydrogenase kinase-4 after short-term dexamethasone is controlled by an Akt-p38-forkhead box other factor-1 signaling axis. Endocrinology. 2010;151:2306–2318. doi: 10.1210/en.2009-1072. [DOI] [PubMed] [Google Scholar]
- 156.Thompson JW, Wei J, Appau K, Wang H, Yu H, Spiga MG, Graham RM, Webster KA. Bnip3 Binds and Activates p300: Possible Role in Cardiac Transcription and Myocyte Morphology. PLoS One. 2015;10:e0136847. doi: 10.1371/journal.pone.0136847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 158.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 159.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 160.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 161.Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29:2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP Augments Promoter-Specific DNA Binding of Nrf2 during the Antioxidant Response. Molecular and Cellular Biology. 2009;29:2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kawai Y, Garduno L, Theodore M, Yang J, Arinze IJ. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J Biol Chem. 2011;286:7629–7640. doi: 10.1074/jbc.M110.208173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, Li RC, Xu Y, Dore S, Cao W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med. 2012;52:928–936. doi: 10.1016/j.freeradbiomed.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E, Gupta MP. SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3beta. Mol Cell Biol. 2015;36:678–692. doi: 10.1128/MCB.00586-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cao D, Wang Z, Zhang CL, Oh J, Xing W, Li S, Richardson JA, Wang DZ, Olson EN. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol Cell Biol. 2005;25:364–376. doi: 10.1128/MCB.25.1.364-376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cao D, Wang C, Tang R, Chen H, Zhang Z, Tatsuguchi M, Wang DZ. Acetylation of myocardin is required for the activation of cardiac and smooth muscle genes. J Biol Chem. 2012;287:38495–38504. doi: 10.1074/jbc.M112.353649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Suzuki T, Kimura A, Nagai R, Horikoshi M. Regulation of interaction of the acetyltransferase region of p300 and the DNA-binding domain of Sp1 on and through DNA binding. Genes Cells. 2000;5:29–41. doi: 10.1046/j.1365-2443.2000.00302.x. [DOI] [PubMed] [Google Scholar]
- 169.Matsumura T, Suzuki T, Aizawa K, Munemasa Y, Muto S, Horikoshi M, Nagai R. The deacetylase HDAC1 negatively regulates the cardiovascular transcription factor Kruppel-like factor 5 through direct interaction. J Biol Chem. 2005;280:12123–12129. doi: 10.1074/jbc.M410578200. [DOI] [PubMed] [Google Scholar]
- 170.Guo P, Dong XY, Zhang X, Zhao KW, Sun X, Li Q, Dong JT. Pro-proliferative factor KLF5 becomes anti-proliferative in epithelial homeostasis upon signaling-mediated modification. J Biol Chem. 2009;284:6071–6078. doi: 10.1074/jbc.M806270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Guo P, Dong XY, Zhao K, Sun X, Li Q, Dong JT. Opposing effects of KLF5 on the transcription of MYC in epithelial proliferation in the context of transforming growth factor beta. J Biol Chem. 2009;284:28243–28252. doi: 10.1074/jbc.M109.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Guo P, Zhao KW, Dong XY, Sun X, Dong JT. Acetylation of KLF5 alters the assembly of p15 transcription factors in transforming growth factor-beta-mediated induction in epithelial cells. J Biol Chem. 2009;284:18184–18193. doi: 10.1074/jbc.M109.007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li X, Zhang B, Wu Q, Ci X, Zhao R, Zhang Z, Xia S, Su D, Chen J, Ma G, Fu L, Dong JT. Interruption of KLF5 acetylation converts its function from tumor suppressor to tumor promoter in prostate cancer cells. Int J Cancer. 2015;136:536–546. doi: 10.1002/ijc.29028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guenther MG, Lane WS, Fischle W, Verdin E, Lazar MA, Shiekhattar R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000;14:1048–1057. [PMC free article] [PubMed] [Google Scholar]
