

ABSTRACT
Darier’s disease (DD, keratosis follicularis: OMIM#124200) is an autosomal dominant skin disorder characterized by multiple dark brown keratotic plaques and warty papules covered by thick crusts. Most cases of DD are caused by mutations in ATP2A2, which is expressed in both the skin and the brain. ATP2A2 encodes the cardiac muscle SERCA2a protein and the ubiquitously expressed SERCA2b. SERCA2 plays an important role as a calcium pump. It is thought that a mutation in ATP2A2 causes dyskeratosis and abnormality of cell-cell adhesion. Here, we report five DD patients from five independent families who presented or were referred to the Nagoya University Hospital in the past five years. We detected five mutations in ATP2A2, including a previously unreported mutation. We observed no apparent genotype/phenotype correlation between types and sites of the ATP2A2 mutations and DD phenotypes in the present series of DD patients. Genetic diagnosis from ATP2A2 mutation search is useful for the definite diagnosis of DD, although it is difficult to predict the severity and prognosis of skin symptoms from the results of the ATP2A2 mutation analysis in DD patients.
Key Words: ATP2A2, Darier’s disease, genotype/phenotype correlation, mutation, SERCA2
INTRODUCTION
Darier’s disease (DD, keratosis follicularis: OMIM#124200) is a genetic keratinization disorder with autosomal dominant inheritance. DD shows almost complete penetrance. Patients with DD have been reported around the world, and the prevalence of DD is estimated to be between 1/36,000 and 1/100,000.1) Disease phenotypes of DD are not seen in infancy in most cases, and the disease frequently occurs in childhood and adolescence.2) Patients with DD show various skin manifestations, including keratotic papules on the chest and the forehead, verrucous papules on the hands and the feet, hemorrhagic vesicles on the acral areas of the extremities and vegetative lesions in the intertriginous regions. DD sometimes shows neuropsychiatric manifestations.3)
Most cases of DD are caused by mutations in ATP2A2. ATP2A2 is expressed in both the skin and the brain and encodes type 2 sarcoendoplasmic reticulum calcium-ATPase (SERCA2). SERCA2 works in establishing ion gradients by pumping ions across biological membranes. Two calcium ions can be transported for each ATP that is hydrolysed, and 2 or 3 hydrogen ions are counter-transported. Active removal of calcium ions from the cytosol is a critical step of signal transduction pathways in animal cells, facilitating the rapid cytosolic calcium ion oscillations that are required for a wide range of physiological functions, such as muscle contraction, neuronal transmission, cellular motility and cell growth. SERCA2b is ubiquitously expressed, but expression is particularly high in keratinocytes. SERCA2b has 11 transmembrane helix domains (TMs), TM1-TM11, and there are 9 calcium binding residues (CBRs) in TM4, 5, 6 and 8. SERCA2b has 3 cytoplasmic globular domains: an actuator or anchor domain (the A domain), a phosphorylation domain (the P domain) and a nucleotide binding domain (the N domain).4) When calcium ions are released and a pathway for entry of new calcium ions is created, three extramembranous domains gather to form a single headpiece, and TMs exhibit large-scale rearrangement.5) Causative abnormalities in the adhesion of epidermal keratinocytes have been proposed from the fact that acantholytic cells show aberrant distribution of desmosomal components.6) A review of the literature on ATP2A2 mutations and DD phenotypes suggested certain genotype/phenotype correlations.3)
Here, we report 5 mutations in 5 DD patients from 5 independent families, including 1 previously unreported mutation in ATP2A2.
MATERIALS AND METHODS
Patients
We analyzed 5 Japanese patients from 5 independent families who presented or were referred to our department in the past 5 years (Table 1). The clinical features and ATP2A2 mutations in Case 3 and Case 4 were reported previously,7, 8) and the clinical features of Case 5 were reported previously.9) Detailed clinical features of each patient are as follows.
Table 1.
