

Abstract
Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) is essential for the exflagellation of male gametocytes. Inhibition of PfCDPK4 is an effective way of blocking the transmission of malaria by mosquitoes. A series of 5-aminopyrazole-4-carboxamide analogues are demonstrated to be potent inhibitors of PfCDPK4. The compounds are also able to block exflagellation of Plasmodium falciparum male gametocytes without observable toxicity to mammalian cells.
Keywords: Plasmodium falciparum, PfCDPK4, Microgametocyte exflagellation, Malaria transmission blocking, 5-Aminopyrazole-4-carboxamide
Graphical abstract
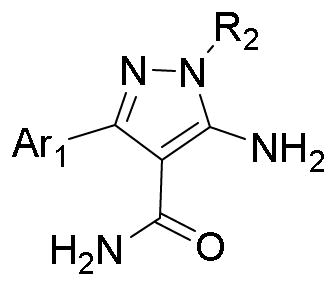
Malaria is one of the most widespread and dangerous infectious diseases in developing countries. Malaria is caused by five Plasmodium species that affect humans, Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale and Plasmodium knowlesi.1 Of these, P. falciparum and P. vivax are the most important public health concern. P. falciparum causes the most deadly form of malaria while P. vivax has a wider global distribution.2 Although efforts to control and eradicate malaria (especially falciparum malaria) through vector control and chemotherapy have provided significant gains, transmission is still continuously occurring and additional control methods are needed. This is reflective of the World Health Organization’s estimated 214 million new cases and 438,000 malaria deaths globally in 2015. The majority of deaths occurred in Sub-Saharan Africa among children under 5 years of age.3 Recent studies concluded that even with years of exposure to intense malaria transmission, individuals living in endemic areas have no evidence for the acquisition of sterile immunity to P. falciparum infection.4
Currently available drugs for treating malaria mainly target asexual blood stages. However, malaria transmission remains high because circulating gametocytes are still infectious to mosquitoes weeks after therapy, which hinders the effective control and eradication of the disease.5 Although two antimalarials, primaquine and artemisinin combination therapy (ACT) may have some antigametocyte activity, they are not sufficient for preventing malaria transmission by mosquitoes. ACTs encompass multiple drugs and combinations with varied gametocytocidal efficacy of each therapeutic agent but the artemisinin component only kills immature gametocytes while primaquine only kills mature gametocytes.6 All gametocytes can be killed through extended primaquine treatment, but this treatment can lead to gastrointestinal intolerance and hemolysis in patients with glucose-6-phosphate dehydrogenase deficiency.7 This emphasizes the need for novel intervention mechanisms that prevent malaria parasite transmission. Thus, the development of new transmission-blocking strategies will aid the control and eradication of malaria.
Plasmodia calcium-dependent protein kinase 4 (PfCDPK4) is a signaling molecule that is necessary for microgametocyte transition into sperm-like, male gametes by a process called exflagellation in the mosquito midgut.8,9 Inhibition of PfCDPK4 blocks Plasmodium microgamete exflagellation, thereby disrupting malaria transmission.9,10 Therefore, PfCDPK4 is a promising drug target for the development of transmission blocking therapies. We have previously reported that a number of pyrazolopyrimidine (PP)-based bumped kinase inhibitors (BKIs), which selectively inhibit Toxoplasma gondii CDPK1 (TgCDPK1) and Cryptosporidium parvum CDPK1 (CpCDPK1), were potent inhibitors of PfCDPK4 and were able to block microgamete exflagellation.9–13 Further research studies to explore alternative scaffold compounds as malaria transmission blocking inhibitors targeting PfCDPK4 are ongoing. Here, we report the exploration of compounds based on the 5-aminopyrazole-4-carboxamide (AC) scaffold as potent inhibitors of PfCDPK4 for use to block P. falciparum exflagellation.
