

Abstract
To cause disease, a microbial pathogen must adapt to the challenges of its host environment. The leading fungal pathogen Candida albicans colonizes nutrient-poor bodily niches, withstands attack from the immune system, and tolerates treatment with azole antifungals, often evolving resistance. To discover agents that block these adaptive strategies, we screened 300,000 compounds for inhibition of azole tolerance in a drug-resistant Candida isolate. We identified a novel indazole derivative that converts azoles from fungistatic to fungicidal drugs by selective inhibition of mitochondrial cytochrome bc1. We synthesized 103 analogs to optimize potency (0.4 μM IC50) and fungal selectivity (28-fold over human). In addition to reducing azole resistance, targeting cytochrome bc1 prevents C. albicans from adapting to the nutrient-deprived macrophage phagosome and greatly curtails its virulence in mice. Inhibiting mitochondrial respiration and restricting metabolic flexibility with this synthetically tractable chemotype provides an attractive therapeutic strategy to limit both fungal virulence and drug resistance.
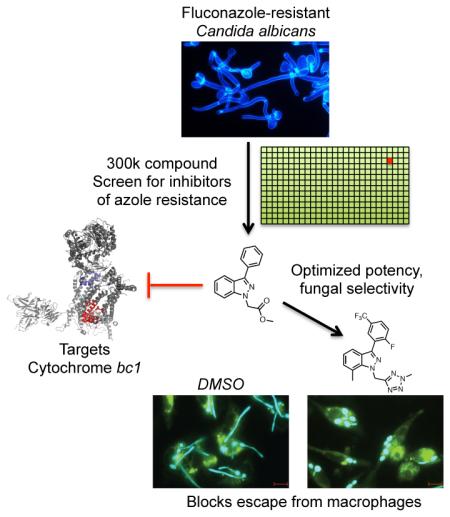
Graphical abstract
INTRODUCTION
Many pathogens have evolved to optimally exploit very particular niches within the host. But rigid specialization comes with several costs. Specialist pathogens struggle to adapt to the severe disparities in nutrient composition, immunological surveillance, and drug exposure found in different host compartments. Because of these challenges, hyper-specialized pathogens fail to disseminate and colonize new niches, an effect which limits their reproductive fitness. Furthermore, when perturbations of host physiology cause dramatic changes in local environmental conditions, pathogen populations that cannot adapt go extinct. In such circumstances, the adaptive plasticity of generalist pathogens provides strong evolutionary benefits (Kussell and Leibler, 2005).
Candida albicans provides a paradigm for the study of flexible and resilient pathogens. It is the most common cause of fungal infection in humans worldwide. In healthy humans, C. albicans thrives as a harmless commensal of the oral cavity and gastrointestinal tract (Calderone et al., 2009). Immunocompetent individuals can suffer from recurrent infections (e.g. diaper rash and vaginal infections), but they typically resolve with minimal morbidity. Mild to moderate immunosuppression often leads to more significant C. albicans infections of the oropharynx, esophagus, and other superficial sites. In severely immunocompromised patients, Candida and other fungal pathogens can invade the bloodstream and internal organs, causing sepsis, multi-organ system failure, and death (Pfaller and Diekema, 2010).
Invasive fungal infections are now responsible for an estimated 1.5 million deaths per year worldwide (Brown et al., 2012). The incidence of such infections is increasing due to climate change and the expansion of human populations into new environments, which exposes them to new pathogenic fungi (Byrnes et al., 2010). Exacerbating the problem, the increasing use of immunosuppressive therapies in an aging population disarms our intrinsic defenses against fungi. Compounding the problem yet further, the relatively recent evolutionary divergence of humans from fungi (relative to their divergence from bacteria) reduces the number of molecular targets available for selective therapeutic intervention. Indeed, only three classes of antifungal drug are broadly used in the clinic—and none is ideal (Roemer and Krysan, 2014).
Azoles are the most widely used antifungals in the clinic due to their safety and good oral bioavailability. They block fungal growth by disrupting ergosterol biosynthesis (through inhibition of the Erg11, lanosterol 14-alpha demethylase enzyme) (Odds et al., 2003). Other synthetic drug classes with single-agent, broad-spectrum antifungal activity have not been approved for systemic or mucosal infections, despite extensive efforts to discover and develop them (Roemer and Krysan, 2014). Though safe and highly effective in certain settings, azoles do have several drawbacks. First, their activity is fungistatic, not fungicidal, against Candida and most other fungal pathogens. This limited activity allows pathogens to persist, creating ideal conditions for the emergence of resistant strains. Indeed, azole resistance is increasing in prevalence, and is typically driven by pump-mediated drug efflux or by mutations in ERG11, the drug target (Cowen et al., 2014).
In the treatment of bacterial infections, combination therapy has proven highly effective and slowed the emergence of resistance. This approach has not been possible with the agents approved for the treatment of fungal infection, due to in large part to their antagonistic antimicrobial activities. Given the safety and efficacy of azoles, agents that render them fungicidal (in combination) and limit the emergence of resistance would have tremendous clinical utility. Promising targets for such intervention include two signal transduction proteins that enable survival of azole-induced membrane stress, calcineurin and protein kinase c (PKC) (Cruz et al., 2002; LaFayette et al., 2010; Sanglard et al., 2003). These metastable proteins require high levels of the molecular chaperone Hsp90 for function (Cowen and Lindquist, 2005; Diezmann et al., 2012). Inhibition of Hsp90, calcineurin, or PKC all render azole treatment fungicidal and dramatically reduce the emergence of resistant strains. However, the human homologs of these highly conserved proteins play critical roles in host stress tolerance and immune responses, necessitating fungal-selectivity if they are to be used in the context of an active infection. Selectivity has been difficult to realize. Other potential strategies for reducing azole resistance (Epp et al., 2010) have either not yet been pursued in drug development or have not proven successful in the clinic.
RESULTS
Identification of novel small molecules that reverse fluconazole resistance in C. albicans
To find new agents that might be useful in antifungal combination therapy, we screened for compounds that are fungicidal in combination with fluconazole and prevent the emergence of fluconazole resistance. We employed a previously reported isolate of C. albicans (referred to as CaCi-2) that had evolved resistance to fluconazole during the treatment of an HIV-infected patient (Redding et al., 1994) (White, 1997). We screened this isolate at 8 μg/ml fluconazole, which slows, but does not stop, its growth (Figure 1A). Of 302,509 compounds screened at 25μM, 1654 compounds (0.5%) inhibited growth >75% in combination with fluconazole (Figure 1B). Hits were filtered for potency (IC50 < 1 μM), lack of toxicity to mouse 3T3 fibroblasts (IC50 > 26μM), and lack of strong single-agent activity without fluconazole (IC50 > 26μM).
Figure 1. Discovery of Inz-1 by high-throughput phenotypic screening.

A) Schematic of the high throughput screen for inhibitors of azole tolerance. B) Triage of hits from the screen, indicating number of compounds that passed each filter. C) Profiling of fungal growth under stress conditions in the presence of Inz-1 and other small molecules. Strain SC5314 was grown for 24hr in the presence of the indicated stressors and small molecule drugs (concentrations indicated in Experimental Procedures). Growth was measured by reading OD600 after 24hr. Results were normalized to the growth under each stressor condition (rows) with addition of DMSO used as negative control and visualized as a heatmap using TreeView. D) Effect of Inz-1 on growth of S. cerevisiae (strain BY4741) and C. albicans (SC5314) in media containing glucose or glycerol as carbon source. Figures in C-D depict data pooled from two independent experiments.