Summary of clinical features and genotypes in the present series of DD patients
| Case | Age (year) | Sex | Nucleotide change | Amino acid change | Mutation site (functional domain) | Age of onset (years) | Severity of skin symptoms | Affected family members |
|---|---|---|---|---|---|---|---|---|
| 1 | 40 | female | c.2255_2257del (previously unreported) | p.Ile752_Tyr753delinsAsn | stalk domain | 7 | moderate | father, sister |
| 2 | 43 | male | c.548A>T | p.Glu183Val | A domain | 33 | moderate | father |
| 3 | 40 | female | c.2721A>C | p.Glu907Asp | transmembrane helix domain (calcium-binding residue) | 40 | mild | none |
| 4 | 66 | male | c.953G>A | p.Cys318Tyr | stalk domain | 64 | moderate | none |
| 5 | 84 | male | c.391C>T | p.Arg131* | A domain (truncation) | 74 | mild | none |
Case 1
A 40-year-old woman presented with multiple brown papules with itching on her entire body surface, especially on the seborrheic areas (Fig. 1). She had been suffering from DD for 5 years. Her father and sister had similar symptoms.
Fig. 1.

Clinical features of Case 1. Multiple brown papules with crusts are present on the abdomen (A), and the hands (B).
Case 2
A 43-year-old man presented with brown papules on his neck, breast and forearms, which had existed for ten years when he began topical adapalene treatment. The papules exacerbated during summer, and they were itchy. His father had similar symptoms. A skin biopsy specimen from the lesion showed acantholysis and abnormal keratinization (Fig. 2). Keratotic tiers and suprabasal cleft formation were also seen.
Fig. 2.
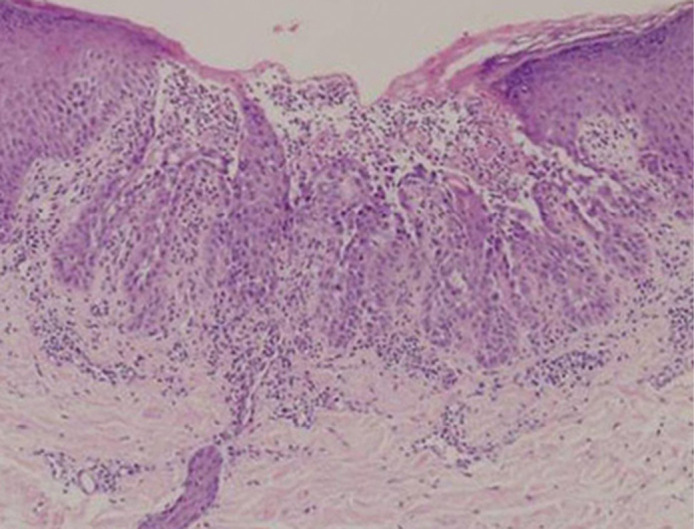
Histopathological features of the skin lesions of DD. A skin biopsy specimen from Case 2 shows typical histopathological features of DD, marked hyperkeratosis, acantholysis, and clefts and dyskeratosis in the suprabasal layers of epidermis. Haematoxylin-eosin stain, original magnification X400.
Case 3
A 40-year-old woman presented with intensely pruritic reddish follicular papules. She had neither family history nor other serious complications. The papules had first appeared less than 1 year previously on the back and the breast, and had exacerbated in the first month after onset. Slight hyperkeratosis was seen on the wrist, and erythema appeared on the hand. One follicular papule was excised and submitted for histopathological examination. High-power microscopy showed suprabasal acantholysis and well-developed corps ronds and grains. We reported this patient elsewhere in 2015.7)
Case 4
A 66-year-old Japanese man presented with multiple brown papules with crusts on the seborrheic areas of the head and face, and on the neck, abdomen, back and legs. Partially exudative hyperkeratotic lesions with odor were seen on the forehead, the nasal wings and the intertriginous areas. He had been diagnosed with schizophrenia and intellectual disability at the age of 63. Light microscopy of a lesional skin biopsy from his neck revealed hyperkeratosis, acantholysis, clefts, grains and corps ronds in the epidermis. This case was reported in 2016 as “DD complicated with schizophrenia”.8)
Case 5
An 84-year-old man had been suffering from DD for 10 years. The age of onset was notably late. He presented with multiple brown papules with crusts, especially on his trunk. His nails showed whitish striae and wedged recesses (so-called “notches”). The clinical features of this case were previously reported in Japanese.9)
Evaluation of disease severity of DD
The severity of DD for each patient was classified according to the methods described in the previous report.1) Briefly, patients with only grouped keratotic papules in the disease predilection areas were classified as having the mild phenotype. Cases showing widespread papules or localized verrucous plaques were judged to have moderate disease severity. Extensive, hyperkeratotic and macerated plaques and/or hypertrophic growth in the intertriginous areas were thought to be signs of the severe phenotype.
