

Abstract
Background & Aims
Lynch syndrome patients have DNA mismatch repair deficiency and up to 80% life-time risk of colorectal cancer. Screening of mutation carriers reduces colorectal cancer incidence and mortality. Selection for constitutional mutation testing relies on family history (Amsterdam and Bethesda Guidelines) and tumour derived biomarkers. Initial biomarker analysis uses mismatch repair protein immunohistochemistry and microsatellite instability. Abnormalities in either identify mismatch repair deficiency but do not differentiate sporadic epigenetic defects, due to MLH1 promoter region methylation (13% of CRCs) from Lynch Syndrome (4% of CRCs). A diagnostic biomarker capable of making this distinction would be valuable. This study compared two biomarkers in tumours with mismatch repair deficiency; quantification of methylation of the MLH1 promoter region using a novel assay and BRAF c.1799T>A, p.(Val600Glu) mutation status in the identification of constitutional mutations.
Methods
Tumour DNA was extracted (FFPE tissue) and pyrosequencing used to test for MLH1 promoter methylation and presence of the BRAF c.1799T>A, p.(Val600Glu) mutation 71 CRCs from individuals with pathogenic MLH1 mutations and 73 CRCs with sporadic MLH1 loss. Specificity and sensitivity was compared.
Findings
Unmethylated MLH1 promoter: sensitivity 94.4% (95% CI 86.2–98.4%), specificity 87.7% (95% CI 77.9–94.2%), Wild-type BRAF (codon 600): sensitivity 65.8% (95% CI 53.7–76.5%), specificity 98.6% (95% CI 92.4–100.0%) for the identification of those with pathogenic MLH1 mutations.
Conclusions
Quantitative MLH1 promoter region methylation using pyrosequencing is superior to BRAF codon 600 mutation status in identifying constitutional mutations in mismatch repair deficient tumours.
Keywords: Lynch Syndrome, MLH1 methylation, BRAF, sensitivity, specificity
Introduction
Lynch syndrome (LS) is responsible for 3–4% of all colorectal cancer (CRC) and is the most common cause of hereditary CRC (1, 2). It is caused by mutations in one of the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6 and PMS2. Mutations result in MSI-H (microsatellite instability high) cancers. Identification of families with LS is necessary to initiate screening and to reduce CRC mortality (3–5).
Diagnosis of LS is complicated by the expense and time-consuming nature of constitutional mutation analysis. Family history criteria and tumour-derived biomarkers are used to pre-screen to select patients for germline testing. The Amsterdam II criteria were designed to select research families for linkage analysis. They are currently used, somewhat inappropriately, for clinical purposes to select individuals at high risk of having a MMR gene mutation. Patients who meet these criteria have at least 60% chance of a mutation (6). These criteria are inherently specific but consequently have low sensitivity. Much work has been done over the last decade to improve the identification of non-Amsterdam Lynch families. The revised Bethesda guidelines described in 2004 (7) are sensitive but have low specificity. They have been criticised for being overly complicated and are little used in clinical practice (8). Tumour microsatellite instability (MSI) and mismatch repair protein immunohistochemistry (MMR IHC) are currently used in conjunction with the revised Bethesda guidelines (or other medium risk criteria). The sensitivity of MSI is 89% for MLH1 and MSH2 but less than 80% for MSH6 and PMS2, with a specificity of 90% for all genes (9). MSI testing is impractical for population-based screening due to the need for a molecular genetics laboratory. MMR IHC may be preferable in patients meeting Bethesda guidelines because of the low sensitivity of MSI for detecting MSH6 and PMS2 gene mutation carriers. MMR IHC has a sensitivity of 100% and a specificity 91.5% for the detection of MLH1 carriers, a sensitivity 87.5% and specificity of 88.5% for the detection of MSH2 carriers (10). Pre-screening of all newly diagnosed CRCs (population-based) is used in some specialist centres in the US and Europe (none in the UK) in order to identify families not meeting clinical criteria for Lynch syndrome. A multi-centre study of over 10,000 newly diagnosed CRC probands, found that MMR tumour testing was the most effective strategy for the identification of mutation carriers (sensitivity 100%, specificity, 93.0%, diagnostic yield 2.2% compared to use of the Bethesda guidelines; sensitivity 87.8% specificity, 97.5%, diagnostic yield, 2.0% P < 0.001) (11).
There are two independent molecular pathways which lead to MSI-H (MMR deficient) CRC (12). MSI-H cancers occur not only in LS but also as a result of epigenetic silencing of the MLH1 gene through hypermethylation of its promoter. This occurs in around 13% of sporadic CRC (12). These cancers are also associated with the BRAF c.1799T>A, p.Val600Glu mutation (13) and are not familial. LS cancers are characterised by MSI-H, a normal (unmethylated) MLH1 gene promoter region, and wild-type BRAF (i.e. c.1799T, p.Val600). MMR IHC is able to effectively identify patients for MSH2, MSH6 testing. Sporadic defects in these genes are rare so protein loss is highly indicative of a constitutional abnormality. However, MSI and MMR IHC are not specific enough to identify MLH1 constitutional mutation carriers because of this large group of sporadic cancers with MLH1 deficiency. A method of differentiating between these groups of cancers is required.
BRAF mutation testing has been suggested. The methodology is well established and is currently in use in some centres. However BRAF testing has low specificity. MLH1 promoter region methylation testing is attractive as a better pre-screen test. Methylation is thought to be the first step in pathogenesis of cancers with sporadic loss of MLH1 and is thought to be rare in Lynch cancers (14). Lack of methylation should, therefore, be more specific for the identification of constitutional mutation carriers. Constitutional MLH1 methylation has been reported as a rare cause of mutation negative Lynch Syndrome (four cases reported) (15–19). This may confound the use of methylation as a pre-screen, but the incidence of this is likely to be extremely low. Tumour MLH1 promoter region methylation has not previously been tested in a large group of patients. Whilst a number of methods for MLH1 methylation analysis have been developed, most are technically difficult (particularly in FFPE tissue) and expensive.