Next we filtered the screen hits for compounds with activity against a clinical isolate that had emerged later in the treatment of the same patient and had higher levels of resistance (CaCi-8; (White, 1997)). We then eliminated chemically reactive compounds, compounds with metal-chelating motifs, and promiscuous compounds that were hits in multiple other assays run using the same chemical library (Baell and Holloway, 2010). Thirty candidates were advanced and retested from dry powders, and 3 were chosen for structure-activity relationship studies. Detailed information on the primary screen is freely available through the PubChem database (Pubchem AID 2007; http://pubchem.ncbi.nlm.nih.gov/bioassay/2007). Three chemical series, piperazinyl quinolines (IC50 0.7 μM), tetracyclic indoles (IC50 0.8μM), and indazoles (IC50 1.6μM) were selected for synthesis of ~30 analogs to explore preliminary structure activity relationships (SAR) as well as biological and chemical properties (Youngsaye et al., 2012; Youngsaye et al., 2011).. Of these three, the indazoles exhibited the best combination of high maximal fungal growth inhibition, solubility, dynamic SAR and chemical tractability. We thus pursued the indazole series (based on Inz-1) for target identification and further chemical development.
Inz-1 Blocks Mitochondrial Respiration by Targeting Cytochrome bc1
To better understand the effects of Inz-1 on fungal cells, we generated a profile of Inz-1-induced phenotypes under a variety of growth conditions and in the presence of various stressors. These conditions included high salt, oxidative stress, nutrient deprivation, and exposure to diverse toxic agents. We then compared the profile of phenotypes induced by 10μM Inz-1 to that of other small molecules that sensitize C. albicans to azoles, including inhibitors of Hsp90, Calcineurin, PKC, histone deacetylases, PKA, and ARF cycling (Cowen and Lindquist, 2005; Epp et al., 2010; LaFayette et al., 2010; Marchetti et al., 2000; Smith and Edlind, 2002). Inz-1 produced a unique phenotypic profile (Figure 1C). Most intriguingly, Inz-1 potently inhibited growth of both C. albicans and the model yeast S. cerevisiae in media containing glycerol as the sole carbon source, while only mildly slowing growth in glucose (Figure 1D). Although yeasts can ferment glucose and other sugars, utilizing glycerol as the sole carbon source requires cellular respiration. This result suggested that Inz-1 interferes with mitochondrial function.
As a way to identify the target of Inz-1, we exploited its potent, single-agent inhibition of respiratory growth to select resistant mutants for whole-genome sequencing. A wild-type S. cerevisiae strain (BY4741) was plated on media containing glycerol as carbon source and 10μM Inz-1. After 7 days of incubation, two colonies with robust growth were isolated. The resistance of these isolates was confirmed and their genomes were sequenced together with that of the parental BY4741 strain. Alignment of mutant and parental genomes followed by polymorphism discovery (Experimental Procedures) revealed that both resistant isolates contained an identical mutation in the cytochrome B (COB) gene of the mitochondrial chromosome (Figure 2A). This mutation replaced phenylalanine 90 with tyrosine (F90Y) in the mitochondrial cytochrome B protein, which is the core enzymatic subunit of cytochrome bc1 (Complex III) of the electron transport chain.
Figure 2. Inz-1 is a fungal-selective inhibitor of mitochondrial cytochrome bc1.

A) Mutations found in cytochrome B (COB) in two Inz-1-resistant isolates, visualized by the Integrative Genomics Viewer (IGV). B) Inhibition of yeast cytochrome B enzymatic activity by Inz-1 in both wild-type and F90Y mutant mitochondria. C) Resistance of F90Y mutant to growth inhibition by Inz-1. D) Fluconazole tolerance of C. albicans CaCi-2 WT, treated with Inz-1, or rip1Δ/Δ deletion mutant. Growth at the indicated concentrations of fluconazole was measured by broth microdilution using OD600 as endpoint. E) Comparison of inhibition of cytochrome B activity in C. albicans and human (HEK293) mitochondria. F) Comparison of the effect of Inz-1 on proliferation of HepG2 and NIH-3T3 cells under respiring conditions (DMEM-galactose) with effect on proliferation of C. albicans in presence of 8 μg/mL fluconazole. Graphs in B-F each represent data pooled from two independent experiments, with error bars depicting S.E.M.
Cytochrome bc1 is an 11-protein complex embedded in the mitochondrial inner membrane that couples the oxidation of ubiquinol to the reduction of cytochrome C (Hunte et al., 2003). The activity of the complex is essential to pump protons from the mitochondrial matrix into the intermembrane space and maintain the proton gradient required for mitochondrial ATP production. Taken together with the specific inhibition of respiratory growth by Inz-1, the resistance conferred by this mutation suggested that cytochrome bc1 might be the direct target of Inz-1.
Inz-1 Inhibits Cytochrome bc1 Activity in Isolated Yeast Mitochondria
To determine whether Inz-1 inhibits cytochrome bc1, we used a well-established in vitro assay of its enzymatic activity using purified yeast mitochondria, permeabilized with Triton X-100 (Gutierrez-Cirlos et al., 2004). This assay exploits a spectral shift in cytochrome C upon its reduction by cytochrome bc1 and ubiquinol. Indeed, Inz-1 exhibited dose-dependent inhibition of cytochrome bc1 from both S. cerevisiae and C. albicans (both isolated by the same procedure) with an IC50 of 2.5 and 8.0 μM, respectively (Figure 2B-E). We suspect that the differences in Inz-1 IC50 between enzymatic and growth assays could be due to the differences between the detergent-containing enzymatic buffer and yeast growth media, high cellular uptake of Inz-1, or a requirement of only mild cytochrome B enzyme inhibition to block growth. However, we cannot fully rule out the contribution of off-target effects. To provide yet stronger proof that cytochrome B is the relevant target of Inz-1, we performed the same assay with mitochondria purified from the indazole-resistant cytochrome b F90Y mutant and asked if the enzymatic activity was resistant to Inz-1. Indeed, a four-fold higher concentration of Inz-1 was required to achieve 50% inhibition of the F90Y mutant enzyme than for WT (Figure 2B). This increased resistance to inhibition of the enzymatic activity of F90Y mutant cytochrome B closely parallels the 4-fold increased resistance to growth inhibition by Inz-1 in this mutant stain (Figure 2C).
To confirm that impairment of cytochrome b function sensitizes C. albicans to azoles, we used genetic tools to disable the complex. Methods to selectively mutate the mitochondrial genome in C. albicans are unavailable. Instead, we deleted a nuclear-encoded gene, RIP1, which encodes a conserved member of the complex that is required for cytochrome bc1 function (Beckmann et al., 1987). We knocked out both copies of the RIP1 gene in the fluconazole-resistant CaCi-2 strain, and reconstituted the gene to control for potentially extraneous effects of the deletion. rip1Δ/Δ mutants were indeed hypersensitive to azole treatment, mimicking the effects of Inz-1 (Figure 2D).
Inz-1 is selective for yeast over human cytochrome bc1
The cytochrome bc1 complex is highly conserved from fungi to humans. To determine the relative fungal selectivity of Inz-1, we tested its effect on mitochondria purified from HEK293 human embryonic kidney cells. Inz-1 inhibited human cytochrome bc1 activity only weakly, with an IC50 of 45.3 μM, 5.6-fold higher than the IC50 for the C. albicans homolog (Figure 2E).
