

Abstract
Nerve injury and inflammation cause up-regulation of an endogenous opioid ligand, dynorphin A (Dyn A), in the spinal cord resulting in hyperalgesia via the interaction with bradykinin receptors (BRs). This is a non-opioid neuroexcitatory effect that cannot be blocked by opioid antagonists. Our systematic structure-activity relationships study on Dyn A identified lead ligands 1 and 4, along with the key structural feature (i.e. amphipathicity) for the BRs. However, the ligands showed very low metabolic stability in plasma (t1/2 < 1 h) and therefore, in order to improve their metabolic stabilities with retained biological activities, various modifications were performed. Cyclization of ligand 4 afforded a cyclic Dyn A analogue 5 that retained the same range of binding affinity as the linear ligand with improved metabolic stability (t1/2 > 5 h) and therefore possesses the potential as a pharmacophoric scaffold to be utilized for drug development.
Keywords: Cyclic ligands, Non-opioid dynorphin A, Amphipathicity, Bradykinin receptors, Structure-activity relationship, Metabolic stability
Graphical abstract

Chronic neuropathic pain is difficult to treat by current methods using opioids due to serious side effects such as tolerance and addiction which is indispensable for treatment.1,2 Long term administration of opioids can even develop different types or more serious pain with prolonged use.3 This is more likely caused by gene expression that is related to treatment attempts: nervous system adaptation/change.4 From this perspective, it seems important to develop drugs through novel approaches considering the possible changes in pain pathways for efficient treatment of chronic pain states. Dynorphin A (Dyn A, H-Tyr1-Gly-Gly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Leu-Lys-Trp-Asp-Asn-Gln17-OH), which is an endogenous ligand for three subtypes (mu, delta, and kappa) of opioid receptors, with a slight preference for the kappa opioid receptor (KOR), has been a good target to investigate possible system changes owing to its very distinctive biological roles in the pain pathway: neuroinhibitory (opioid) effects and neuroexcitatory (non-opioid) effects.5–7 While Dyn A’s neuroinhibitory effects are well-known with the opioid receptors, its non-opioid neuroexcitatory effects are not yet established despite its frequent observation in animal models. It was shown that under nerve injury and inflammation, up-regulated Dyn A or its fragments, specially [des-Tyr1]-Dyn A analogues, interact with the bradykinin receptors (BRs) in the central nervous system (CNS) to cause hyperalgesia.8–13
Therefore, in our earlier studies, we pursued systematic structure-activity relationship (SAR) studies to gain key insights into the structure for the central BRs and to utilize them to understand the uncertain non-opioid effects via the BRs.14–17 Based on the results, ligands 1 and 4 were identified as lead ligands with good affinity along with a key structural feature for the BRs: amphipathicity. In in vivo tests, ligand 4 blocked Dyn A(2–13)-induced hyperalgesia and motor impairments in naïve animals and showed anti-hyperalgesic effects in L5/L6 spinal nerve ligation (SNL) animals in a dose-dependent manner.14 Ligand 4 also inhibited Dyn A(2-13) induced wide dynamic range (WDR) neuronal response in naïve animals and modulated WDR neuronal response to innocuous and noxious mechanical stimuli in SNL animals.18 All of these effects were considered to be localized in the CNS because no peripheral activity was shown in in vivo tests via intraplantar (i. pl.) administration.14 These results demonstrated that Dyn A structure-based BR antagonists can be developed for a therapeutic purpose to treat abnormal pain which can be caused by up-regulation of Dyn A in the CNS in chronic neuropathic pain states.
Even with high potency and affinity, ligand 4 showed very low metabolic stability in plasma and was completely degraded within 4 h of incubation (t1/2 = 0.7 h). In an effort to improve metabolic stability and blood brain barrier (BBB) permeability, various modifications were performed on our lead ligands 1 and 4. Here we report SAR results of cyclic Dyn A analogues at the BRs (Table 1).
Table 1.
Binding affinities of cyclic Dyn A analogues at BRs in rat brain.
| no | Structure | Ring size | BR, [3H]BKa
|
|
|---|---|---|---|---|
| Log[IC50]b | IC50 (nM) | |||
| 1c | H-Nle-Lys-Pro-Lys-Nle-Lys-OH | – | −7.11+0.14 | 78 |
| 2 | H-c1,5(-cisCH=CH-)[Ala-Lys-Pro-Lys-Ala]-Lys-OH | 17 | – | n.c. |
| 3 | H-c1,5(-CH2CH2-)[Ala-Lys-Pro-Lys-Ala]-Lys-OH | 17 | – | n.c. |
| 4c | H-Phe-Leu-Arg-Ile-Arg-Pro-Lys-OH | – | −7.16+0.09 | 69 |
| 5 | cN,6[Phe-Leu-Arg-Ile-Arg-Glu]-Lys-OH | 20 | −6.72+0.13 | 191 |
| 6 | cN,6[Lys-Leu-Arg-Ile-Arg-Glu]-Lys-OH | 20 | −6.52+0.15 | 302 |
| 7 | cN,5[Leu-Arg-Ile-Arg-Glu]-Lys-OH | 17 | −5.82+0.09 | 1510 |
Radioligand competition assays were carried out using [3H]BK in rat brain membranes at pH 6.8.