Guidelines for constitutional mutation testing for cancer susceptibility genes suggest a threshold of 10% risk (20). Using Bayes theorem, specificity and sensitivity of any pre-screen test can be applied to individuals with differing risk determined by their family history of cancer. Individuals who fulfil Amsterdam II criteria have a pre-test probability of harbouring a mutation of 60% (6). Individuals who fulfil the revised Bethesda guidelines and have loss of MLH1 in their tumour have a pre-test probability of at least 10.5% (21–23). Patients from the general population who have MLH1 loss (tumour) have a pre-test probability of 4.0% (23–26). We have previously shown that MSI testing alone is not an appropriate pre-screening tool in Amsterdam criteria (I and II) positive families. Even if their tumour is microsatellite stable, the risk of having a mutation remains greater than 10% (27). Given that a recent Health Technology Assessment study has recommended that all colorectal cancers in patients aged 60 years of age or younger should be pre-screened for tumour mismatch repair deficiency (MMRd) (28), strategies need to be developed to deal with the large number of MMRd tumours most of which will be the result of MLH1 promoter methylation in the tumour and not be caused by constitutional mutations.
The aim of this study was to: 1) develop a simple, cheap, reproducible method for quantitative MLH1 promoter region methylation analysis in FFPE (formalin fixed, paraffin embedded) tissue, 2) compare this with BRAF c.1799T>A p.Val600Glu mutation testing in patients whose CRCs demonstrate loss of MLH1 protein expression and 3) assess additional benefit of adding a methylation assay to BRAF testing in order to select patients for constitutional MLH1 mutation testing.
Methods
Ethical approval was obtained from South Manchester (UK) Research Ethics Committee.
Participants
To compare tumour MLH1 promoter methylation testing and BRAF c.1799T>A p.Val600Glu somatic mutation testing for selecting patients for constitutional MLH1 mutation analysis, two groups of patients with MLH1 deficient tumours were identified; those patients with pathogenic constitutional MLH1 mutations and patients with MLH1 promoter hypermethylated CRCs with MLH1 loss.
CRCs from patients with known pathogenic constitutional MLH1 mutations were identified from the Familial Colorectal Cancer Registry (Central Manchester University Hospitals NHS Foundation Trust, UK n=22). Additional cases were provided by The Jeremy Jass Memorial Pathology Tissue Bank of the Australasian Colorectal Cancer Family Registry (ACCFR: U01 CA097735)(29). MMR IHC to identify CRCs with loss of MLH1 protein expression was performed (by the ACCFR) (30). Screening for constitutional mutations in MLH1, MSH2, MSH6 and PMS2 was performed for all probands recruited from high-risk clinic and for population-based probands who had a CRC with evidence of MSI or loss of MMR protein expression by IHC. Mutation testing was performed as previously described (31, 32). 49 ACCFR CRC cases with loss of MLH1 protein expression from patients with known pathogenic MLH1 mutations (n=49) were included.
Semi-quantitative MMR Immunohistochemistry (IHC), as previously described (10), was conducted on 86 consecutive right sided CRCs (sporadic MSI-H tumours occur more frequently in the right colon (12)) from patients aged over 50 years who did not fulfil Amsterdam or Bethesda criteria identified at Manchester Royal Infirmary. Patients known to have LS, FAP or inflammatory bowel disease were excluded. Those with MLH1 loss were considered to be sporadic MLH1 loss cancers (n=33). MMR deficiency was confirmed by MSI analysis (MSI Analysis Version 1.2 (Promega, USA)). Additional MLH1 loss cases were provided by ACCFR(29). CRC cases with MLH1 loss were classified as sporadic (n=40) based on the presence of the BRAF mutation and/or methylation of the MLH1 gene promoter and did not harbour a pathogenic mutation in the MLH1 gene. Detection of BRAF mutation was determined on CRC tissue DNA using an allele-specific PCR assay as previously described (33). Methylation of the MLH1 gene promoter region was assayed using MethyLight qPCR on sodium bisulphite converted tissue DNA where samples with a percent of methylated reference (PMR) greater than or equal to 10 were classified as positive for MLH1 methylation (13, 14).
Cancer specimens
Manchester samples: H&E slides were reviewed by an experienced Consultant Gastrointestinal Pathologist (RM) and areas which contained at least 70% cancer cells were selected. 10µm thick slices were taken from the corresponding FFPE block for DNA extraction. ACCFR samples: approximately six 4µm thick FFPE slices mounted on microscope slides were obtained per cancer. Each slice was reported to contain at least 70% tumour tissue. The tissue was manually removed from the slides and placed into 1.5ml Eppendorf tubes for DNA extraction.
MLH1 methylation analysis
MLH1 promoter methylation was quantified using a novel pyrosequencing assay developed to UK Good Laboratory Practice standard. This assay relies on sodium bisulphite conversion. The EpiTect plus FFPE Bisulphite kit® (Qiagen, UK) was used as per manufacturer’s instructions, with an additional overnight tissue lysis step, to extract and bisulphite modify genomic tumour DNA from approximately 10µm thick sections of FFPE tissue. An area of the MLH1 promoter from −248 to −178, known to be functionally significant (34) was amplified (see Figure 1). Each sample was tested in triplicate. A CpGenome Universal Methylated DNA control (Millipore™, cat no. S7821) was used.
Figure 1.

MLH1 promoter region and BRAF polymerase chain reaction (PCR) conditions
The amplicons were sequenced using the Pyrosequencer (PSQ 96MA). Sequencing primer: GAATTAATAGGAAGAG. Pyrograms were analysed independently by two blinded scientists. Greater than 10% methylation at each cytosine (35), in at least two of the three triplicates was considered significant. Figure 2 show a typical pyrogram of methylated MLH1 tumour DNA. Genotype: CmGGACAGCmGATTTTTAACmGCmG (methylated).
Figure 2.