Sanger sequencing and whole exome sequencing
Ethical approval was obtained and all research was performed in accordance with the Declaration of Helsinki principles. Genomic DNA from the patient’s peripheral blood leukocytes was used for Sanger sequencing analysis and whole exome sequencing analysis, as described previously.10, 11) Briefly, for whole exome sequencing, genomic DNA from the proband was enriched for protein-coding sequences with the SureSelect Human All Exon V5 kit (Agilent Technologies, Santa Clara, CA, USA). Massively parallel sequencing was performed with the HiSeq 2500 platform with 100 bp paired-end reads (Illumina, San Diego, CA, USA). Candidate germline variants were identified through our in-house pipeline for exome-sequencing analysis with minor modifications. The obtained sequences were aligned with the UCSC Genome Browser hg19 by using the Burrows-Wheeler Aligner.
RESULTS
Mutation detection
ATP2A2 mutations detected in the present DD cases and their consequences are summarized in Table 1.
Case 1
Whole exome sequencing analysis revealed the previously unreported deletion mutation c.2255_2257del. DNA samples from the peripheral blood of the patient and her parents and sister were analyzed by Sanger sequencing to confirm the mutation. This revealed that the patient was heterozygous for the deletion mutation (Fig. 3). This deletion results in the deletion/insertion p.Ile752_Tyr753delinsAsn. Her father and sister were also heterogeneous for the identical mutation in ATP2A2, although her mother had only wild-type alleles.
Fig. 3.
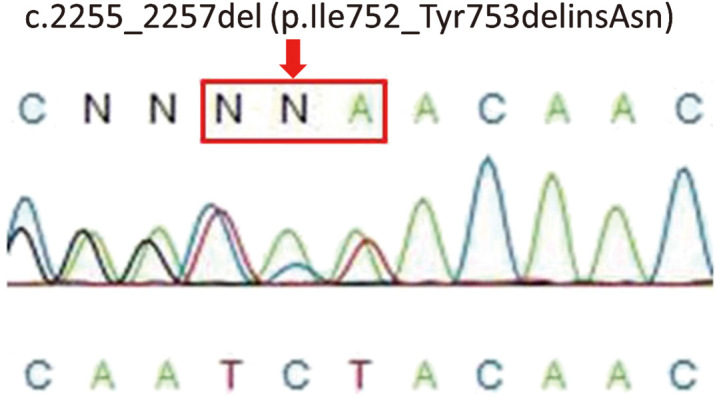
Sanger sequencing of ATP2A2 in Case 1. Direct sequencing reveals a heterozygous mutation within ATP2A2, c.2255_2257del (p.Ile752_Tyr753delinsAsn) in the patient.
Case 2
Sanger sequencing of the entire coding regions of ATP2A2 revealed the missense mutation c.548A>T (p.Glu183Val), which was previously reported in a Japanese patient with DD.12) Glu183 is in the A domain, which works as a lever when calcium ions go through SERCA2.
Case 3
Sanger sequencing of the entire coding regions of ATP2A2 revealed the missense mutation c.2721A>C (p.Glu907Asp). The mutation analysis in this patient was previously reported elsewhere.7) This mutation resulted in an alteration of a calcium-binding residue.
Case 4
By Sanger sequencing, the missense mutation c.953G>A (p.Cys318Tyr) was found in the patient’s ATP2A2.8) The mutation changes an amino acid in the stalk 4 domain of the molecule. The stalk domain connects the P domain and TM4.
Case 5
Sanger sequencing of the entire coding regions of ATP2A2 revealed the nonsense mutation c391C>T (p.Arg131*).13) This nonsense mutation is located in the A domain, one of the three major domains on the cytoplasmic surface of SERCA2. The mutation site is close to the N-terminus of SERCA2, and p.Arg131* results in a serious truncation of the SERCA2 peptide, causing SERCA2 to lose most of its structure, including the cytoplasmic P domain and the N domain. In addition, there is a possibility that nonsense-mediated mRNA decay might lead to a loss of mutant SERCA2 products in the present patient.