Logarithmic values determined from the nonlinear regression analysis of data collected from at least two independent experiments in duplicate using GraphPad Prism 6.
Reference #14, [3H]DALKD.
n.c. : no competition.
To take advantage of cyclic peptide ligands, analogues 2 and 3 were first designed based on the structure of ligand 1, which showed good binding affinity (IC50 = 78 nM) at the BRs, and in these structures, two Nle residues of ligand 1 were replaced by two allyl glycine residues to form a 17-membered carba ring via ring metathesis (Figure 1).19 The cyclization consumed, and thus buried, two hydrophobic chains in the ring, while allocating three Lys residues to position their side amino groups to be exposed due to the important role of positive charges in the BRs recognition. In contrast, cyclic ligands 5–7 retained hydrophobic alkyl chains exposed after a ring formation, which fulfill amphipathicity that is critical for interaction with the BRs. Originally, these cyclic ligands were designed based on the structure of linear ligand 4, and a turn making Pro residue was replaced with a Glu residue for ring formation. The N-terminal amino group, which was shown not to be critical, was consumed for ring formation with a Glu residue. A central idea in the design was to enhance the turn structure around the Pro residue and to properly expose both positive charges and hydrophobic groups to satisfy ligand amphipathicity.
Figure 1.
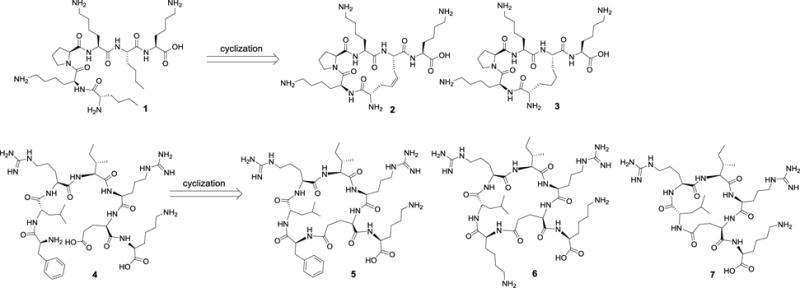
Cyclizations of amphipathic Dyn A analogues.
For the synthesis of the cyclic analogues, chain elongations were performed by standard solid phase peptide synthesis (SPPS) using Nα-fluorenylmethyloxycarbonyl (Fmoc) chemistry with 2-phenylisopropyl (Pip), 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf), and t-butyloxycarbonyl (Boc) group as a side protecting group for Glu, Arg, and Lys, respectively, on Fmoc-Lys(Boc)-attached Wang resin in high yields (overall yields > 40%). Each coupling reaction used 3 equiv 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HBTU) / 3 equiv N-hydroxybenzotriazole (HOBt) / 6 equiv diisopropylethylamine (DIPEA) for 50 min at rt and each Fmoc-deprotection used 20% piperidine / N,N-dimethylformamide (DMF) for 20 min at rt. After chain elongation, ring closing metathesis was performed under microwave (100 °C) using second generation Grubbs catalyst in dichloromethane (DCM) containing 10% of 0.4 M LiCl in DMF solution for 1 h. (Scheme 1)20 Crude dicarba cyclic peptide-resin, of which the half portion was cleaved by a 90% trifluoroacetic acid (TFA) cocktail solution containing 5% thioanisole, 3% ethanedithiol, and 2% anisole and purified by preparative reversed-phase high performance liquid chromatography (RP-HPLC, 10–50% of acetonitrile in 20 min) to afford pure dicarba cyclic analogue 2. The other half was reduced by Wilkinson’s hydrogenation method using cat. Rh(PPh3)3Cl in 90% DCM and 10% MeOH at rt for 1 day, cleaved by the TFA cocktail, and finally purified by RP-HPLC to afford more than 95% pure saturated dicarba cyclic analogue 3.20 For the synthesis of cyclic analogues 5–7, a Pip group on a Glu residue was first deprotected after chain elongation by 5% TFA, and the resulting acid was coupled with a N-terminal amino group using 5 equiv DIC / 5 equiv HOBt. The cyclic analogues were then cleaved by the same method as before and purified by RP-HPLC to afford more than 98% pure cyclic Dyn A analogues. Owing to their hydrophilic characters (aLogPs < 0), all analogues showed very short retention time (<15 min) (Table 2). The purified analogues were validated by analytical RP-HPLC and mass spectroscopy.