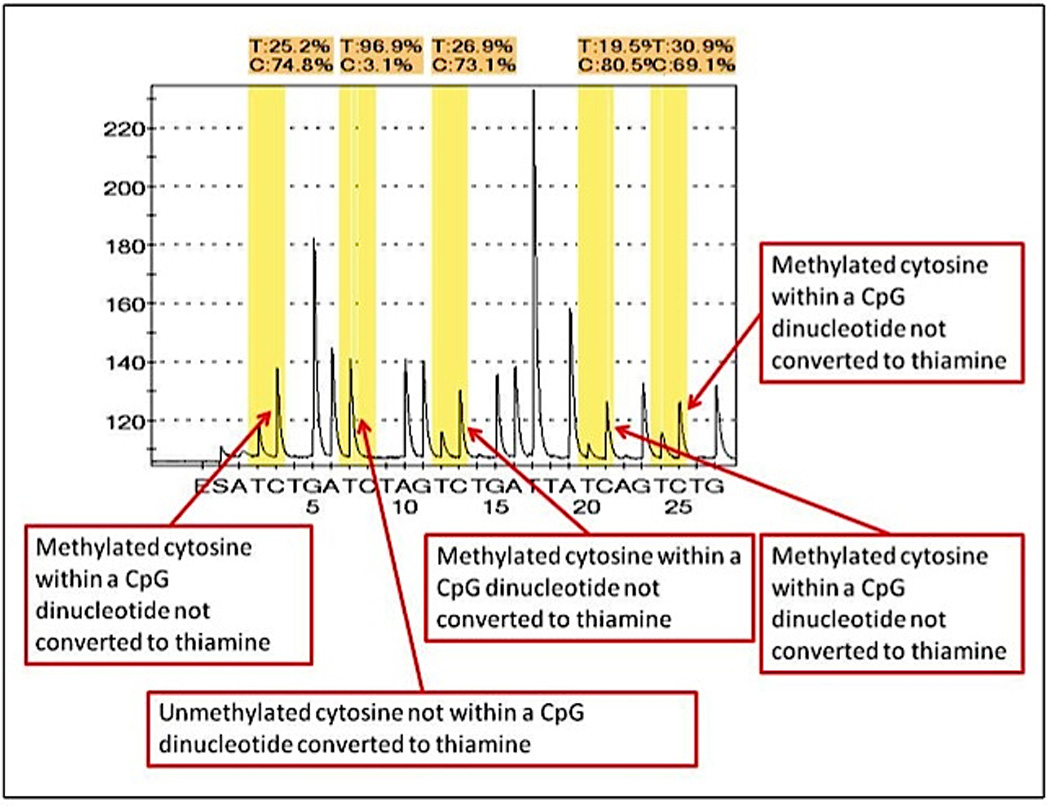
Pyrogram of methylated MLH1 promoter region of tumour DNA
BRAF c.1799T>A p.Val600Glu mutation analysis
The region of BRAF codon 600 (exon 15) was sequenced using the Pyrosequencer (PSQ 96MA) and associated software (Qiagen, UK). Tumour DNA was extracted from the FFPE tissue using the Qiagen EZ1 robot in conjuction with the Tissue Extraction kit (Qiagen, UK) as per the manufacturer’s guidelines. Codon 600 was amplified using a non-nested 25µl polymerase chain reaction. Each sample was tested in triplicate. The amplicons were sequenced using the Pyrosequencer (PSQ 96MA) and associated software. Sequencing primer 5’-TGATTTTGGTCTAGCTACA-3’. Pyrograms were genotyped by two independent blinded scientists.
Statistical Methods
The performance characteristics of unmethylated MLH1 promoter region and wild-type BRAF c.1799T>A p.Val600Glu and for the identification of cancers from the individuals with a constitutional MLH1 mutation were analysed using Diagnostic test two by two tables (StatsDirect Ltd, Cheshire UK). Each test was analysed separately and in conjunction by applying the ‘either positive’ and ‘both positive’ rules. A Bayesian calculation was used to calculate the post-test risk of harbouring a constitutional MLH1 mutation in groups of patients with differing a priori risk. This was calculated from all relevant published studies. In this setting the post-test risk of an individual harbouring an MLH1 mutation can be calculated taking into account the pre-test (priori) risk which is determined by their family history, and also their test result.
Results
71 CRCs from pathogenic constitutional MLH1 mutation carriers, and 73 sporadic cancers with MLH1 protein loss were analysed. Somatic MLH1 promoter region methylation was found in 4/71 (5.6%) tumours from constitutional MLH1 mutation carriers and 64/73 (87.7%) sporadic cancers with MLH1 loss. Somatic BRAF mutation was found in 1/71 (1.4%) tumours from constitutional MLH1 mutation carriers and 48/73 (65.8%) sporadic cancers with MLH1 loss (See table 1). 33/73 sporadic MLH1 loss cancers were known to be wild-type (wt) for constitutional MLH1 gene mutations. Of these 28/33 (84.8%) had somatic MLH1 methylation and 18/33 (54.5%) had somatic BRAF mutation. Of the 40 that had not been tested for constitutional mutations, 36/40 (90.0%) had somatic MLH1 methylation and 30/40 (75.0%) had somatic BRAF mutation.
Table 1.
BRAF c.1799T>A p.Val600Glu mutation and MLH1 promoter region methylation in MLH1 mutation carriers and sporadic MLH1 loss cancers
| Tumour Status | Mutant BRAF AND MLH1 methylation |
wt BRAF AND normal MLH1 |
Mutant BRAF AND normal MLH1 |
wt BRAF AND MLH1 methylation |
|---|---|---|---|---|
| MLH1 mutation carriers tumours n = 71 | 0 | 66 (92.9%) | 1 (1.4%) | 4 (5.6%) |
| MMR deficient sporadic tumours n = 73 | 46 (63.0%) | 7 (9.6%) | 2 (2.7%) | 18 (24.7%) |
The mean percentage of methylation (of the four CpGs examined in triplicate for each sample) of the methylated tumours was 75.8%, and the median 81.5% (range 19.3–100%). There was no difference in the level of methylation between the MLH1 methylated tumours from the sporadic group and those tumours that demonstrated MLH1 methylation from constitutional MLH1 mutation carriers.
We have demonstrated that a normal (unmethylated) MLH1 promoter region has a higher specificity (87.67% [95% CI 77.88–94.2%]) than wt BRAF (65.75% [95% CI 53.72–76.47%]) and similar sensitivity (normal MLH1 promoter region 94.37% [95% CI 86.2–98.44%], wt BRAF (98.59% [92.4–99.96%]) for the identification of MLH1 mutation carriers.
When used in combination, a wt BRAF OR normal MLH1 promoter region result has the highest sensitivity for the identification of MLH1 mutation carriers (100% [95% CI 94.94% to 100%]), but has the lowest specificity (63.01% [50.91% to 74.03%]). wt BRAF AND normal MLH1 has the lowest sensitivity (92.96% [95% CI 84.33% to 97.67%]) but the highest specificity (90.41% [81.24% to 96.06%]).
Applying MLH1 methylation and/or BRAF mutation analysis to tumour DNA from patients who have a dMMR cancer and fulfil Amsterdam II criteria does not significantly improve diagnostic prediction of MLH1 mutation. Although the identification of wt BRAF and/or normal MLH1 promoter region strongly suggests that a constitutional MLH1 mutation is present, mutant BRAF and/or MLH1 hypermethylation does not bring the (post-test) risk of a mutation to below 10%.
Applying MLH1 methylation and/or BRAF mutation analysis to tumour DNA from patients who fulfil the revised Bethesda guidelines and have loss of MLH1 is informative. An unmethylated MLH1 promoter region suggests a risk of having a constitutional MLH1 mutation of 31.4% while the identification of wild-type BRAF suggests a post-test risk of 19.0%. The finding of MLH1 methylation gives a risk of 1.2%. The finding of mutant BRAF gives a risk of 1.6%.