Apicomplexan signaling enzymes TgCDPK1, CpCDPK1 and PfCDPK4 share a unique structural similarity within their ATP binding cavities. These kinases contain small amino acid residues at the “gatekeeper” position. This feature is rarely found in mammalian kinases. PfCDPK4 is predicted to have a very similar binding pocket to that of TgCDPK1, with the most substantial difference being a serine gatekeeper in PfCDPK4 versus a glycine gatekeeper in TgCDPK1.9 Because serine is smaller than most gatekeeper residues found in mammalian kinases, it is also possible to develop highly selective inhibitors targeting PfCDPK4. As an alternative to the PP scaffold, 5-aminopyrazole-4-carboxamide based compounds were generated and demonstrated to be potent and selective inhibitors of Tg/CpCDPK1.14 Further, the binding model of the AC scaffold with TgCDPK1 is consistent with that of the PP scaffold, which has been confirmed by crystal structures.15 Based upon this, the successful development of the PP scaffold as PfCDPK4 inhibitors was taken into consideration to develop the AC scaffold compounds as selective PfCDPK4 inhibitors as well.
We tested a focused AC library of 100 compounds for their ability to inhibit recombinant PfCDPK4 in vitro. We also tested the compounds against PfCDPK1, as PfCDPK1 is predicted to have a very similar binding pocket to that of PfCDPK4 and TgCDPK1, with the major difference being a slightly bulkier threonine gatekeeper.16 Inhibition data was obtained indirectly by measuring changes in initial ATP concentration after enzymatic phosphorylation of peptide substrate via luminescence with the Kinaseglo© luciferase reagent (Promega, Madison, WI) as previously described.9–12 Compounds were also tested for potential off-target effects against the mammalian kinase Src, which contains a threonine gatekeeper, one of the smallest amino acid side chains present at this position in human kinases. Assays for inhibition of Src phosphorylation activity also relied on indirect measurement of ATP consumption in the reaction via luminescence with Kinaseglo© as described.16
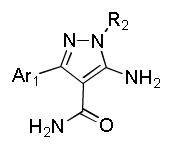
Among the 100 compounds tested against PfCDPK4, 28 were found to inhibit PfCDPK4 with an IC50 of <100 nM and a further 7 compounds with IC50 <50 nM (Table 1). The results demonstrate that some of the AC compounds could fit the binding pocket of PfCDPK4 and inhibit its ability to phosphorylate peptide substrate. We analyzed the inhibitors with various substituted naphthyls and quinolines as Ar1 substructures, while fixing the R2 as a t-butyl group and found that in general, the larger the substituent on Ar1 (cyclobutoxy > cycloproxy > ethoxy > methoxy > hydroxyl > hydrogen) the weaker its activity was against PfCDPK4 (Table 1). It should be noted that compounds with smaller Ar1 groups are liable to inhibit Src because it (the Ar1) is directed towards the gatekeeper residue. The above results indicate that bumped Ar1 groups likely impact activity against both PfCDPK4 and mammalian kinases, therefore affecting the selectivity of PfCDPK4 over mammalian kinases. We also noticed that the activities of AC compounds against PfCDPK4 are not as potent as they were against TgCDPK1. Even for the top 28 PfCDPK4 inhibitors shown in Table 1, all of the compounds except 2 displayed a >5 fold lower IC50 for TgCDPK1 than PfCDPK4, and 12 compounds were >10 fold more potent against TgCDPK1. In contrast, all the inhibitors in Table 1 are more potent against PfCDPK4 than PfCDPK1. Twenty (20) out of the 28 compounds differ in IC50 by more than 5 fold and a further 7 compounds are >10 fold more potent against PfCDPK4. The above results demonstrate that the enlarged ATP binding pocket of TgCDPK1 is more favorable for the AC compounds than that of PfCDPK4, while PfCDPK4 is more favorable than PfCDPK1. The results are not surprising and are consistent with previous observations, suggesting that the size of the gatekeeper residue side chain could be the most dominant determinant of BKI inhibition of a given CDPK.16 The significant difference in sensitivities caused by the small differences in the gatekeeper hints that it is highly likely for the AC compounds to achieve selectivity against mammalian kinases with larger gatekeeper residues.