We then interrogated the toxicity of Inz-1 to respiring mammalian cells. In tissue culture, most mammalian cell lines do not require mitochondrial respiration for proliferation. To impose a dependence on respiration for growth, we cultured cell lines in media containing galactose instead of glucose (Marroquin et al., 2007). We used a luminescent assay of cellular ATP content as a stringent test for inhibition of mitochondrial respiration (the dominant source of ATP production in these conditions) in HepG2 hepatocellular carcinoma cells and NIH-3T3 mouse fibroblasts. Inz-1 inhibited proliferation under forced respiration conditions only at 32 μM (the solubility limit), and only mildly at that concentration (Figure 2F). These results indicate that the inhibition of respiration by Inz-1 is at least moderately selective for fungal over human cells.
Mutational and Structural Analysis Suggests Inz-1 Binds the Cytochrome B Qo Site
Cytochrome b is a large protein with multiple deep potential drug-binding pockets. To investigate regions critical for Inz-1 activity, we isolated additional Inz-1-resistant mutants and sequenced their mitochondrial cytochrome B genes. From these selections, we identified P270L and L275S mutations that confer resistance to Inz-1 (Figure 3A). Along with F90Y, P270L and L275S map to a small region of the previously solved crystal structure of the protein, known as the Quinol-oxidizing (Qo) pocket (Figure 3B) (Hunte et al., 2003). This site is the target of the antimalarial drug atovaquone, which, unlike Inz-1, is an ubiquinol substrate analog (Kessl et al., 2003).
Figure 3. Structural analysis of Inz-1 binding to cytochrome bc1.

A) Minimal Inhibitory Concentration (MIC) of Inz-1 for indicated cytochrome B mutants under respiratory growth conditions in S. cerevisiae. MIC’s were concordant from two independent experiments. B) Structure of yeast cytochrome B complex (PDB:1EZV) indicating location of Qi and Qo pockets. C) Structural alignment of yeast (PDB:1P84) and bovine (PDB:1SQV) cytochrome B showing Qo site and indicating location of two mutations that confer resistance to Inz-1. D) Computational docking of Inz-1 into Qo pocket of yeast cytochrome bc1 (PDB:1EZV) with location of resistance-conferring mutations overlaid.
An alignment of the structures of the Qo site from the yeast and mammalian (bovine) homologs revealed several key differences that could be exploited for selective inhibition (Figure 3C). Most prominently, L275 in the fungal enzyme is replaced in the mammalian enzyme with a bulkier phenylalanine residue, which protrudes into of the central cavity of the Qo site. To determine whether this residue is involved in the fungal selectivity of Inz-1, we tested a previously described yeast L275F mutant (Hill et al., 2003; Kessl et al., 2003). This mutation conferred ~4-fold resistance to Inz-1. We also noted that the previously described G143A mutant conferred strong resistance to Inz-1 (Figure 3A). From these findings, we conclude that the Qo pocket is the most likely site for Inz-1 binding. Moreover, differential interactions with fungal L275 and human F275 residues in this site might be partially responsible for the fungal selectivity observed (Figure 3A).
To explore the binding mode of Inz-1, we used computational docking of the compound onto the structure of cytochrome b. Inz-1 was docked into a 16 Å grid surrounding the Qo site, using a previously solved structure of the cytochrome bc1 complex (PDB: 1EZV). After sampling possible conformations of Inz-1 and their position within the active site, a lowest energy structure was computed (Figure 3D). Notably, in this proposed conformation, the indazole-2-nitrogen, which is required for activity in all analogs (figure S1) is positioned 2.5Å from L275; mutation of this residue confers strong resistance to Inz-1. In addition, the α-carbon of the ester, another essential element of the pharmacophore, is positioned 2.4Å from P270, while the carbonyl oxygen is 2.5 Å from G143; mutation of either of these residues confers resistance to Inz-1 (figure 3A). The phenyl group fits into a large hydrophobic pocket formed by I125, I147, L150, L275, M295, F296, and I299 (Figure 3A).
Taken together, these data very strongly suggest that the Qo pocket is the binding site for Inz-1. Although this site is targeted by several agricultural fungicides, including the widely used strobilurins, none of these compounds have been pursued for the treatment of human fungal infections due to their lack of selectivity for fungi, poor pharmacological properties, or inactivity against relevant human pathogens (Esser et al., 2004). The low molecular weight and chemical tractability of Inz-1, combined with its potency and fungal selectivity, made it an attractive lead for further development through medicinal chemistry efforts.
Optimization of Inz-1 Through a Structure-Activity Relationship Study
A total of 103 analogs of Inz-1 were synthesized and assayed in an effort to explore structure-activity relationships and identify a more potent and selective inhibitor of the cytochrome bc1 enzyme (a complete list of compounds is provided in Table S1). We first evaluated various analogs presenting different substitution patterns on the top aromatic ring (Table S2). Interestingly, the combination of a fluorine atom in position 2 as well as a trifluoromethyl group in position 5 proved optimal and induced a significant enhancement of potency against the fungal enzyme (Inz-2, Figure 4; see also Inz-10 and Inz-19, Table S2). A concomitant improvement of the selectivity index was also noticed. Unfortunately, Inz-2 appears highly lipophilic with a clogP value of 4.7, which typically results in low aqueous solubility and high metabolic clearance (Lipinski et al., 2001). Unfortunately, all attempts to introduce polar substituents or heterocycles invariably resulted in compounds with lower antifungal activity (Table S2). Next, modification of the indazole core of the molecule was investigated (Table S3). Addition of a methyl group in position 7 was beneficial to both the activity and the selectivity of the resulting compound (Inz-3, Figure 4). Remarkably, a molecule combining the fluorinated aromatic ring previously identified together with a methyl indazole further decreased the IC50 value down to 0.026 μM, while improving the selectivity index to more than 398-fold in favor of the Candida versus human HepG2 cells (Inz-4, Figure 4). It is worth noting that the successive structural modifications leading to Inz-4 coincided with a noticeable gain in ligand efficiency as compared with Inz-1 (from 0.36 to 0.41, Table S3) (Hopkins et al., 2014).
Figure 4. Chemical optimization of Inz-1.

104 analogs of Inz-1 were synthesized and tested for inhibition of yeast and human cytochrome B. Data for selected analogs are shown; results for the remaining compounds are shown in Table S1. Each data point represents the average of at least 2 independent experiments. Experiments were performed as described in Figure 2. Candida growth inhibition indicates the IC50 for growth inhibition of CaCi-2 by compounds in the presence of 8 μg/mL fluconazole. Selectivity was calculated by dividing growth inhibition IC50 of HepG2 cells by IC50 for growth inhibition of C. albicans.
Unfortunately, all of these compounds contained a highly labile ester group susceptible to hydrolysis in the presence of serum esterases. Plasma stability testing indeed demonstrated that Inz-1 is fully degraded after an incubation of 1 hour at 370 C in human plasma (Table S5). Such instability in serum might contribute to their lack of toxicity to cultured HepG2 cells and hence lead to an overestimation of their selectivity index, and moreover limit their utility as probes (Inz-1 to Inz-4, Figure 4). To address this problem, many analogs of Inz-1 containing surrogates of the methyl ester were prepared and evaluated in vitro (Table S4). Among them, two nitrogen-rich heterocycles, methyltetrazole (Inz-34) and pyrazine (Inz-37) retained activity. When combined with the structural determinants exemplified in Inz-4, the methyltetrazole analog Inz-5 proved to be the most suitable compound showing sub-micromolar activity against Candida proliferation as well as an acceptable selectivity index of 28 (Table S3 and Figure 4, see also Inz-38 and Inz-39). A detailed description of the synthetic route towards Inz-5 is provided in the supporting information.