Applying MLH1 promoter region methylation analysis to tumour DNA from patients from the general population who have a tumour with MLH1 loss is informative. The finding of normal MLH1 promoter region suggests a risk of a constitutional MLH1 mutation of 14.0% compared with the finding of methylated MLH1 promoter region, which suggests a risk of 0.4%. However, applying BRAF mutation analysis to tumour DNA from patients from the general population who have MLH1 loss is uninformative. Wt BRAF only indicates a risk of 7.7%, so constitutional testing would not necessarily be indicated. Applying both BRAF and MLH1 analysis is only informative if both are wt/normal.
The ACCFR had performed somatic MLH1 promoter methylation and BRAF analysis on a proportion of their sporadic MLH1 loss CRCs. MLH1 methylation result was consistent in 21/22 cases and BRAF result was consistent in 39/39 cases with the pyrosequencing assays performed in this study.
Discussion
This study is the first large-scale assessment of specificity and sensitivity of pre-testing MLH1 CRC loss for the presence of a constitutional MLH1 mutation. We have demonstrated the utility of pyrosequencing-based testing of CRC tumour DNA for MLH1 promoter methylation in a large cohort of CRCs. A novel MLH1 promoter region methylation assay has been developed to GLP standards and its clinical utility demonstrated in the assessment of patients meeting Bethesda guidelines and in population based pre-screening for Lynch Syndrome. A single assay is time and cost-effective and may encourage the introduction of pre-screening into routine clinical practice. Unmethylated MLH1 has a sensitivity of 94.4% and a specificity of 87.7% for the identification of MLH1 mutation carriers from a group of cancers with MLH1 loss mismatch repair deficiency. As such methylation is more effective than BRAF testing.
MMR protein immunohistochemistry should be used as the first-line LS pre-screening test in patients meeting Bethesda guidelines and cancers with typical Lynch syndrome features histologically. If MSH2, MSH6 or PMS2 proteins are absent this indicates high risk of LS and the individual should be tested for constitutional mutations in the relevant gene. If MLH1 protein is absent, tumour DNA should be subjected to MLH1 promoter methylation testing. If methylation is absent, this indicates high risk (>10%) of LS and constitutional MLH1 mutation analysis should be conducted.
It was previously unknown whether BRAF mutation or MLH1 promoter region methylation or both, is best able to distinguish between sporadic MLH1 loss CRCs and cancers from patients with constitutional MLH1 mutations. Previous studies have examined small numbers of patients and thus there is no guide for clinical practice (36–38).
Sporadic MMR deficient cancers with loss of MLH1 (or MSI-high) are associated with MLH1 gene silencing through the epigenetic effect of promoter region methylation. In this study MLH1 promoter region methylation was found in 64/73 (87.7%) sporadic MMR deficient colorectal cancers. 33/73 (45.0%) of the sporadic MMR deficient CRCs are known to be negative for constitutional MLH1 mutations. 28/33 (84.8%) were found to have MLH1 promoter methylation. Methylation was consistent across all four cytosine residues in the functional area of the promoter region as described by Deng et al (34). Pyrosequencing allows accurate quantification of methylation. 9/73 (12.3%) sporadic MMR deficient CRCs were found to have normal MLH1 promoter region. Two were found to have BRAF mutation. Of the seven that were wild-type BRAF and normal MLH1 promoter region, four had been tested for constitutional MLH1 mutations by the ACCFR and were found to be negative. The remaining three had been classified as sporadic MMR deficient due to the patient’s age (over 50 years) and lack of family history. The aetiology of these cancers without promoter region hypermethylation is unclear but possible factors include loss of protein expression, somatic mutation of MLH1 and loss of heterozygosity (39). It is feasible that the MLH1 promoter displayed mosaic or heterogeneous patterns of methylation for the CpGs dinucleotides captured in the pyrosequencing amplicon but enough of the surrounding CpGs dinucleotides were methylated to result in loss of MLH1 protein expression. Alternatively, the three untested patients may be carriers of constitutional MLH1 mutations.
It has been thought that MLH1 promoter methylation is found exclusively in sporadic MMR deficient CRCs (24, 34, 40–42). The current study is the largest dataset of MLH1 mutation carriers tested for MLH1 promoter region methylation. MLH1 promoter methylation was found in 4/71 (5.6%) mutation carriers (see Table 4). Of these 1/22 (4.5%) was a Manchester patient (from an Amsterdam family), and 3/49 (6.1%) were from the Australasian Colon Cancer Family Registry (two from Amsterdam families) A first degree relative of the Manchester patient (with the same constitutional MLH1 mutation) has an unmethylated somatic MLH1.
Table 4.
MLH1 promoter region methylation in MLH1 mutation heterozygotes
| Sample number |
Level of methylation |
Clinical details | Family History | MLH1 Mutation |
|---|---|---|---|---|
| 11005300 | 29–38% | Carcinoma in situ in adenoma, aged 40 | Amsterdam | c.405insA |
| 11007868 | 19–60% | CRC aged 44 | Amsterdam | c.1852_1854delAAG p.Lys618del |
| 11007938 | 70–80% | CRC aged 53 | *FDR with endometrial cancer | MLH1 c.1668-1G>A r.spl? p.? |
| 11007944 | 67–82% | CRC aged 39 | Amsterdam | MLH1 del x6 |
First degree relative
This low MLH1 promoter methylation frequency in mutation carriers is supported by a recent literature review and meta-analysis (43) which found eight positively methylated tumours in MLH1 mutation carriers taken from 12 studies (5.56%). It has been suggested that sporadic inactivation of the second normal MLH1 allele by hypermethylation may be the ‘second hit’ event in mutation carriers (44). Whilst somatic MLH1 promoter region methylation is an infrequent event in constitutional MLH1 mutation carriers, this data demonstrates that it is not rare and supports the hypothesis that it may function as the second hit event. These findings also suggest that the discovery of MLH1 hypermethylation does not exclude the diagnosis of LS. Whilst the sensitivity of MLH1 hypermethylation testing is adequate for low/moderate risk individuals, it is not for high risk patients (3/4 MLH1 mutation carriers that had MLH1 promoter methylation were from Amsterdam families).