Table 1.
Enzymatic PfCDPK4, PfCDPK1, TgCDPK1 and Src inhibition (IC50) results. All results are the averages of at least three assays. N/T= not tested.
| ||||||
|---|---|---|---|---|---|---|
| Ar1 | R2 | IC50 (μM)
|
||||
| PfCDPK4 | PfCDPK1 | TgCDPK1 | Src | |||
| 1 |
|
|
0.039 (±0.012) | 0.115 (± 0.063) | 0.002 (±0.0004) | 1.32 |
| 2 |
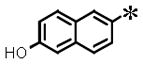
|
0.031 (±0.003) | 0.142 (± 0.011) | 0.007 (±0.0003) | N/T | |
| 3 |
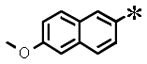
|
0.036 (±0.004) | 0.114 (± 0.001) | 0.002 (±0.001) | 6.93 | |
| 4 |
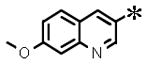
|
0.057 (±0.007) | 1.128 (± 0.105) | 0.005 (±0.003) | >30 | |
| 5 |
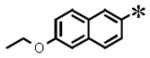
|
0.074 (±0.033) | 0.287 (± 0.048) | 0.004 (±0.001) | 7.27 | |
| 6 |
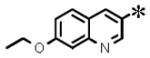
|
0.037 (±0.005) | 0.733 (± 0.082) | 0.002 (±0.0003) | >30 | |
| 7 |
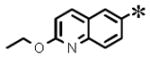
|
0.058 (±0.022) | 0.307 (± 0.071) | 0.006 (±0.0007) | >10 | |
| 8 |
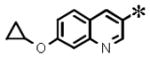
|
0.082 (±0.021) | 0.225 (± 0.028) | 0.010 (±0.002) | 4.75 | |
| 9 |
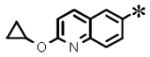
|
0.075 (±0.015) | 0.480 (± 0.183) | 0.005 (±0.0009) | >10 | |
| 10 |
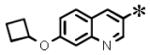
|
0.083 (±0.030) | 0.188 (± 0.085) | 0.007 (±0.003) | 10 | |
|
| ||||||
| 11 |
|
|
0.094 (±0.042) | 0.401 (± 0.100) | 0.006 (±0.003) | N/T |
| 12 |
|
0.077 (±0.043) | 0.999 (± 0.429) | 0.004 (±0.002) | 0.98 | |
|
| ||||||
| 13 |
|
|
0.082 (±0.006) | 1.542 (± 0.354) | 0.013 (±0.005) | >10 |
| 14 |
|
0.044 (±0.004) | 0.496 (± 0.133) | 0.008 (±0.003) | >10 | |
| 15 |
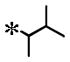
|
0.102 (±0.033) | 0.628 | 0.018 (± 0.009) | N/T | |
| 16 |
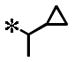
|
0.123 (±0.043) | 0.916 | 0.014 (± 0.006) | N/T | |
| 17 |
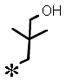
|
0.081 (±0.011) | 1.74 (± 0.331) | 0.011 (± 0.002) | >10 | |
| 18 |
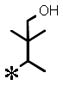
|
0.113 (± 0.073) | >2 | 0.024 (± 0.016) | N/T | |
| 19 |
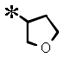
|
0.082 (± 0.003) | 0.910 (± 0.145) | 0.012 (± 0.002) | >10 | |
| 20 |
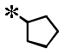
|
0.090 (±0.031) | 0.635 (± 0.048) | 0.011 (± 0.002) | >10 | |
| 21 |
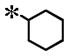
|
0.094 (±0.040) | 0.506 (± 0.078) | 0.012 (± 0.007) | >10 | |
| 22 |
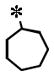
|
0.079 (±0.032) | 0.589 (± 0.107) | 0.006 (± 0.002) | >10 | |
|
| ||||||
| 23 |
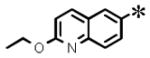
|
|