Scheme 1.

Synthesis of olefin (or alkyl) bridged cyclic Dyn A analogues.
Table 2.
Analytical data of cyclic Dyn A analogues
| no | molecular formula | MSa | HPLCb (tR,min) |
Purity (%) | aLogPsc | |
|---|---|---|---|---|---|---|
| calculated | observed | |||||
| 2 | C31H55N9O7 | 666.4 | 666.4 | 15.0 | 95 | −2.25 |
| 3 | C31H57N9O7 | 668.4 | 668.5 | 11.6 | 95 | −2.17 |
| 5 | C44H74N14O9 | 943.6 | 943.7 | 14.1 | 98 | −2.24 |
| 6 | C41H77N15O9 | 924.6 | 924.5 | 11.9 | 98 | −2.35 |
| 7 | C35H65N13O8 | 796.5 | 796.5 | 11.7 | 98 | −2.74 |
(M + H)+, ESI method (Finnigan, Thermoelectron, LCQ classic).
Performed on a Hewlett Packard 1100 (C-18, Microsorb-MV™, 4.6 mm × 250 mm, 5 μm) using gradient system (10–100% of acetonitrile containing 0.1% TFA within 45 min, 1 mL/min).
The purified analogues were tested for their binding affinities by competition assays using [3H]BK in rat brain membranes where non-specific binding is defined by 10 μM kallidin (Table 1).14 Data was analyzed by nonlinear least-squares analysis using GraphPad Prism 6 and IC50 values were determined from nonlinear regression analysis of data collected from at least two independent experiments.
Ligands 2 and 3, which are lacking exposed hydrophobic alkyl moieties, lost affinity (no competition) at the BRs even with a C-terminal basic amino acid residue and an additional positive charge (net charge: + 3) which is required for strong electrostatic interactions with the BRs. Our earlier studies showed that Dyn A ligands’ binding affinities exhibit pH dependence (lower pH, higher binding affinity), and based on those results, electrostatic interactions were considered to be a main interaction between ligands and the BRs.14 It was also shown that proper allocation of positive charges in linear ligands, which was accomplished by the insertion of hydrophobic amino acid residues, and thus resulted in amphipathic property, is another key feature for receptor recognition. Here in these ligands 2 and 3, the hydrophobic amino acid residues were consumed to form a ring and thus no alkyl chain was exposed outside of the ring. Therefore, even with the exposed three amino groups, ligands 2 and 3 lost their binding affinities at the BRs due to the lack of hydrophobic moieties. A comparison of linear ligand 1 with 2 and 3 clearly indicated an important role of hydrophobic alkyl residues for receptor recognition. On the basis of the SAR results, new cyclic ligands 5–7 were designed to uncover two hydrophobic amino acid residues, Leu and Ile. In the design, the N-terminal amino group was consumed for ring formation with a Glu residue to afford 20- and 17-membered rings. Cyclic ligands 5 (IC50 = 191 nM) and 6 (IC50 = 302 nM) with a 20-membered ring retained the same range of binding affinity at the BRs as linear ligand 4 (IC50 = 69 nM). Even with the slight loss of affinities, it is clear that the cyclization between the Pro residue position and the N-terminus is well tolerated for the receptor. However, ligand 7 with a 17-membered ring reduced the affinity 22-fold, suggesting that the 20-membered ring is an optimum ring size for the BRs. The reduction of affinity may be caused by its constrained structure inhibiting proper location of amphipathic moieties from basic and hydrophobic amino acid residues.
Peptides are degraded mainly by proteolytic enzymes in plasma, and therefore to validate metabolic stability, cyclic ligand 5 was tested for its half-life in rat plasma at 37 °C. After certain points of the incubation time (1 h, 2 h, 4 h, 8 h), quenched sample was analyzed by RP-HPLC. The half-life was calculated by the equation (t1/2=0.693/b, b is the slope of linear fit). As expected, cyclic ligand 5 was found to be more stable (t1/2 = 5.5 h) than linear ligand 4 (t1/2 = 0.7 h) in rat plasma. The cyclization resulted in ligand 5 that retains the same high affinity at the BRs with improved metabolic stability compared to 4.
In conclusion, it was demonstrated that amphipathicity is a key structural feature for cyclic ligands to interact with the BRs, and a 20-membered ring is an ideal size for the ligands to allocate their hydrophobic and basic moieties appropriately. Importantly, the cyclic ligand 4 with high affinity had improved metabolic stability and therefore demonstrated the potential as a pharmacophoric scaffold for drug development to treat abnormal chronic neuropathic pain states.
Acknowledgments
This work has been supported by U.S.Public Health Services, NIH, and NIDA P01DA006284 and R01DA013449.