BRAF gene mutations are found in 5–15% of all CRCs (45, 46). They are more frequent in cancers from Jass’s subtypes 1 (MSI-H, chromosome stable, CIMP high, methylated MLH1 promoter region; 13% of all CRCs) and 2 (MSI-low or stable, chromosome stable, CIMP-high, partial MLH1 methylation; 8% of all CRCs) (12). Both are thought to originate in serrated lesions. BRAF mutation is thought to be an unequivocal marker of the serrated neoplasia pathway. The discovery of a BRAF mutation is thought to rule out LS (47). In the current study, BRAF mutation was found in 48/73 (66%) sporadic MLH1 loss CRCs. This is consistent with previous studies (42, 48). A BRAF mutation was detected in 1/71 (1.4%) CRCs from MLH1 mutation carriers. BRAF mutations have previously been reported as a rare finding in patients with LS (49, 50), and are thought to represent a mixed lineage of cancer predisposition. Walsh et al have reported two families with evidence of LS and probable additional constitutional factors causing serrated neoplasia (47). Senter et al investigated 99 probands with Lynch spectrum cancers that demonstrated loss of PMS2 on IHC. Constitutional PMS2 mutations were detected in 62%, and three (one exon 10 deletion, two c.736_741del6ins11) of these were found to have tumour BRAF mutation (31). It is likely that BRAF mutation is a rare finding in LS, and that its occurrence represents the influence of other molecular pathways, as suggested by Walsh (47).
It has been suggested that in MMR deficient cancers, BRAF mutation is a surrogate marker for MLH1 promoter methylation. However, there is now evidence that BRAF mutation occurs in only 50–75% of sporadic MLH1 loss cancers. In a series of 270 CRCs, Wang et al found BRAF mutations in 42/123 (34%) MMR deficient cases. BRAF was closely associated with MLH1 methylation (30/36 [83.3%] MLH1 hypermethylated cases also had a BRAF mutation) (48). In a large population-based study, Woods et al examined 68 MSI-H CRCs for BRAF and MLH1 methylation in order to pre-screen for constitutional mutation testing. BRAF mutation was closely but not exclusively associated with MLH1 methylation. 31/40 (78%) of the hypermethylated tumours had BRAF mutations. In the current study 46/73 (63.0%) sporadic MLH1 loss cancers had both BRAF mutation and MLH1 methylation, but 18/73 (24.7%) had only MLH1 methylation. This is consistent with previous studies (42, 48).
A recent HTA report has established that it would be cost-effective for the NHS to introduce systematic testing for Lynch Syndrome of all colorectal cancers up to age 70 (28). This report addressed the issue of excluding sporadic MLH1 cases from requiring unnecessary referral to clinical genetics services. The cost effectiveness analysis allows for the increased costs of performing additional tests and the inherent reduction in sensitivity when more tests are performed serially in an attempt to increase specificity. Our data suggest that only 65% of cases without constitutional mutations will be identified by using BRAF alone. This is increased to 90% by adding a MLH1 methylation test. There are around 16800 new cases of colorectal cancer up to age 70 each year in the UK (51). Around 2200 (13%) of these will be sporadic MLH1 cases. The increased specificity of additional MLH1 methylation testing (90%) rather than BRAF alone (65%) would reduce the number of cases requiring genetic counselling and testing from around 780 to 220, a reduction of 550 cases each year. In our laboratory MLH1 mutation testing costs around £483, MLH1 methylation testing costs around £138 and BRAF testing around £69. On average in the UK, a new person appointment with a genetic counselor or physician costs around £500 and a follow up appointment around £350. Adding MLH1 methylation testing into systematic testing would cost around £300,000 per year. The cost saving each year would be around £700,000 (£450,000 for counseling and £250,000 for constitutional MLH1 analysis).
There are some limitations to the current study. A proportion of the sporadic samples did not undergo constitutional testing for MLH1. Even full sequencing and a dosage test of MLH1 may miss mutations such as deep intronic splicing mutations and sensitivity may therefore be reduced. Clendenning et al have reported the discovery of an intronic MSH2 mutation, 478bp upstream from exon 2 causing LS (52). As such some of the ‘sporadic’ MLH1 loss CRCs may have had an undetected constitutional mutation. However, the rate of non-methylated MLH1 and wt BRAF in the 40 (4/40, 10%) sporadic tumours with mutation testing was not different to that in the 33 (3/33, 9.1%) untested sporadic cases. In addition, the untested sporadic patients may harbour constitutional methylation.
Constitutional MLH1 promoter region methylation has been described as a rare (33 reported cases) finding in CRC (16–18, 53–57). It is thought that this epimutation is usually erased in the gametes but inheritance has been demonstrated in four cases (15–17, 19). A recent study from the German HNPCC consortium investigated 32 mutation negative suspected Lynch cases with MSI-H and MLH1 loss CRC. They report one case of heritable partial MLH1 promoter methylation, which is induced by a large genomic duplication including the complete MLH1 gene and the promoter (15). This suggests that even in mutation negative Lynch cases, the finding of constitutional methylation is low. Constitutional MLH1 methylation is likely a rare cause for CRC tumour DNA MLH1 promoter region methylation, and a rare cause for LS, although the true incidence is unknown. In 10–15% of suspected Lynch cases, no disease causing mechanism can be detected. In these cases it may be prudent to test for constitutional MLH1 promoter methylation.
Schofield et al reported a population based screening programme utilising IHC, MSI and BRAF testing in CRCs in patients aged below 60. In the cohort of 270, 70 were MSI-H. 82 had loss of MMR protein expression. BRAF testing was conducted on 76 tumours. 25 mutant BRAF tumours were excluded from further testing. 45 ‘Red Flag’ cases were identified (MSI-H and loss of MSH2 or MSH6, OR MSH-H and loss of MLH1/PMS2 and wtBRAF). 31 were tested for constitutional mutations. 15 mutation carriers (7 MLH1, 2 MSH2, 3 PMS2 and 3 MSH6) were identified. The incidence of constitutional mutation in their ‘Red Flag’ cases is 48%. Our study has demonstrated that IHC followed by MLH1 methylation testing is likely to have a higher ‘hit’ rate due to the higher specificity of MLH1 methylation compared to BRAF (88% versus 66% in our study). Utilising IHC as the initial test avoids additional expense of MSI and allows the appropriate gene to be targeted for constitutional testing.
Identification of families with LS is vital to enable reduction in morbidity and mortality with screening. The use of population-based pre-screening has been hampered by a lack of evidence for the specificity and sensitivity of MLH1 promoter region methylation analysis for the detection of mutation carriers. It is hoped that this current study provides that evidence.
Conclusion
MLH1 promoter region methylation analysis is simple, reproducible and cheap. It can be used in conjunction with mismatch repair IHC in the pre-screening of low and moderate risk patients for Lynch Syndrome mutation testing. This will be a vital addition to BRAF testing when population assessment of MMR deficiency is introduced. Amsterdam criteria CRCs should be tested for constitutional mutation regardless of CRC pre-screening status.