0.048 (±0.013) | 0.338 (± 0.062) | 0.007 (± 0.002) | 3.33 |
| 24 |
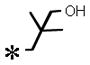
|
0.049 (± 0.020) | 0.391 (± 0.026) | 0.007 (± 0.003) | >10 | |
| 25 |
|
0.103 (± 0.016) | 0.468 | 0.009 (± 0.004) | 10 | |
| 26 |
|
0.094 (± 0.004) | 0.478 (± 0.488) | 0.006 (± 0.002) | >10 | |
|
| ||||||
| 27 |
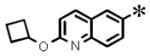
|
|
0.074 (± 0.023) | 0.159 (± 0.051) | 0.008 (± 0.002) | 3.33 |
| 28 |
|
|
0.071 (± 0.008) | 0.647 (± 0.244) | 0.010 (± 0.002) | >10 |
To evaluate the target selectivity of the AC compounds, 23 compounds in Table 1 were tested for inhibition of the mammalian kinase Src. Some of the Src inhibition data have been reported together with the CpCDPK1 and TgCDPK1 inhibition data previously,14,15 where high selectivity was observed of TgCDPK1 over Src. Similarly, significant selectivity of PfCDPK4 over Src was also observed. Sixteen (16) out of 23 tested compounds in Table 1 exhibited Src inhibition activity of >10 μM, and 17 out of 23 compounds displayed greater than 100-fold selectivity for PfCDPK4 over Src. Analogues that contain smaller 2-naphthyl C-3 Ar1 substituents, notably 1 and 12, are more potent against Src. Though compounds 1 and 12 display IC50s of 1.32 and 0.98 μM, the selectivity of PfCDPK4 over Src is still 33-fold and 12-fold, respectively. Moreover, the size of the R2 substituent also plays a significant role on Src activity.16 Two of the small R2 (isopropyl) inhibitors (23 and 27) exhibited relatively high Src activity at 3.33 μM. Therefore, with optimal Ar1 and R2 groups, high selectivity of PfCDPK4 over Src could be achieved, and the potential off-target effects could be avoided. As an indicator of selectivity at the cellular level, we measured the cytotoxicity of a number of potent inhibitors using human liver (HepG2) and lymphocyte (CRL-8155) cell lines (Table 2). Assays were performed following previously reported procedures.13,16 None of the compounds tested showed significant inhibition of cell growth at concentrations up to 30 μM. Furthermore, they were tested against the S147M recombinant PfCDPK4 to show their selectivity for the smaller gatekeeper enzyme. All the tested compounds are not effective against this mutated enzyme.
Table 2.
P. falciparum gametocyte exflagellation inhibition (%), enzymatic assay (IC50) results for a methionine gatekeeper mutant of PfCDPK4 (Ser147Met), growth inhibition of human cell lines (HepG2 and CRL-8155), and hERG inhibition (IC50). All results are the averages of at least two assays. N/T = not tested.