Abbreviations
- BBB
blood brain barrier
- BK
bradykinin
- Boc
t-butyloxycarbonyl
- BR
bradykinin receptor
- CNS
central nervous system
- DALKD
[des-Arg10, Leu9]-kallidin
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- Dyn A
dydnorphin A
- Fmoc
9-fluorenylmethylcarboxy
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate
- HOBt
N-hydroxybenzotriazole
- i.pl.
intraplantar
- MS
Mass spectroscopy
- Pbf
2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- RP-HPLC
reversed-phase high performance liquid chromatography
- SNL
spinal nerve ligation
- SPPS
solid phase peptide synthesis
- Pip
2-phenylisopropyl
- TFA
trifluoroacetic acid
- TIS
triisopropylsilane
- WDR
wide dynamic range
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Neurosignals. 2005;14:194. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- 2.Foley KM. Anticancer Drugs. 1995;6:4. [Google Scholar]
- 3.Vanderah TW, Ossipov MH, Lai K, Malan TP, Porreca F. Pain. 2001;92:5. doi: 10.1016/s0304-3959(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 4.Hruby VJ, Porreca F, Yamamura HI, Tollin G, Agnes R, Lee YS, Cai M, Alves I, Cowell S, Varga E, Davis P, Salamon Z, Roeske W, Vanderah TW, Lai J. AAPS J. 2006;8:E450. doi: 10.1208/aapsj080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai J, Ossipov M, Vanderah TW, Malan TP, Jr, Porreca F. Mol Interventions. 2001;1:160. [PubMed] [Google Scholar]
- 6.Koetzner L, Hua X-Y, Lai J, Porreca F, Yaksh TJ. Neurosci. 2004;24:1451. doi: 10.1523/JNEUROSCI.1517-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai J, Luo MC, Chen Q, Ma S, Gardell LR, Ossipov M, Porreca F. Nat Neurosci. 2006;9:1534. doi: 10.1038/nn1804. [DOI] [PubMed] [Google Scholar]
- 8.Ruda MA, Iadarola MJ, Cohen LV, Young WS. Proc Natl Acad Sci USA. 1988;85:622. doi: 10.1073/pnas.85.2.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draisci G, Kajander KC, Dubner R, Bennett GJ, Iadarola MJ. Brain Res. 1991;560:186. doi: 10.1016/0006-8993(91)91231-o. [DOI] [PubMed] [Google Scholar]
- 10.Malan TP, Jr, Ossipov MH, Ibrahim M, Bian DI, Lai J, Porreca F. Pain. 2000;86:185. doi: 10.1016/s0304-3959(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 11.Vanderah TW, Cardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong C, Zhang E-T, Malan TP, Jr, Ossipov MH, Lai J, Porreca FJ. Neurosci. 2000;20:7074. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altier C, Zamponi GW. Nat Neurosci. 2006;9:1465. doi: 10.1038/nn1206-1465b. [DOI] [PubMed] [Google Scholar]
- 13.Gardell LR, Ibrahim M, Wang R, Wang Z, Ossipov MH, Malan TP, Jr, Porreca F, Lai J. Neurosci. 2004;123:43. doi: 10.1016/j.neuroscience.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 14.Lee YS, Muthu D, Hall SM, Ramos-Colon C, Rankin D, Hu J, Sandweiss A, Felice MD, Xie JY, Vanderah TW, Porreca F, Lai J, Hruby VJ. J Am Chem Soc. 2014;136:6608. doi: 10.1021/ja501677q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YS, Rankin D, Hall SM, Ramos-Colon C, Ortiz JJ, Kupp R, Porreca F, Lai J, Hruby VJ. Bioorg Med Chem Letts. 2014;24:4976. doi: 10.1016/j.bmcl.2014.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee YS, Hall SM, Ramos-Colon C, Michael R, LeBaron L, Nguyen A, Rankin D, Kupp R, Porreca F, Lai J, Hruby VJ. Bioorg Med Chem Letts. 2015;25:30. doi: 10.1016/j.bmcl.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YS, Kupp R, Remesic M, Ramos-Colon C, Hall SM, Chan C, Rankin D, Lai J, Porreca F, Hruby VJ. Chem Biol Drug Des. doi: 10.1111/cbdd.12789. published online June 6, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bannister K, Lee YS, Goncalves L, Porreca F, Lai J, Dickenson AH. Neuropharmacol. 2014;85:375. doi: 10.1016/j.neuropharm.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim YW, Grossmann TN, Verdine GL. Nat Protocols. 2011;6:761. doi: 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- 20.Robinson AJ, Elaridi J, Van Lierop BJ, Mujcinovic S, Jackson WR. J Pept Sci. 2007;13:280. doi: 10.1002/psc.840. [DOI] [PubMed] [Google Scholar]