Table 2.
Diagnostic Test (2 by 2 tables) analysis for the identification of MLH1 mutation carriers
| Sensitivity (95% CI) | Specificity (95% CI) | |
|---|---|---|
| Normal MLH1 promoter region | 94.37% (86.20–98.44%) | 87.67% (77.88–94.2%) |
| wt BRAF p.V600E | 98.59% (92.4–99.96%) | 65.75% (53.72–76.47%) |
| wt BRAF c.1799T>A p.Val600Glu OR Normal MLH1 promoter region | 100% (94.94% to 100%) | 63.01% (50.91% to 74.03%) |
| wt BRAF c.1799T>A p.Val600Glu AND Normal MLH1 promoter region | 92.96% (84.33% to 97.67%) | 90.41% (81.24% to 96.06%) |
Table 3.
Pre- and Post-test probabilities of being an MLH1 mutation carrier for three a priori risk groups
| *ACII | *BG & *1dMMR tumour |
General population & *1dMMR tumour |
||
|---|---|---|---|---|
| Pre-test probability | 60% (6) | 10.5% (21–23) | 4.0% (23–26) | |
| Post-test normal MLH1 promoter region | True positive | 85.5% | 31.4% | 14.0% |
| False negative | 12.6% | 1.2% | 0.4% | |
| Post-test wt BRAF | True positive | 74.9% | 19.0% | 7.7% |
| False negative | 17.5% | 1.6% | 0.6% | |
| Post-test wt BRAF OR normal MLH1 | True positive | 71.6 | 18.5 | 7.7 |
| False negative | 13.1 | 1.31 | 0.6 | |
| Post-test wt BRAF AND normal MLH1 | True positive | 86.9 | 33.5 | 15.5% |
| False negative | 22.9 | 2.3 | 0.8% | |
AC = Amsterdam criteria and BC = Bethesda guidelines
dMMR tumour = MLH1 loss on immunohistochemistry or MSI-H
Acknowledgments
Keeling C and Griffiths-Davies L (Department of Histopathology, Manchester Royal Infirmary, Central Manchester University Hospitals NHS Foundation Trust) for conducting immunohistochemistry.
Newton K, Jorgensen NM, Lalloo, F, Hill J, and Evans DG are supported, by Manchester NIHR BRC.
Biospecimens were provided by The Jeremy Jass Memorial Pathology Bank, Australasian Colorectal Cancer Family Registry (U01 CA097735)
“This work was supported by the National Cancer Institute, National Institutes of Health under RFA # CA-95-011 and through cooperative agreements with members of the Colon Cancer Family Registry and P.I.s. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CFRs, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the CFR”
Footnotes
Author contributions: Newton K, Wallace AJ, Hill J, Lalloo F, Evans DG contributed to the study concept, study design, acquisition of data, analysis and interpretation of data, drafting of the manuscript and critical revision of the manuscript for important intellectual content. Jorgensen NM contributed to acquisition of data, analysis and interpretation of data, drafting of the manuscript and critical revision of the manuscript for important intellectual content. Buchanan DD contributed to acquisition of data, analysis and interpretation of data and critical revision of the manuscript for important intellectual content
No disclosures.
References
- 1.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525–2538. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler JM, Bodmer WF, Mortensen NJ. DNA mismatch repair genes and colorectal cancer. Gut. 2000;47(1):148–153. doi: 10.1136/gut.47.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 4.de Jong AE, Hendriks YM, Kleibeuker JH, de Boer SY, Cats A, Griffioen G, Nagengast FM, Nelis FG, Rookus MA, Vasen HF. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology. 2006;130(3):665–671. doi: 10.1053/j.gastro.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 5.Stupart DA, Goldberg PA, Algar U, Ramesar R. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Colorectal Dis. 2009;11(2):126–130. doi: 10.1111/j.1463-1318.2008.01702.x. [DOI] [PubMed] [Google Scholar]
- 6.Park JG, Vasen HF, Park YJ, Park KJ, Peltomaki P, de Leon MP, Rodriguez-Bigas MA, Lubinski J, Beck NE, Bisgaard ML, Miyaki M, Wijnen JT, Baba S, Lindblom A, Madlensky L, Lynch HT. Suspected HNPCC and Amsterdam criteria II: evaluation of mutation detection rate, an international collaborative study. Int J Colorectal Dis. 2002;17(2):109–114. doi: 10.1007/s003840100348. [DOI] [PubMed] [Google Scholar]
- 7.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. Jama. 2006;296(12):1507–1517. doi: 10.1001/jama.296.12.1507. [DOI] [PubMed] [Google Scholar]
- 9.EGAPP. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41. doi: 10.1097/GIM.0b013e31818fa2ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrow E, Jagger E, Brierley J, Wallace A, Evans G, Hill J, McMahon R. Semiquantitative assessment of immunohistochemistry for mismatch repair proteins in Lynch syndrome. Histopathology. 2011;56(3):331–344. doi: 10.1111/j.1365-2559.2010.03485.x. [DOI] [PubMed] [Google Scholar]
- 11.Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, Hopper JL, Le Marchand L, Gallinger S, Newcomb PA, Haile R, Thibodeau SN, Gunawardena S, Jenkins MA, Buchanan DD, Potter JD, Baron JA, Ahnen DJ, Moreno V, Andreu M, Ponz de Leon M, Rustgi AK, Castells A. Identification of Lynch syndrome among patients with colorectal cancer. Jama. 2012;308(15):1555–1565. doi: 10.1001/jama.2012.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50(1):113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 13.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 14.Poynter JN, Siegmund KD, Weisenberger DJ, Long TI, Thibodeau SN, Lindor N, Young J, Jenkins MA, Hopper JL, Baron JA, Buchanan D, Casey G, Levine AJ, Le Marchand L, Gallinger S, Bapat B, Potter JD, Newcomb PA, Haile RW, Laird PW. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol Biomarkers Prev. 2008;17(11):3208–3215. doi: 10.1158/1055-9965.EPI-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morak M, Koehler U, Schackert HK, Steinke V, Royer-Pokora B, Schulmann K, Kloor M, Hochter W, Weingart J, Keiling C, Massdorf T, Holinski-Feder E, German Hc. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J Med Genet. 2011;48(8):513–519. doi: 10.1136/jmedgenet-2011-100050. [DOI] [PubMed] [Google Scholar]