| AC-BKI | P. falciparum cells | P. yoelii | S147M PfCDPK4 IC50 (μM) | CRL8155 EC50 (μM) | HepG2 EC50 (μM) | hERG EC50 (μM) | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Exflagellation at 0.1 μM (% inhibition) | blood stage (μM) | Exflagellation at 0.1 μM (% inhibition) | |||||
| 2 | 64% | 7.2 | N/T | >2 | >30 | >30 | >10 |
| 4 | 73% | >20 | N/T | >2 | >30 | >30 | >30 |
| 5 | 79% | 18.8 | 55% | >2 | >30 | >30 | >30 |
| 6 | 89% | 9.2 | 31% | >2 | >30 | >30 | >30 |
| 8 | 84% | >20 | 31% | >2 | >30 | >30 | 22.8 |
| 12 | 75% | >20 | N/T | >2 | >30 | >30 | >30 |
| 14 | 80% | >20 | 29% | >2 | >30 | >30 | 18.8 |
| 17 | 81% | >20 | 43% | >2 | >30 | >30 | >30 |
| 26 | 39% | 7.5 | N/T | >2 | >30 | >30 | 14.1 |
| 27 | 69% | N/T | N/T | >2 | >30 | >30 | >30 |
Transmission blocking efficacy of 10 selected AC analogues was determined by measuring inhibition of P. falciparum exflagellation center of movement at a final compound concentration of 1 μM and 0.1 μM. All but one (26) of the selected leads had potent inhibition of P. falciparum microgametocyte exflagellation of >50% at a concentration of 0.1 μM (Table 2). Average inhibition of exflagellation at the 1 μM screening was 87% for all the compounds. Screening for inhibition of exflagellation at these low concentrations was chosen to give unbiased assessments of potential in vivo exposure of the AC-BKIs. In order to determine if these compounds may broadly affect multiple Plasmodium species, five AC analogues were similarly tested for their ability to inhibit P. yoelii gametocyte activation (Table 2). As seen with P. falciparum, these compounds reduced the number of centers of movement but some compounds were less effective on P. yoelii than P. falciparum. P. falciparum and P. yoelii CDPK4 share a serine “gatekeeper” residue and >98% amino acid identity in the active site which would suggest similar responses to BKIs. However, the reduced impact of BKIs on development of microgametocyte exflagellation in P. yoelii as observed in this study might suggest some degree of species to species differences in drug uptake.
Inhibition of P. falciparum exflagellation center of movement in this series of compound at the screening concentration 0.1 μM was more potent than expected based on the PfCDPK4 enzyme inhibition data. This may indicate the presence of other protein kinase targets found in the malaria parasites that are sensitive to the AC scaffold compounds but that are absent in the mammalian host. We have previously observed the presence of a similar phenomenon with this compound series when it was tested against T. gondii.14 Examples of potential plasmodia off-target kinase candidates that may be impacted by AC-BKIs include PfCDPK1 and PfPKG. These 2 kinases have threonine at their gatekeeper position which could potentially be inhibited by AC-BKIs. Evidence for inhibition by AC-BKIs of kinases with a threonine gate keeper residue is presented in the IC50 values in this case against PfCDPK1 (Table 1). Previous studies suggested that PfCDPK1 functions specifically in host cell lysis steps prior to the emergence of exflagellating microgametocytes18 while PfPKG controls gametocyte differentiation, or “rounding up”, an early event in P. falciparum gametogenesis.19 Both of these kinases modulate processes that occur prior to the steps catalyzed by PfCDPK4 and might have been impacted by AC-BKIs. There still exists the possibility that the potent inhibition of exflagellation by the AC-BKIs is due to a yet unknown plasmodia enzyme target(s). It is also possible that the AC-BKIs are relatively concentrated in the male gametocyte or that they (AC-BKIs) are not competing with as much ATP in the male gametocyte. Specific inhibition of malaria parasite exflagellation by the AC compounds