In silico relevance analysis of the novel mutation in Case 1
The novel deletion/insertion mutation p.Ile752_Tyr753delinsAsn was not found in the Human Genetic Variation Database, including in 1,208 exome datasets from normal controls (http//www.genome.med.kyoto-u.ac.jp/SnpDB/index.html). The mutation was analyzed using PROVEAN (http://provean.jcvi.org/). The PROVEAN program predicts that the present mutation p.Ile752_Tyr753delinsAsn has “deleterious” effects on the SERCA2 structure/function.
Evaluation of skin phenotype severity in the DD patients
The severity of skin symptoms in the present DD patients is summarized in Table 1. Three of the five patients had moderate skin symptoms, and the other two showed the mild skin phenotype.
DISCUSSION
Light microscopy of a skin biopsy reveals hyperkeratosis, acantholysis, cleft, grains and corps ronds in the epidermis in DD patients. Such morphological features indicate cell-cell adhesion abnormality and keratinocyte differentiation abnormality in DD. In fact, acantholytic cells show aberrant distribution of desmosomal components.6) Thus, pathogenic defects of keratinocyte structure and adhesion have been proposed as the cause of DD.
SERCA2b has 11 TMs. There are 10 CBRs in TMs in the transmembrane region.4) SERCA2b has three major cytoplasmic globular domains, A domain, P domain and N domain. The three cytoplasmic globular domains play an important role when calcium ions bind and change their positions.
Genotype/phenotype correlations between ATP2A2 mutations and DD phenotypes are still unknown. We found five distinct, heterozygous mutations in five patients from five independent families. The mutations consisted of three missense, one nonsense and one deletion mutations (Table 1. Four mutations out of five were previously reported7, 8, 12, 13) and the other one has not been reported previously. The mutations and their consequences are summarized in Table 1.
In Case 1, we detected a previously unreported three-base deletion mutation c.2255_2257del (p.Ile752_Tyr753delinsAsn) in ATP2A2. The amino acids altered by the mutation localize in a stalk domain behind N domain and the hinge domain. Other three-base deletion mutations in ATP2A2 were reported previously in the literature.1, 14) In the present study, the PROVEAN program predicts that the deletion mutation is “deleterious”. The PROVEAN (protein variation effect analyzer) is a software which forecasts whether a missense or an indel mutation affects the function of a protein.15) The PROVEAN is a powerful tool to screen out functionally significant nonsynonymous or indel variants.15) In Case 1, the novel mutation c.2255_2257del (p.Ile752_Tyr753delinsAsn) in exon 15 affects both isoforms, SERCA2a and SERCA2b. SERCA2a is known to be expressed favorably in skeletal and cardiac muscles. However, the patient had no apparent symptom other than the skin manifestations.
Case 3 had the missense mutation of one calcium binding residue which is important for the function of SERCA2. Case 5 had the serious truncation mutation, losing most part of the SERCA2 peptide. Thus, both in Case 3 and Case 5, the mutations are thought to have serious effects on the SERCA2 function. However, DD skin symptoms of both patients were mild. Especially, Case 5 had had no apparent DD phenotype until over 70 years of age, indicating that DD skin manifestations were mild in this patient.
In Case 4, we detected the missense mutation p.Cys318Tyr in a stalk domain which is thought to be not so important for the SERCA2 function. However, the skin symptoms were moderate in Case 4 and, in addition, the patient had schizophrenia, which might be a complication of DD. In a recent Swedish cohort study, 6 of 770 DD patients (0.8%) were found to have schizophrenia.16) It was determined that individuals with DD had a 2.3 times higher risk of being diagnosed with schizophrenia than matched individuals from the general population.
Ahn et al. reported that several mutants which cause severe phenotypes of DD inhibited the activity of the endogeneous and the co-expressed wild type SERCA2b in vivo.17) If this is the case, it is difficult to speculate the total SERCA2 activity in the patients’ keratinocytes, simply from the nature and sites of the ATP2A2 mutations. In the present DD cases, there was no significant genotype/phenotype correlation between the ATP2A2 mutation sites and nature and the severity of DD phenotypes.
ACKNOWLEDGMENTS
The authors thank Ms. Haruka Ozeki and Ms. Yuka Terashita for their technical help in analyzing mutations of ATP2A2. This study was supported by JSPS KAKENHI Grant Numbers 15H04887, 15K15415, 15H04886, 15K15414 and 15H06280. The work was also supported by the Japan Intractable Diseases Research Foundation.