Plasma stability assessment demonstrated that Inz-5 was fully recovered after 3 hours of incubation in human serum, confirming its superiority over Inz-1 (Table S5). Inz-1 is also unstable in mouse liver microsomes, with <1% of the compound remaining after 15 minute incubation at 370 C (table S5). Inz-5 demonstrated modestly improved microsomal stability (19.5% remaining after 15 minutes), but still requires further optimization to limit metabolism by hepatic CYP enzymes and achieve adequate systemic exposure in an animal model.
Interestingly, Inz-5 achieved complete inhibition of C. albicans cytochrome bc1 activity (Figure S1), whereas the original hit Inz-1 allowed significant residual activity of the enzyme (Figure 1C). Although the fungal selectivity of Inz-5 may require further optimization, we note that the clinically useful antimalarial drug atovaquone inhibited human cytochrome bc1 with an IC50 of 1.8 μM in our assay and was toxic to respiring HepG2 cells with an IC50 of 12.7 μM (Figure 4), a concentration that may be reached in patients, at least transiently (although it has low nanomolar potency against the malarial cytochrome B (Hughes et al., 1998). Therefore, clinical experience with atovaquone indicates that partial inhibition of the Qo site of cytochrome bc1 may be well-tolerated in humans. Encouraged by such considerations, Inz-5 was chosen as a tool compound for further biological investigations.
Inhibition of Cytochrome bc1 Prevents the Emergence of Fluconazole Resistance and Renders Fluconazole Fungicidal
The potency, selectivity, and improved stability of Inz-5 allowed us to investigate the role of mitochondrial respiration in diverse aspects of fungal pathogenesis. We first interrogated the effect of cytochrome B inhibition on the emergence of resistance to fluconazole. We plated C. albicans on media containing 64 mg/L fluconazole, 10 μM Inz-5, or both drugs (Figure 5A). As expected, resistant colonies emerged at a relatively high rate (~0.5%) on the plates containing fluconazole alone. Plates containing Inz-5 alone exhibited reduced colony size, but no reduction in the total number of colonies. However, the combination of Inz-5 with fluconazole completely abrogated the emergence of resistant colonies (Figure 5A).
Figure 5. Inz-5 renders fluconazole fungicidal and reduces the emergence of resistance.

A) 2×103 C. albicans wild-type (SC5314) cells were plated on media containing 64 mg/L fluconazole (top right), 10μm Inz-5 (bottom left) or both compounds in combination. Images were acquired after 4d incubation at 37C. B) 5×103 C. albicans SC5314 cells were incubated in the presence of 32 mg/L fluconazole, 10 μM Inz-5, or both compounds while C. albicans rip1Δ/Δ were incubated in the presence and absence of 32 mg/L fluconazole. To measure clonogenic survival, dilutions were plated on YPD after 24 and 48 hr. Data are pooled from two independent experiments; error bars indicate S.E.M.
Up to this point, it was unclear whether Inz-5 merely enhances the fungistatic activity of fluconazole, or if the combination renders cells inviable. To distinguish between these possibilities, we treated liquid cultures of C. albicans with 32 mg/L fluconazole, 10 μM Inz-5, or both compounds, and measured viable colony forming units recoverable from the culture over time. As expected, fluconazole alone slowed growth of the wild-type strain but did not cause a decrease in viability (Figure 5B). Inz-5 also partially slowed growth alone. Importantly, combination of fluconazole with Inz-5 reduced the number of viable colonies by 97%. The fungicidal effect of fluconazole was even stronger against the rip1Δ/Δ mutant (in which cytochrome b function is completely eliminated) (Figure 5B). Thus, Inz-5 both prevents the de novo emergence of new resistance to fluconazole and transforms fluconazole from a fungistatic compound to a fungicidal one.
Inhibiting Cytochrome B with Inz-5 Restricts Carbon Source Utilization
Having established the effects of Inz-5 in combination with fluconazole, we used this compound as a tool to probe the role of cytochrome bc1 in the physiology and virulence of C. albicans. A key function of mitochondrial respiration conserved across eukaryotes is to support ATP production and biomass expansion when sugar sources are limiting. In the laboratory, fungal pathogens are typically studied in high glucose conditions (2%), in which ATP can be readily produced from glycolytic fermentation without respiration. However, in host environments, sugars are much scarcer. In the bloodstream, readily fermentable sugars are present at 0.1-0.2%, and in other niches they are present at lower concentrations or completely absent (Brown et al., 2014; Lorenz et al., 2004; Ramirez and Lorenz, 2007). To infect humans, therefore, Candida must rely on utilization of suboptimal carbon sources, including lactate, acetate, fatty acids and amino acids (Ene et al., 2014). Might Inz-5 restrict the metabolic flexibility of C. albicans by preventing utilization of these carbon sources?
Under conditions of high glucose (and thus fermentative metabolism), discs containing 10 μg Inz-5 caused very little growth inhibition in a diffusion halo assay on agar plates (Figure 6A). On agar plates with 80% fetal bovine serum as the nutrient source (~0.1-0.2% glucose), Inz-5 caused slight growth inhibition. And in media containing the non-optimal sugar galactose, which is abundant in the GI tract. Inz-5 also caused moderate growth inhibition. But when non-fermentable carbon sources were tested, the inhibitory effect of Inz-5 was dramatically stronger. On media containing amino acids, lactate, or acetate as the carbon source, Inz-5 exhibited potent and profound growth inhibition (Figure 6A, bottom panel).
Figure 6. Inhibition of cytochrome bc1 by Inz-5 prevents adaptation to certain carbon sources.

A) Growth of ~104 C. albicans SC5314 cells on agar plates containing various carbon sources in the presence of a filter disc containing DMSO (top) or 10μg Inz-5 dissolved in DMSO (bottom). B) Co-culture of BFP-labeled C. albicans and GFP-tubulin-labeled mouse bone-marrow-derived macrophages. C) Quantification of effects on filamentation observed in (B); data from two independent experiments; error bars indicate S.E.M. D) Relative fungal survival quantified by resazurin dye reduction after 14 hr co-culture at MOI of 0.125 in the presence of mouse bone marrow-derived macrophages. Data from two independent experiments; error bars indicate S.E.M.
Inhibition of Cytochrome bc1 Sensitizes C. albicans to Attack by Macrophages
Perhaps the most severe nutrient limitation that C. albicans encounters is upon engulfment into the macrophage phagosome (Lorenz and Fink, 2002). Upon phagocytosis, C. albicans induces the glyoxylate cycle of 2-carbon metabolism and activates a morphogenetic program of filamentation to enable escape. To determine if inhibition of fungal cytochrome B might impair this process, we co-cultured C. albicans engineered to express blue fluorescent protein (BFP) with mouse bone-marrow-derived macrophages, with or without 5 μM Inz-5. As expected, the macrophages phagocytosed the fungi within 30 minutes, in the presence or absence of the compound (Figure 6B). The fungi showed filamentous growth and escaped the macrophages within 2 hours in the absence of Inz-5. In contrast, with Inz-5 present, C. albicans remained trapped in the phagosome (Figure 6B-C). Similar effects were observed when macrophages were co-cultured with the rip1Δ/Δ mutant, confirming that loss of cytochrome B function is responsible for this phenotype (Figure 6B-C).