however, does not appear to translate into automatic inhibition of the asexual blood stages of Plasmodium. Most of the initial asexual blood stage activity was lost from our BKI series during optimization for specificity. We specifically designed the compounds to not be therapeutic in blood stages of malaria to reduce the chances of acquired resistance from the large number of parasites encountered in the blood stage of the disease.9 However, few AC-BKIs (2, 5, 6 and 26) showed some observable anti-erythrocytic stage inhibition (Table 2). These compounds were tested for inhibitory activity against recombinant PfPKG. Kinaseglo© luciferase reagent (Promega, Madison, WI) based ATP depletion assay with luminescence readout was used as an indicator of catalytic activity. The IC50 values are 2.52 μM, 2.37 μM, 2.22 μM and 2.36 μM for AC-BKIs 2, 5, 6 and 26 respectively. Inhibition of PfPKG by a class of pyrimidine based compounds was recently shown to killed malaria parasites at late schizogony stage.20 Hence, there is a potential for redirecting AC-BKI chemical analogues for polypharmacology with distinct asexual and transmission stage targets. This would preserve the decreased selection pressure for drug-resistant parasites during the sexual stage and provide some activity against the asexual stage to aid in the clinical and parasitological cure. We have previously shown that BKIs with transmission blocking activity (BKI-1 and 1294) are orally available and have low toxicity in initial mouse studies.9,10 Recently, 1294 was found to have hERG inhibitory activity at therapeutic concentrations in two in vitro screening assays. hERG inhibition has been associated with long Q-T syndrome (cardiotoxicity).10,17 In the AC series of BKIs, we have used iterative testing to direct us away from this liability as hERG activity was not seen at >10 μM in all the analogues examined. In in vivo toxicity and pharmacokinetic studies of AC-BKIs, we have demonstrated that therapeutic plasma levels of compounds can be achieved after oral dosing in mice without toxicity.14 Based on these findings, we conclude that AC-BKIs can be delivered as a prophylatic formulation with a blood stage acting antimalarial like ACT. The benefit of this would include stopping widespread transmission of artemisinin-resistant and nonresistant malaria strains while alleviating clinical symptoms. Plasmodia CDPK4 activity is mostly activated in the mosquito host during the sexual stage of development; hence, evolution of resistance to AC-BKIs should be low in comparison with typical antimalarial drugs.
Acknowledgments
Research work described in this study was supported by the National Institute of Allergy and Infectious Diseases and National Institute of Child Health and Human Development of the National Institutes of Health under the award numbers R01AI089441, R01AI111341, R01GM086858, and R01HD080670, and by start-up funds from the Pennsylvania State University. The work was also supported by awards # 2014-06183 from the United States Department of Agriculture National Institute of Food and Agriculture.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.White NJ. Clin Infect Dis. 2008;46:172. doi: 10.1086/524889. [DOI] [PubMed] [Google Scholar]
- 2.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. Nature. 2005;434:214. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. World Malaria Report 2015. Geneva, Switzerland: Publications of the World Health Organization; 2015. WHO World malaria report 2015; pp. 1–280. [Google Scholar]
- 4.Tran TM, Li S, Doumbo S, Doumtabe D, Huang CY, Dia S, Bathily A, Sangala J, Kone Y, Traore A, Niangaly M, Dara C, Kayentao K, Ongoiba A, Doumbo OK, Traore B, Crompton PD. Clin Infect Dis. 2013;57:40. doi: 10.1093/cid/cit174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guidelines for the Treatment of Malaria. 3. WHO; Geneva: 2015. http://apps.who.int/iris/bitstream/10665/162441/1/9789241549127_eng.pdf?ua=1. [Google Scholar]