CONFLICT OF INTEREST
The authors declare that there is no duality of interest.
REFERENCES
- 1).Ringpfeil F, Raus A, DiGiovanna JJ, Korge B, Harth W, Mazzanti C, et al Darier disease-novel mutations in ATp2A2 and genotype-phenotype orrelation. Exp Dermatol, 2001; 10: 19–27. [DOI] [PubMed]
- 2).Burge SM, Wilkinson J D. Darier–White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol, 1992:27:40–50. [DOI] [PubMed]
- 3).Ruiz-Perez VL, Carter SA, Healy E, Todd C, Rees JL, Steijlen PM, et al ATP2A2 mutations in Darier’s disease: variant cutaneous phenotypes are associated with missense mutations, but neuropsychiatric features are independent of mutation class. Hum Mol Genet, 1999; 8: 1621–1630. [DOI] [PubMed]
- 4).ClausenD,Vandecaetsbeek I, Wuytack F, Vangheluwe P, Andersen J. Distinct roles of the C-terminal 11th transmembrane helix and luminal extension in the partial reactions determining the high Ca affinity of Sarco (endo) plasmic reticulum Ca-ATPase isoform 2b (SERCA2b). J Biol Chem, 2012; 287: 39460–39469. [DOI] [PMC free article] [PubMed]
- 5).Toyoshima C, Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature, 2002; 18: 605–611. [DOI] [PubMed]
- 6).Burge SM, Garrod DR. An immunohistological study of desmosomes in Darier’s disease and Hailey-Hailey disease. Br J Dermatol, 1991; 124: 242–251. [DOI] [PubMed]
- 7).Kaibuchi-Noda K, Sugiura K, Takeichi T, Miura S, Kagami S, Takama H, et al. Darier’s disease: a novel ATP2A2 missense mutation at one of the calcium-binding residues. Acta Derm Venereol, 2015; 95: 362–363. [DOI] [PubMed]
- 8).Takeichi T, Sugiura K, Nakamura Y, Fujio Y, Konohana I, Akiyama M. Darier’s disease complicated with schizophrenia caused by a novel ATP2A2 mutation. Acta Derm Venereol, 2016; doi: 10.2340/00015555-2422. [DOI] [PubMed]
- 9).Otani A, Suzuki T, Hukuda E, Takeshige Y, Mukai H, Takeichi T, et al The case of late-onset Darier Disease. Rinsho Derma (Tokyo), 2013; 55: 1087–1090 (in Japanese).
- 10).Takeichi T, Nanda A, Liu L, Salam A, Campbell P, Fong K, et al Impact of next generation sequencing on diagnostics in a genetic skin disease clinic. Exp Dematol, 2013; 22: 825–831. [DOI] [PubMed]
- 11).Kunishima S, Okuno Y, Yoshida K, Shiraishi Y, Sanada M, Muramatsu H, et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet, 2013; 92: 431–438. [DOI] [PMC free article] [PubMed]
- 12).Ikeda S, Mayuzumi N, Ogawa H. Mutations in ATP2A2 in patients with Darier’s disease. J Invest Dematol, 2003; 121: 475–477. [DOI] [PubMed]
- 13).Amichai B, Karpati M, Goldman B, and Peleg L. Novel mutations in two families with Darier’s disease. Int J Dermatol, 2007; 46: 64–67. [DOI] [PubMed]
- 14).Tsuruta D, Akiyama M, Ishida-Yamamoto A, Imanishi H, Mizuno N, Sowa J, et al. Three-base deletion mutation c,120-122del GTT in ATP2A2 leads to the unique phenotype of comedonal Darier’s disease. Br J Dermatol, 2010; 162: 687–689. [DOI] [PubMed]
- 15).Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 2015; 31: 2745–2747. [DOI] [PMC free article] [PubMed]
- 16).Cederlof M, Bergen SE, Langstrom N, Larsson H, Boman M, Craddock N, et al The association between Darier disease, bipolar disorder, and schizophrenia revisited: a population-based family study. Bipolar Disord, 2015; 17: 340–344. [DOI] [PubMed]
- 17).Ahn W, Lee MG, Kim KH, Muallem S. Multiple effects of SERCA2b mutations associatiated with Darier’s disease. J Biol Chem, 2003; 278: 20795–20801. [DOI] [PubMed]