We also asked if Inz-5 would sensitize C. albicans to growth inhibition by macrophages. During a more prolonged 14-hour co-culture, untreated wild-type C. albicans evaded macrophage attack and grew robustly (Figure 6D). Inhibition of cytochrome bc1 by 5 μM Inz-5 (or deletion of RIP1) sharply curtailed fungal growth in the presence of macrophages. These results suggest that inhibition of cytochrome bc1 dramatically increases the ability of host macrophages to control the growth of C. albicans.
Cytochrome bc1 Is Required for Fungal Virulence and Drug Resistance in Mice
Given the effects of cytochrome bc1 inhibition on properties key to fungal pathogenesis, we asked if its function is required for fungal virulence in a whole animal model. Unfortunately, pilot experiments indicated that Inz-5 will require further optimization of its systemic exposure and metabolic stability to achieve sufficient target engagement for use in mice (data not shown). As a surrogate for the effects of the compound, we examined our rip1Δ/Δ mutant that was created in the CaCi-2 fluconazole-resistant background. Both CaCi-2 (and the rip1Δ/Δ:RIP1 re-integrant strain) killed mice within several days, as expected (Figure 7A). In stark contrast, the same inoculum of the rip1Δ/Δ mutant did not cause severe disease in the mice for over two weeks.
Figure 7. Genetic disruption of cytochrome bc1 severely reduces fungal virulence and fluconazole tolerance, but increases brain colonization.

A) Kaplan-Meier analysis of survival after tail-vein infection of Balb/C mice with 5×105 fungal cells of indicated strains. N = 7 mice per strain, data pooled from two independent experiments. B) Viable fungal burden isolated from brain 4d after infection with 6×104 CFU of indicated strains. N = 6-7 mice per strain; data pooled from two independent experiments. C) Kaplan-Meier analysis of survival for mice infected with 106 CFU of CaCi-2 then treated with fluconazole (24 mg/kg IP) once daily X 3d. N = 6-10 mice per strain; data pooled from two independent experiments. D) Mice were infected with 6×104 CFU of the indicated strains, then treated with fluconazole (24 mg/kg IP, once daily X3). After treatment mice were monitored for 4 more days, then sacrificed and fungal burden in kidney and brain quantified. Data are pooled from two independent experiments.
However, after 17-20 days, mice infected with the rip1Δ/Δ strain exhibited agitated behavior and a head-tilt, suggestive of a possible central nervous system (CNS) infection, and were promptly euthanized. Indeed, necropsy revealed extensive fungal burden within the central nervous system (CNS) of these mice. To quantitatively test whether cytochrome bc1 compromise increases invasion of the CNS, we infected mice with a low inoculum of WT, rip1Δ/Δ, and the RIP1 reconstituted strain. We allowed this infection to proceed for four days, then sacrificed the completely asymptomatic mice and quantified fungal burden in the brain and kidney. While the rip1Δ/Δ mutant was almost completely defective in kidney colonization, it exhibited approximately 5-fold higher fungal burden in the brain than the otherwise isogenic wild-type or re-integrant strain (Figure 7B). Thus, while the rip1Δ/Δ mutant does not kill mice through the typical pattern of systemic organ system invasion and compromise by C. albicans, this mutant has an increased capacity to invade and proliferate within the CNS.
Azoles are not the treatment of choice for disseminated candidiasis, as their fungistatic activity is insufficient to fully eradicate the fungus. We tested whether the fungicidal effects of disabling cytochrome bc1 in the presence of azoles would provide more durable disease control and act as a curative therapy. We infected mice with a high inoculum of fungus and then treated them for 3 days with a standard dose of fluconazole that controls but does not eradicate the infection. After discontinuation of therapy, we monitored the mice for recurrence of the infection over the next four weeks. In mice infected with CaCi-2, severe infection recurred within 1-2 weeks of stopping treatment. Only 10% of these mice survived (Figure 7C). In mice infected with the rip1Δ/Δ mutant, 90% survived, with only one mouse succumbing to CNS infection after more than 3 weeks.
Given the increase in brain colonization in mice infected with strains lacking cytochrome function, we tested if this adverse event was eliminated by fluconazole treatment. We infected mice with a low inoculum of CaCi-2 or rip1Δ/Δ, allowed infection to proceed for four days, treated for 3 days with fluconazole, and then waited four more days before sacrificing mice. Quantification of fungal burden in both the brain and kidney established that fluconazole treatment eliminated brain colonization in these mice (Figure 7D). We conclude that even if Candida lacking cytochrome bc1 function were to succeed in colonizing the brain, they remain highly susceptible to fluconazole. Thus, these two antifungal strategies strongly potentiate each other to provide far more efficacious control of Candida infection.
Inz-5 Inhibits Fungal Pathogens Separated by Over a Billion Years of Evolution
The Qo binding site for Inz-5 is strictly conserved across evolutionarily distant fungal pathogens, including the critical L275 residue, which dictates the fungal selectivity of this scaffold (Figure S2A). Consequently, we anticipated that Inz-5 might have activity against a broad range of species. As a preliminary assessment of its spectrum, we tested its effects on Scedosporium prolificans, Aspgerillus terreus, and Rhizopus oryzae. These filamentous molds cause life-threatening invasive infections of the lungs and central nervous system that respond poorly to available agents. These species are also closely related to agricultural pathogens that cause devastating crop failures. As a single agent, Inz-5 exhibited promising growth inhibition of all three fungi, which was enhanced under respiratory growth conditions (Figure S2B).
DISCUSSION
The evolution of mechanisms that permit flexibility and resilience in the face of environmental challenges has rendered C. albicans a formidable pathogen. Here we demonstrate that mitochondrial respiration presents a pharmacologically targetable vulnerability required for these adaptive strategies. We discovered and optimized a potent and fungal selective inhibitor of cytochrome bc1 that abrogates the ability of C. albicans to tolerate exposure to triazole drugs and evolve resistance. Disabling cytochrome bc1 also prevents fungal adaptation to nutrient deprivation, sensitizes C. albicans to attack by macrophages, and curtails virulence in mice. Restricting the adaptive plasticity of fungal pathogens by inhibiting respiration may present a broadly useful approach to the development of sorely-needed combination therapies.
Considerable progress has been made in identifying pathogen-specific pathways, such as the glyoxylate cycle, that enable survival in the face of carbon starvation (Barelle et al., 2006; Ene et al., 2014; Lorenz and Fink, 2002). However, given the formidable metabolic flexibility of C. albicans, pharmacological targeting of any individual pathway may prove ineffective as a therapeutic strategy. In support of this notion, mutants lacking a functional glyoxylate cycle exhibit only partially reduced virulence (Barelle et al., 2006). In contrast, the mitochondrial electron transport chain is a central molecular node in the cellular metabolic network, and is required for many pathways supporting metabolic adaptation. It is thus unlikely that fungi can readily tolerate or bypass its inhibition. Our work with Inz-5 suggests that therapeutic efforts in this area need not focus on pathways unique to fungal pathogens. Indeed, mitochondrial proteins with conserved human homologs such as cytochrome bc1 can provide excellent targets for intervention. We anticipate that mitochondria will prove essential to many other fungal virulence processes, as others have begun to reveal (Grahl et al., 2015; Shingu-Vazquez and Traven, 2011; Sun et al., 2013).