- 6.Bousema T, Drakeley C. Clin Microbiol Rev. 2011;24:377. doi: 10.1128/CMR.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. Am J Trop Med Hyg. 2006;75:402. [PubMed] [Google Scholar]
- 8.Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, Brinkmann V. Cell. 2004;117:503. doi: 10.1016/s0092-8674(04)00449-0. [DOI] [PubMed] [Google Scholar]
- 9.Ojo KK, Pfander C, Mueller NR, Burstroem C, Larson ET, Bryan CM, Fox AM, Reid MC, Johnson SM, Murphy RC, Kennedy M, Mann H, Leibly DJ, Hewitt SN, Verlinde CL, Kappe S, Merritt EA, Maly DJ, Billker O, Van Voorhis WC. J Clin Invest. 2012;122:2301. doi: 10.1172/JCI61822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ojo KK, Eastman RT, Vidadala R, Zhang Z, Rivas KL, Choi R, Lutz JD, Reid MC, Fox AM, Hulverson MA, Kennedy M, Isoherranen N, Kim LM, Comess KM, Kempf DJ, Verlinde CL, Su XZ, Kappe SH, Maly DJ, Fan E, Van Voorhis WC. J Infect Dis. 2014;209:275. doi: 10.1093/infdis/jit522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ojo KK, Larson ET, Keyloun KR, Castaneda LJ, Derocher AE, Inampudi KK, Kim JE, Arakaki TL, Murphy RC, Zhang L, Napuli AJ, Maly DJ, Verlinde CL, Buckner FS, Parsons M, Hol WG, Merritt EA, Van Voorhis WC. Nat Struct Mol Biol. 2010;17:602. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Merritt EA, Van Voorhis WC, Maly DJ. ACS Med Chem Lett. 2010;1:331. doi: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidadala RS, Ojo KK, Johnson SM, Zhang Z, Leonard SE, Mitra A, Choi R, Reid MC, Keyloun KR, Fox AM, Kennedy M, Silver-Brace T, Hume JC, Kappe S, Verlinde CL, Fan E, Merritt EA, Van Voorhis WC, Maly DJ. Eur J Med Chem. 2014;74:562. doi: 10.1016/j.ejmech.2013.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Ojo KK, Zhang Z, Rivas K, Vidadala RS, Scheele S, DeRocher AE, Choi R, Hulverson MA, Barrett LK, Bruzual I, Siddaramaiah LK, Kerchner KM, Kurnick MD, Freiberg GM, Kempf D, Hol WG, Merritt EA, Neckermann G, de Hostos EL, Isoherranen N, Maly DJ, Parsons M, Doggett JS, Van Voorhis WC, Fan E. ACS Med Chem Lett. 2015;6:1184. doi: 10.1021/acsmedchemlett.5b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, Ojo KK, Vidadala R, Huang W, Geiger JA, Scheele S, Choi R, Reid MC, Keyloun KR, Rivas K, Siddaramaiah LK, Comess KM, Robinson KP, Merta PJ, Kifle L, Hol WG, Parsons M, Merritt EA, Maly DJ, Verlinde CL, Van Voorhis WC, Fan E. ACS Med Chem Lett. 2014;5:40. doi: 10.1021/ml400315s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keyloun KR, Reid MC, Choi R, Song Y, Fox AM, Hillesland HK, Zhang Z, Vidadala R, Merritt EA, Lau AO, Maly DJ, Fan E, Barrett LK, VAN Voorhis WC, Ojo KK. Parasitology. 2014;141:1499. doi: 10.1017/S0031182014000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollard CE, Abi Gerges N, Bridgland-Taylor MH, Easter A, Hammond TG, Valentin JP. Br J Pharmacol. 2010;159:12. doi: 10.1111/j.1476-5381.2009.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sebastian S, Brochet M, Collins MO, Schwach F, Jones ML, Goulding D, Rayner JC, Choudhary JS, Billker O. Cell Host Microbe. 2012;12:9. doi: 10.1016/j.chom.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McRobert L, Taylor CJ, Deng W, Fivelman QL, Cummings RM, Polley SD, Billker O, Baker DA. PLoS Biol. 2008;6:e139. doi: 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green JL, Moon RW, Whalley D, Bowyer PW, Wallace C, Rochani A, Kumar R, Howell SA, Grainger M, Jones HM, Ansell KH, Chapman TM, Taylor DL, Osborne SA, Baker AB, Tatu U, Holder AA. Antimicrob Agents Chemother. 2016;60:1464. doi: 10.1128/AAC.01748-15. [DOI] [PMC free article] [PubMed] [Google Scholar]