- 16.Morak M, Schackert HK, Rahner N, Betz B, Ebert M, Walldorf C, Royer-Pokora B, Schulmann K, von Knebel-Doeberitz M, Dietmaier W, Keller G, Kerker B, Leitner G, Holinski-Feder E. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur J Hum Genet. 2008;16(7):804–811. doi: 10.1038/ejhg.2008.25. [DOI] [PubMed] [Google Scholar]
- 17.Crepin M, Dieu MC, Lejeune S, Escande F, Boidin D, Porchet N, Morin G, Manouvrier S, Mathieu M, Buisine MP. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat. 2011;33(1):180–188. doi: 10.1002/humu.21617. [DOI] [PubMed] [Google Scholar]
- 18.Goel A, Nguyen TP, Leung HC, Nagasaka T, Rhees J, Hotchkiss E, Arnold M, Banerji P, Koi M, Kwok CT, Packham D, Lipton L, Boland CR, Ward RL, Hitchins MP. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. 2011;128(4):869–878. doi: 10.1002/ijc.25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hitchins MP, Wong JJ, Suthers G, Suter CM, Martin DI, Hawkins NJ, Ward RL. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356(7):697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- 20.ASCO. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol. 2003;21(12):2397–2406. doi: 10.1200/JCO.2003.03.189. [DOI] [PubMed] [Google Scholar]
- 21.Julie C, Tresallet C, Brouquet A, Vallot C, Zimmermann U, Mitry E, Radvanyi F, Rouleau E, Lidereau R, Coulet F, Olschwang S, Frebourg T, Rougier P, Nordlinger B, Laurent-Puig P, Penna C, Boileau C, Franc B, Muti C, Hofmann-Radvanyi H. Identification in daily practice of patients with Lynch syndrome (hereditary nonpolyposis colorectal cancer): revised Bethesda guidelines-based approach versus molecular screening. Am J Gastroenterol. 2008;103(11):2825–2835. doi: 10.1111/j.1572-0241.2008.02084.x. quiz 36. [DOI] [PubMed] [Google Scholar]
- 22.Wright DM, Arnold JL, Parry B, Hulme-Moir M, Winship IM, Parry S. Immunohistochemistry to detect hereditary nonpolyposis colorectal cancer in young patients: the 7-year Auckland experience. Dis Colon Rectum. 2011;54(5):552–558. doi: 10.1007/DCR.0b013e31820e3265. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Carbonell L, Ruiz-Ponte C, Guarinos C, Alenda C, Paya A, Brea A, Egoavil CM, Castillejo A, Barbera VM, Bessa X, Xicola RM, Rodriguez-Soler M, Sanchez-Fortun C, Acame N, Castellvi-Bel S, Pinol V, Balaguer F, Bujanda L, De-Castro ML, Llor X, Andreu M, Carracedo A, Soto JL, Castells A, Jover R. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut. 2011 doi: 10.1136/gutjnl-2011-300041. [DOI] [PubMed] [Google Scholar]
- 24.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, Comeras I, de la Chapelle A. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352(18):1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 25.Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, Xicola RM, Rodriguez-Moranta F, Paya A, Jover R, Bessa X. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. Jama. 2005;293(16):1986–1994. doi: 10.1001/jama.293.16.1986. [DOI] [PubMed] [Google Scholar]
- 26.Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, Chadwick RB, Kaariainen H, Eskelinen M, Jarvinen H, Mecklin JP, de la Chapelle A. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338(21):1481–1487. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]
- 27.Evans GD, Lalloo F, Mak T, Speake D, Hill J. Is it time to abandon microsatellite instability as a pre-screen for selecting families for mutation testing for mismatch repair genes? J Clin Oncol. 2006;24(12):1960–1962. doi: 10.1200/JCO.2005.05.3207. author reply 2–3. [DOI] [PubMed] [Google Scholar]
- 28.2014 http://www.nets.nihr.ac.uk/projects/hta/102801. [Google Scholar]
- 29. [Accessed 2011]; http://epi.grants.cancer.gov/CFR/about_colon.html. Colon cancer family registry.
- 30.Walsh MD, Cummings MC, Buchanan DD, Dambacher WM, Arnold S, McKeone D, Byrnes R, Barker MA, Leggett BA, Gattas M, Jass JR, Spurdle AB, Young J, Obermair A. Molecular, pathologic, and clinical features of early-onset endometrial cancer: identifying presumptive Lynch syndrome patients. Clin Cancer Res. 2008;14(6):1692–1700. doi: 10.1158/1078-0432.CCR-07-1849. [DOI] [PubMed] [Google Scholar]
- 31.Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM, Young J, Winship I, Dowty JG, White DM, Hopper JL, Baglietto L, Jenkins MA, de la Chapelle A. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135(2):419–428. doi: 10.1053/j.gastro.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Southey MC, Jenkins MA, Mead L, Whitty J, Trivett M, Tesoriero AA, Smith LD, Jennings K, Grubb G, Royce SG, Walsh MD, Barker MA, Young JP, Jass JR, St John DJ, Macrae FA, Giles GG, Hopper JL. Use of molecular tumor characteristics to prioritize mismatch repair gene testing in early-onset colorectal cancer. J Clin Oncol. 2005;23(27):6524–6532. doi: 10.1200/JCO.2005.04.671. [DOI] [PubMed] [Google Scholar]
- 33.Buchanan DD, Sweet K, Drini M, Jenkins MA, Win AK, English DR, Walsh MD, Clendenning M, McKeone DM, Walters RJ, Roberts A, Pearson SA, Pavluk E, Hopper JL, Gattas MR, Goldblatt J, George J, Suthers GK, Phillips KD, Woodall S, Arnold J, Tucker K, Muir A, Field M, Greening S, Gallinger S, Perrier R, Baron JA, Potter JD, Haile R, Frankel W, de la Chapelle A, Macrae F, Rosty C, Walker NI, Parry S, Young JP. Risk factors for colorectal cancer in patients with multiple serrated polyps: a cross-sectional case series from genetics clinics. PLoS One. 2010;5(7):e11636. doi: 10.1371/journal.pone.0011636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng G, Bell I, Crawley S, Gum J, Terdiman JP, Allen BA, Truta B, Sleisenger MH, Kim YS. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(1 Pt 1):191–195. doi: 10.1158/1078-0432.ccr-1118-3. [DOI] [PubMed] [Google Scholar]
- 35.Vaughn CP, Wilson AR, Samowitz WS. Quantitative evaluation of CpG island methylation in hyperplastic polyps. Mod Pathol. 2010;23(1):151–156. doi: 10.1038/modpathol.2009.150. [DOI] [PubMed] [Google Scholar]