Though promising, inhibiting fungal cytochrome bc1 must overcome several challenges to be useful as a therapeutic strategy. As Inz-5 shows, the optimization of agents that can access and occupy a hydrophobic pocket in the inner mitochondrial membrane but retain sufficient aqueous solubility to achieve adequate levels in the blood poses a formidable challenge. In addition, a high degree of fungal selectivity may be necessary for safety in patients. Nevertheless, the successful use of the cytochrome bc1 inhibitor atovaquone in antimalarial therapy suggests that both of these challenges can be overcome to yield a safe and effective therapeutic. In addition, recent advances in the co-crystallization of inhibitors (including atovaquone) with cytochrome bc1 could aid in the structure-based design of improved agents (Birth et al., 2014).
The increased brain colonization of the rip1Δ/Δ mutant is also a cause for concern, and future research will be needed to determine if it proceeds through previously described mechanisms of fungal trafficking to the brain (Liu et al., 2011). However, the clinical relevance of this phenomenon in low-virulence mutants is unclear (Ariyachet et al., 2013), and the brain infections caused by rip1Δ/Δ respond strongly to azole treatment. Another critical challenge will be to determine if mitochondrial inhibitors can be effective against Candida glabrata, in which loss of mitochondrial respiration appears to increase azole resistance through induction of drug efflux (Ferrari et al., 2011). But even if targeting of mitochondrial respiration is ineffective against C. glabrata, it may be effective in the treatment of emerging molds that are refractory to treatment with current agents. The extremely broad conservation of the Qo pocket of cytochrome bc1 across fungi suggests that these inhibitors could have a very broad spectrum of activity against emerging fungal pathogens, for which effective agents are urgently needed (Brown et al., 2012).
Despite extensive efforts, target-based cell-free screening of synthetic chemical libraries has failed to deliver any new classes of antifungals or antibacterials in clinical practice (Payne et al., 2007; Roemer and Krysan, 2014). In part, this is due to the fact that synthetic compounds with good activity in cell-free assays fail to traverse microbial cell walls and membranes. Most (arguably all) classes of antibiotics and antifungals of the 20th century were identified (either intentionally or serendipitously) by phenotypic screening of natural product extracts and synthetic libraries, assaying simply for single-agent microbial growth inhibition. Yet, novel classes of natural product antimicrobials are increasingly hard to discover, and their development into drugs poses substantial challenges (Roemer et al., 2011). Screening synthetic chemical libraries for microbial growth inhibition suggests that the few classes with useful activity as single agents have already been discovered (Payne et al., 2007). Phenotypic screening in combination with older, now less effective antimicrobials might uncover therapeutic value in vast regions of chemical space that have previously been ignored (Ejim et al., 2011; Spitzer et al., 2011). Indeed, phenotypic screens can provide an excellent means for the discovery and validation of disease-relevant druggable proteins that can motivate future target-based screens. To this end, our discovery and optimization of Inz-5 as an antifungal provides a broadly applicable blueprint for similar projects against other medically challenging microbial pathogens.
EXPERIMENTAL PROCEDURES
Chemical Synthesis and Analysis
All details of chemical reagents, synthesis, and analytical methods are provided in the Supplemental Experimental Procedures.
High Throughput Small Molecule Screen
Detailed methods used in the high throughput small molecule screen performed at the Broad Institute MLPCN center are freely available online at: http://www.ncbi.nlm.nih.gov/books/NBK98920/
A daily cutoff of Z’>0.5 was applied to all primary screening data as a quality control metric. Hit selection and triage are described in the Supplemental Experimental Procedures.
Fungal Growth Assays, Drug Treatments and MIC Determination
C. albicans and S. cerevisiae were maintained under standard conditions, and growth assays were performed as described previously (Vincent et al., 2013). Phenotypic profiling experiments were performed in 96-well flat-bottom plates by diluting overnight YPD cultures to a starting OD of 0.0003, then assaying growth in drug and stress conditions after 24 or 48 hours (as indicated) by reading OD600. In these experiments, drugs and concentrations included 0.9M NaCl, 0.05μg/mL aureobasidin, 80 μM bathophenanthroline disulfonic acid, 0.5μg/mL cerulenin, 2mM tert-butyl peroxide, 40 mg/L calcofluor white, 2 μg/mL tunicamycin, 0.1μg/mL caspofungin, 25 μM simvastatin, and 0.008% SDS. Small molecule inhibitor drugs were used at either 2.5 μM (Brefeldin A), 5 μM (Geldanamycin, FK-506, cercosporamide), 10 μM (Trichostatin A, Inz-1), 15 μM (MDL-12,330A), or 20 μM (rotenone). Synthetic media used a base of Difco Yeast-Nitrogen-Base (YNB) with complete amino acid supplement, and addition of 2% glucose or 2% glycerol. Growth results were quantitatively displayed in heatmaps using Treeview as described previously (Vincent et al., 2013). Minimal inhibitory concentration (MIC) assays were performed at 2 fold concentration increments in 96-well plates over 24-72 hours as described in figure legends. The MIC was defined as the lowest concentration that results in no visible fungal growth.
Selection of resistant mutants and whole genome sequencing
Selection of mutants resistant to Inz-1 was performed by plating 107 cells per plate on YNB-glycerol-agar plates containing Inz-1. Plates were incubated for 7 days before picking colonies and retesting their resistance in liquid media. Isolates were retested for resistance to cycloheximide to eliminate nonspecific efflux mutants. DNA extraction, whole genome sequencing and polymorphism detection was performed as described before (Vincent et al., 2013).
Cytochrome B enzyme assay
The cytochrome B enzyme assay was adapted from previous studies (Gutierrez-Cirlos et al., 2004). Briefly, compounds were diluted in a 2x reaction buffer containing 100mM Tris-Cl pH 7.4, 10mM sodium azide, 0.02% BSA, and 0.1% Tween-20. Mitochondria (~5 μg protein) and cytochrome C (final concentraion 50μM) were then added, and the reaction was initiated by the addition of ubiquinol (50μM), reduced from ubiquinone with dithionite. Reaction plates were incubated at room temperature for 10 minutes before reading absorbance at 550nm. Absolute IC50’s were calculated in Graphpad Prism 6.0. Antimycin A (200nM) was included on every plate as a control for 100% inhibition.
Mammalian Cell Respiration Toxicity
Compounds were tested for toxicity to respiring mammalian cells as described in (Marroquin et al., 2007). Cells were initially passaged in High-Glucose DMEM, then washed and resuspended in glucose-free DMEM with 10mM galactose and added to 96-well white tissue culture plates at 10,000 cells per well. After 72 hours, ATP production was used as a surrogate for growth and assayed using Cell-titer-glo (Promega) at 1:5 dilution.
Bone-Marrow Derived Macrophage Co-Culture
Macrophages were isolated from GFP-tubulin Balb/c mice using standard methods (Strijbis et al., 2013). After 7-9 days in culture, macrophages were added to 12-well plates containing glass cover slips at 3×105 cells/well, and fungi were added at an MOI of 4 and briefly centrifuged. Cells were fixed with 3% formalin before visualization by fluorescence microscopy. Fungal growth over 14hr in the presence of macrophages was assayed by Alamar Blue dye reduction (control experiments demonstrated very low background macrophage dye signal).