- 177.Dwivedi PP, Muscat GE, Bailey PJ, Omdahl JL, May BK. Repression of basal transcription by vitamin D receptor: evidence for interaction of unliganded vitamin D receptor with two receptor interaction domains in RIP13delta1. J Mol Endocrinol. 1998;20:327–335. doi: 10.1677/jme.0.0200327. [DOI] [PubMed] [Google Scholar]
- 178.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 179.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, Schulman IG, Glass CK. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wong MM, Guo C, Zhang J. Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am J Clin Exp Urol. 2014;2:169–187. [PMC free article] [PubMed] [Google Scholar]
- 181.Thiagarajan D, Ananthakrishnan R, Zhang J, O’Shea KM, Quadri N, Li Q, Sas K, Jing X, Rosario R, Pennathur S, Schmidt AM, Ramasamy R. Aldose Reductase Acts as a Selective Derepressor of PPARgamma and the Retinoic Acid Receptor. Cell Rep. 2016;15:181–196. doi: 10.1016/j.celrep.2016.02.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 183.Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, Ho E, Dashwood RH. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer. 2011;10:68. doi: 10.1186/1476-4598-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhang X, Jeyakumar M, Petukhov S, Bagchi MK. A nuclear receptor corepressor modulates transcriptional activity of antagonist-occupied steroid hormone receptor. Mol Endocrinol. 1998;12:513–524. doi: 10.1210/mend.12.4.0089. [DOI] [PubMed] [Google Scholar]
- 185.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 186.Fujitaka K, Otani H, Jo F, Jo H, Nomura E, Iwasaki M, Nishikawa M, Iwasaka T, Das DK. Modified resveratrol Longevinex improves endothelial function in adults with metabolic syndrome receiving standard treatment. Nutr Res. 2011;31:842–847. doi: 10.1016/j.nutres.2011.09.028. [DOI] [PubMed] [Google Scholar]
- 187.Venkatasubramanian S, Noh RM, Daga S, Langrish JP, Joshi NV, Mills NL, Hoffmann E, Jacobson EW, Vlasuk GP, Waterhouse BR, Lang NN, Newby DE. Cardiovascular effects of a novel SIRT1 activator, SRT2104, in otherwise healthy cigarette smokers. J Am Heart Assoc. 2013;2:e000042. doi: 10.1161/JAHA.113.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Baksi A, Kraydashenko O, Zalevkaya A, Stets R, Elliott P, Haddad J, Hoffmann E, Vlasuk GP, Jacobson EW. A phase II, randomized, placebo-controlled, double-blind, multidose study of SRT2104, a SIRT1 activator, in subjects with type 2 diabetes. Br J Clin Pharmacol. 2014;78:69–77. doi: 10.1111/bcp.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. 2006;113:51–59. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- 191.Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, Jiang N, Jessen ME, Warner JJ, Lavandero S, Gillette TG, Turer AT, Hill JA. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Aune SE, Herr DJ, Mani SK, Menick DR. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J Mol Cell Cardiol. 2014;72:138–145. doi: 10.1016/j.yjmcc.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Baeza J, Smallegan MJ, Denu JM. Mechanisms and Dynamics of Protein Acetylation in Mitochondria. Trends Biochem Sci. 2016;41:231–244. doi: 10.1016/j.tibs.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12:534–541. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, Denu JM. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 2015;21:637–646. doi: 10.1016/j.cmet.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 197.Still AJ, Floyd BJ, Hebert AS, Bingman CA, Carson JJ, Gunderson DR, Dolan BK, Grimsrud PA, Dittenhafer-Reed KE, Stapleton DS, Keller MP, Westphall MS, Denu JM, Attie AD, Coon JJ, Pagliarini DJ. Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. J Biol Chem. 2013;288:26209–26219. doi: 10.1074/jbc.M113.483396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]