- 36.Jensen LH, Dysager L, Lindebjerg J, Kolvra S, Byriel L, Cruger DG. Molecular biology from bench-to-bedside - which colorectal cancer patients should be referred for genetic counselling and risk assessment. Eur J Cancer. 2010;46(10):1823–1828. doi: 10.1016/j.ejca.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Bouzourene H, Hutter P, Losi L, Martin P, Benhattar J. Selection of patients with germline MLH1 mutated Lynch syndrome by determination of MLH1 methylation and BRAF mutation. Fam Cancer. 2010;9(2):167–172. doi: 10.1007/s10689-009-9302-4. [DOI] [PubMed] [Google Scholar]
- 38.Perez-Carbonell L, Alenda C, Paya A, Castillejo A, Barbera VM, Guillen C, Rojas E, Acame N, Gutierrez-Avino FJ, Castells A, Llor X, Andreu M, Soto JL, Jover R. Methylation analysis of MLH1 improves the selection of patients for genetic testing in Lynch syndrome. J Mol Diagn. 2010;12(4):498–504. doi: 10.2353/jmoldx.2010.090212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuismanen SA, Holmberg MT, Salovaara R, de la Chapelle A, Peltomaki P. Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am J Pathol. 2000;156(5):1773–1779. doi: 10.1016/S0002-9440(10)65048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yearsley M, Hampel H, Lehman A, Nakagawa H, de la Chapelle A, Frankel WL. Histologic features distinguish microsatellite-high from microsatellite-low and microsatellite-stable colorectal carcinomas, but do not differentiate germline mutations from methylation of the MLH1 promoter. Hum Pathol. 2006;37(7):831–838. doi: 10.1016/j.humpath.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Nakagawa H, Nagasaka T, Cullings HM, Notohara K, Hoshijima N, Young J, Lynch HT, Tanaka N, Matsubara N. Efficient molecular screening of Lynch syndrome by specific 3' promoter methylation of the MLH1 or BRAF mutation in colorectal cancer with high-frequency microsatellite instability. Oncol Rep. 2009;21(6):1577–1583. doi: 10.3892/or_00000390. [DOI] [PubMed] [Google Scholar]
- 42.Woods MO, Younghusband HB, Parfrey PS, Gallinger S, McLaughlin J, Dicks E, Stuckless S, Pollett A, Bapat B, Mrkonjic M, de la Chapelle A, Clendenning M, Thibodeau SN, Simms M, Dohey A, Williams P, Robb D, Searle C, Green JS, Green RC. The genetic basis of colorectal cancer in a population-based incident cohort with a high rate of familial disease. Gut. 2010;59(10):1369–1377. doi: 10.1136/gut.2010.208462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49(3):151–157. doi: 10.1136/jmedgenet-2011-100714. [DOI] [PubMed] [Google Scholar]
- 44.Rahner N, Friedrichs N, Steinke V, Aretz S, Friedl W, Buettner R, Mangold E, Propping P, Walldorf C. Coexisting somatic promoter hypermethylation and pathogenic MLH1 germline mutation in Lynch syndrome. J Pathol. 2008;214(1):10–16. doi: 10.1002/path.2263. [DOI] [PubMed] [Google Scholar]
- 45.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 46.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(6901):934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 47.Walsh MD, Buchanan DD, Walters R, Roberts A, Arnold S, McKeone D, Clendenning M, Ruszkiewicz AR, Jenkins MA, Hopper JL, Goldblatt J, George J, Suthers GK, Phillips K, Young GP, Macrae F, Drini M, Woods MO, Parry S, Jass JR, Young JP. Analysis of families with Lynch syndrome complicated by advanced serrated neoplasia: the importance of pathology review and pedigree analysis. Fam Cancer. 2009;8(4):313–323. doi: 10.1007/s10689-009-9238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, Boardman LA, Burgart LJ, McDonnell SK, Schaid DJ, Thibodeau SN. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63(17):5209–5212. [PubMed] [Google Scholar]
- 49.Lagerstedt Robinson K, Liu T, Vandrovcova J, Halvarsson B, Clendenning M, Frebourg T, Papadopoulos N, Kinzler KW, Vogelstein B, Peltomaki P, Kolodner RD, Nilbert M, Lindblom A. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst. 2007;99(4):291–299. doi: 10.1093/jnci/djk051. [DOI] [PubMed] [Google Scholar]
- 50.Vandrovcova J, Lagerstedt-Robinsson K, Pahlman L, Lindblom A. Somatic BRAF-V600E mutations in familial colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(11):2270–2273. doi: 10.1158/1055-9965.EPI-06-0359. [DOI] [PubMed] [Google Scholar]
- 51. [Accessed 18.08.2014]; http://www.cancerresearchuk.org/cancer-info/cancerstats/types/bowel/incidence/uk-bowel-cancer-incidence-statistics - By2. [Google Scholar]
- 52.Clendenning M, Buchanan DD, Walsh MD, Nagler B, Rosty C, Thompson B, Spurdle AB, Hopper JL, Jenkins MA, Young JP. Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam Cancer. 2011;10(2):297–301. doi: 10.1007/s10689-011-9427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valle L, Carbonell P, Fernandez V, Dotor AM, Sanz M, Benitez J, Urioste M. MLH1 germline epimutations in selected patients with early-onset non-polyposis colorectal cancer. Clin Genet. 2007;71(3):232–237. doi: 10.1111/j.1399-0004.2007.00751.x. [DOI] [PubMed] [Google Scholar]
- 54.Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002;62(14):3925–3928. [PubMed] [Google Scholar]
- 55.Niessen RC, Hofstra RM, Westers H, Ligtenberg MJ, Kooi K, Jager PO, de Groote ML, Dijkhuizen T, Olderode-Berends MJ, Hollema H, Kleibeuker JH, Sijmons RH. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48(8):737–744. doi: 10.1002/gcc.20678. [DOI] [PubMed] [Google Scholar]
- 56.van Roon EH, van Puijenbroek M, Middeldorp A, van Eijk R, de Meijer EJ, Erasmus D, Wouters KA, van Engeland M, Oosting J, Hes FJ, Tops CM, van Wezel T, Boer JM, Morreau H. Early onset MSI-H colon cancer with MLH1 promoter methylation, is there a genetic predisposition? BMC Cancer. 10:180. doi: 10.1186/1471-2407-10-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, Polly P, Goldblatt J, Ward RL. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5'UTR. Cancer Cell. 2011;20(2):200–213. doi: 10.1016/j.ccr.2011.07.003. [DOI] [PubMed] [Google Scholar]