Mouse infection experiments
All mouse experiments were performed under MIT protocol 0312-024-15 and in accordance with NIH standard for the ethical treatment of animals. Mouse experiments were performed as described previously with the specific inoculum used noted in the figure legend (Vincent et al., 2013). 7-12 week-old female Balb/C mice were used in all experiments. Fluconazole was prepared at 1 mg/mL solution in PBS and delivered intraperitoneally.
Supplementary Material
Highlights.
300,000 compound screen identifies indazole that reverses fluconazole resistance
Target identified as mitochondrial cytochrome bc1
Optimized potency and fungal selectivity through chemical synthesis
Impairing cytochrome bc1 restricts carbon source utilization and virulence
SIGNIFICANCE.
Developing more effective treatments for invasive fungal infections would save thousands of lives worldwide. Three key roadblocks preventing the clinical advance of new agents include the lack of tractable chemical matter with fungal-selective activity, the inability to combat fungal drug-resistance mechanisms, and our limited understanding of mechanisms by which fungi adapt to host environments. We address all three of these limitations with our discovery and optimization of the fungal-selective cytochrome bc1 inhibitor Inz-5. Our work demonstrates the potential of unbiased phenotypic chemical screening to both uncover new therapeutic strategies and provide tools for the dissection of pathogenic traits that underlie virulence and drug resistance.
eTOC blurb.
Vincent et al. use a phenotypic screen to discover and optimize a series of indazoles that reverse resistance to triazole antifungals in the pathogenic fungus C. albicans. The indazoles target cytochrome bc1, restricting carbon source utilization, macrophage escape, and virulence.
ACKNOWLEDGEMENTS
The authors acknowledge J. Love, T. Volkert and S. Gupta from the (Whitehead genome technology core) for assistance with genome sequencing, and E. Freinkman (Whitehead metabolomics core) for assistance in performing stability testing of compounds. We also thank B. Meunier for providing S. cerevisiae cytochrome B mutants, and V. Vyas and G. Fink for assistance with Candida strains, K. Strijbis for assistance with macrophage experiments, and C McLellan for helpful discussions. J.B. Langlois acknowledges the Swiss National Science Foundation for a Postdoctoral Fellowship (PBGEP2-145546). BMV acknowledges support from the NSF Graduate Research Fellowship, Howard Hughes Medical Institute, and the Mathers foundation. JBL and SLB thank the National Institutes GM46059 for support. Assay development and secondary screening studies were supported by NIH grant R03 MH086456-01 (SLL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
BMV, LW, and SLL designed the study; BMV, LW, and RSS and AKL performed biological studies, RS performed docking studies under supervision of BT; JBL performed synthetic chemical studies under supervision of SLB; BMV, JBL and SLL wrote the paper.
REFERENCES
- Ariyachet C, Solis NV, Liu Y, Prasadarao NV, Filler SG, McBride AE. SR-like RNA-binding protein Slr1 affects Candida albicans filamentation and virulence. Infection and immunity. 2013;81:1267–1276. doi: 10.1128/IAI.00864-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. Journal of medicinal chemistry. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Barelle CJ, Priest CL, Maccallum DM, Gow NA, Odds FC, Brown AJ. Niche-specific regulation of central metabolic pathways in a fungal pathogen. Cellular microbiology. 2006;8:961–971. doi: 10.1111/j.1462-5822.2005.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann JD, Ljungdahl PO, Lopez JL, Trumpower BL. Isolation and characterization of the nuclear gene encoding the Rieske iron-sulfur protein (RIP1) from Saccharomyces cerevisiae. The Journal of biological chemistry. 1987;262:8901–8909. [PubMed] [Google Scholar]
- Birth D, Kao WC, Hunte C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nature communications. 2014;5:4029. doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Brown GD, Netea MG, Gow NA. Metabolism impacts upon Candida immunogenicity and pathogenicity at multiple levels. Trends in microbiology. 2014 doi: 10.1016/j.tim.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. Hidden killers: human fungal infections. Science translational medicine. 2012;4:165rv113. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- Byrnes EJ, 3rd, Li W, Lewit Y, Ma H, Voelz K, Ren P, Carter DA, Chaturvedi V, Bildfell RJ, May RC, et al. Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS pathogens. 2010;6:e1000850. doi: 10.1371/journal.ppat.1000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone R, Odds FC, Boekhout T. Candida albicans: fundamental research on an opportunistic human pathogen. FEMS yeast research. 2009;9:971–972. doi: 10.1111/j.1567-1364.2009.00585.x. [DOI] [PubMed] [Google Scholar]
- Cowen LE, Lindquist S. Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science. 2005;309:2185–2189. doi: 10.1126/science.1118370. [DOI] [PubMed] [Google Scholar]
- Cowen LE, Sanglard D, Howard SJ, Rogers PD, Perlin DS. Mechanisms of Antifungal Drug Resistance. Cold Spring Harbor perspectives in medicine. 2014 doi: 10.1101/cshperspect.a019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz MC, Goldstein AL, Blankenship JR, Del Poeta M, Davis D, Cardenas ME, Perfect JR, McCusker JH, Heitman J. Calcineurin is essential for survival during membrane stress in Candida albicans. The EMBO journal. 2002;21:546–559. doi: 10.1093/emboj/21.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diezmann S, Michaut M, Shapiro RS, Bader GD, Cowen LE. Mapping the Hsp90 genetic interaction network in Candida albicans reveals environmental contingency and rewired circuitry. PLoS genetics. 2012;8:e1002562. doi: 10.1371/journal.pgen.1002562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejim L, Farha MA, Falconer SB, Wildenhain J, Coombes BK, Tyers M, Brown ED, Wright GD. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nature chemical biology. 2011;7:348–350. doi: 10.1038/nchembio.559. [DOI] [PubMed] [Google Scholar]
- Ene IV, Brunke S, Brown AJ, Hube B. Metabolism in Fungal Pathogenesis. Cold Spring Harbor perspectives in medicine. 2014 doi: 10.1101/cshperspect.a019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epp E, Vanier G, Harcus D, Lee AY, Jansen G, Hallett M, Sheppard DC, Thomas DY, Munro CA, Mullick A, et al. Reverse genetics in Candida albicans predicts ARF cycling is essential for drug resistance and virulence. PLoS pathogens. 2010;6:e1000753. doi: 10.1371/journal.ppat.1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L, Quinn B, Li YF, Zhang M, Elberry M, Yu L, Yu CA, Xia D. Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc(1) complex. Journal of molecular biology. 2004;341:281–302. doi: 10.1016/j.jmb.2004.05.065. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Sanguinetti M, De Bernardis F, Torelli R, Posteraro B, Vandeputte P, Sanglard D. Loss of mitochondrial functions associated with azole resistance in Candida glabrata results in enhanced virulence in mice. Antimicrobial agents and chemotherapy. 2011;55:1852–1860. doi: 10.1128/AAC.01271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahl N, Demers EG, Lindsay AK, Harty CE, Willger SD, Piispanen AE, Hogan DA. Mitochondrial Activity and Cyr1 Are Key Regulators of Ras1 Activation of C. albicans Virulence Pathways. PLoS pathogens. 2015;11:e1005133. doi: 10.1371/journal.ppat.1005133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Cirlos EB, Merbitz-Zahradnik T, Trumpower BL. Inhibition of the yeast cytochrome bc1 complex by ilicicolin H, a novel inhibitor that acts at the Qn site of the bc1 complex. The Journal of biological chemistry. 2004;279:8708–8714. doi: 10.1074/jbc.M311805200. [DOI] [PubMed] [Google Scholar]
- Hill P, Kessl J, Fisher N, Meshnick S, Trumpower BL, Meunier B. Recapitulation in Saccharomyces cerevisiae of cytochrome b mutations conferring resistance to atovaquone in Pneumocystis jiroveci. Antimicrobial agents and chemotherapy. 2003;47:2725–2731. doi: 10.1128/AAC.47.9.2725-2731.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL, Keseru GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nature reviews Drug discovery. 2014;13:105–121. doi: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Hughes W, Dorenbaum A, Yogev R, Beauchamp B, Xu J, McNamara J, Moye J, Purdue L, van Dyke R, Rogers M, et al. Phase I safety and pharmacokinetics study of micronized atovaquone in human immunodeficiency virus-infected infants and children. Pediatric AIDS Clinical Trials Group. Antimicrobial agents and chemotherapy. 1998;42:1315–1318. doi: 10.1128/aac.42.6.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C, Palsdottir H, Trumpower BL. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS letters. 2003;545:39–46. doi: 10.1016/s0014-5793(03)00391-0. [DOI] [PubMed] [Google Scholar]
- Kessl JJ, Lange BB, Merbitz-Zahradnik T, Zwicker K, Hill P, Meunier B, Palsdottir H, Hunte C, Meshnick S, Trumpower BL. Molecular basis for atovaquone binding to the cytochrome bc1 complex. The Journal of biological chemistry. 2003;278:31312–31318. doi: 10.1074/jbc.M304042200. [DOI] [PubMed] [Google Scholar]
- Kussell E, Leibler S. Phenotypic diversity, population growth, and information in fluctuating environments. Science. 2005;309:2075–2078. doi: 10.1126/science.1114383. [DOI] [PubMed] [Google Scholar]
- LaFayette SL, Collins C, Zaas AK, Schell WA, Betancourt-Quiroz M, Gunatilaka AA, Perfect JR, Cowen LE. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS pathogens. 2010;6:e1001069. doi: 10.1371/journal.ppat.1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Mittal R, Solis NV, Prasadarao NV, Filler SG. Mechanisms of Candida albicans trafficking to the brain. PLoS pathogens. 2011;7:e1002305. doi: 10.1371/journal.ppat.1002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz MC, Bender JA, Fink GR. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryotic cell. 2004;3:1076–1087. doi: 10.1128/EC.3.5.1076-1087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz MC, Fink GR. Life and death in a macrophage: role of the glyoxylate cycle in virulence. Eukaryotic cell. 2002;1:657–662. doi: 10.1128/EC.1.5.657-662.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti O, Moreillon P, Glauser MP, Bille J, Sanglard D. Potent synergism of the combination of fluconazole and cyclosporine in Candida albicans. Antimicrobial agents and chemotherapy. 2000;44:2373–2381. doi: 10.1128/aac.44.9.2373-2381.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicological sciences : an official journal of the Society of Toxicology. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- Odds FC, Brown AJ, Gow NA. Antifungal agents: mechanisms of action. Trends in microbiology. 2003;11:272–279. doi: 10.1016/s0966-842x(03)00117-3. [DOI] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nature reviews Drug discovery. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Critical reviews in microbiology. 2010;36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- Ramirez MA, Lorenz MC. Mutations in alternative carbon utilization pathways in Candida albicans attenuate virulence and confer pleiotropic phenotypes. Eukaryotic cell. 2007;6:280–290. doi: 10.1128/EC.00372-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding S, Smith J, Farinacci G, Rinaldi M, Fothergill A, Rhine-Chalberg J, Pfaller M. Resistance of Candida albicans to fluconazole during treatment of oropharyngeal candidiasis in a patient with AIDS: documentation by in vitro susceptibility testing and DNA subtype analysis. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 1994;18:240–242. doi: 10.1093/clinids/18.2.240. [DOI] [PubMed] [Google Scholar]
- Roemer T, Krysan DJ. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harbor perspectives in medicine. 2014;4 doi: 10.1101/cshperspect.a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T, Xu D, Singh SB, Parish CA, Harris G, Wang H, Davies JE, Bills GF. Confronting the challenges of natural product-based antifungal discovery. Chemistry & biology. 2011;18:148–164. doi: 10.1016/j.chembiol.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Sanglard D, Ischer F, Marchetti O, Entenza J, Bille J. Calcineurin A of Candida albicans: involvement in antifungal tolerance, cell morphogenesis and virulence. Molecular microbiology. 2003;48:959–976. doi: 10.1046/j.1365-2958.2003.03495.x. [DOI] [PubMed] [Google Scholar]
- Shingu-Vazquez M, Traven A. Mitochondria and fungal pathogenesis: drug tolerance, virulence, and potential for antifungal therapy. Eukaryotic cell. 2011;10:1376–1383. doi: 10.1128/EC.05184-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, Edlind TD. Histone deacetylase inhibitors enhance Candida albicans sensitivity to azoles and related antifungals: correlation with reduction in CDR and ERG upregulation. Antimicrobial agents and chemotherapy. 2002;46:3532–3539. doi: 10.1128/AAC.46.11.3532-3539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer M, Griffiths E, Blakely KM, Wildenhain J, Ejim L, Rossi L, De Pascale G, Curak J, Brown E, Tyers M, et al. Cross-species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Molecular systems biology. 2011;7:499. doi: 10.1038/msb.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strijbis K, Tafesse FG, Fairn GD, Witte MD, Dougan SK, Watson N, Spooner E, Esteban A, Vyas VK, Fink GR, et al. Bruton's Tyrosine Kinase (BTK) and Vav1 contribute to Dectin1-dependent phagocytosis of Candida albicans in macrophages. PLoS pathogens. 2013;9:e1003446. doi: 10.1371/journal.ppat.1003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Fonzi W, Chen H, She X, Zhang L, Zhang L, Calderone R. Azole susceptibility and transcriptome profiling in Candida albicans mitochondrial electron transport chain complex I mutants. Antimicrobial agents and chemotherapy. 2013;57:532–542. doi: 10.1128/AAC.01520-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent BM, Lancaster AK, Scherz-Shouval R, Whitesell L, Lindquist S. Fitness trade-offs restrict the evolution of resistance to amphotericin B. PLoS biology. 2013;11:e1001692. doi: 10.1371/journal.pbio.1001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TC. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrobial agents and chemotherapy. 1997;41:1482–1487. doi: 10.1128/aac.41.7.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngsaye W, Dockendorff C, Vincent B, Hartland CL, Bittker JA, Dandapani S, Palmer M, Whitesell L, Lindquist S, Schreiber SL, et al. Overcoming fluconazole resistance in Candida albicans clinical isolates with tetracyclic indoles. Bioorganic & medicinal chemistry letters. 2012;22:3362–3365. doi: 10.1016/j.bmcl.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngsaye W, Vincent B, Hartland CL, Morgan BJ, Buhrlage SJ, Johnston S, Bittker JA, MacPherson L, Dandapani S, Palmer M, et al. Piperazinyl quinolines as chemosensitizers to increase fluconazole susceptibility of Candida albicans clinical isolates. Bioorganic & medicinal chemistry letters. 2011;21:5502–5505. doi: 10.1016/j.bmcl.2011.06.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.