

Abstract
Aldehyde dehydrogenase typically performs oxidation of aldehydes to their corresponding carboxylic acid while reducing NAD(P)+ to NAD(P)H via covalent catalysis mediated by an active-site cysteine residue. One member of this superfamily, the enzyme 2-aminomuconate-6-semialdehyde dehydrogenase (AMSDH), is a component of the kynurenine pathway, which catabolizes tryptophan in mammals and certain bacteria. AMSDH catalyzes the NAD+-dependent oxidation of 2-aminomuconate semialdehyde to 2-aminomuconate. We recently determined the first crystal structure of AMSDH and several catalytic cycle intermediates. A conserved asparagine in the oxyanion hole, Asn-169, is found to be H-bonded to substrate-derived intermediates in the active site of AMSDH during catalysis, including both the covalently bound thiohemiacetal and thioacyl intermediates. To better interrogate the significance of the hydrogen bond provided by Asn-169 to the reaction mechanism of AMSDH, we created Ala, Ser, Asp, and Gln mutants and studied them using biochemical, kinetic, crystallographic, and computational studies. The in crystallo chemical reaction of the primary substrate with the co-crystalized complex of the N169D mutant and NAD+ led to the successful trapping of a new catalytic intermediate that was not previously seen. The structural and computational data are consistent with a substrate imine/enol tautomer intermediate being formed prior to the formation of the covalent bond between the substrate and the active-site cysteine. Thus, AMSDH surprisingly includes an isomerization process within its known catalytic mechanism. These data establish a hidden intrinsic isomerization activity of the dehydrogenase and allow us to propose a pitcher-catcher type of catalytic mechanism for the isomerization.
Keywords: biodegradation, crystal structure, decarboxylase, dehydrogenase, enzyme mechanism, enzyme structure, tryptophan, bifunctional enzyme, isomerization, metabolic pathway, kynurenine
Introduction
The kynurenine pathway is the catabolic route for tryptophan degradation in mammals and certain bacteria. In mammals, the pathway has been found to produce neuroactive compounds that correlate with depression and neurodegenerative disease states such as Alzheimer's, Parkinson's, and Huntington's diseases (1–3). Moreover, the kynurenine pathway is a de novo biosynthetic route to produce the coenzyme NAD+/NADH, which is involved in many fundamental biological processes as an energy carrier and redox mediator. In the kynurenine pathway, tryptophan metabolites are partitioned by both enzymatic and non-enzymatic reactions (4). Three consecutive enzymes of the pathway, 3-hydroxyanthranilate dioxygenase (HAO),3 2-amino-3-carboxymuconate-6-semi-aldehyde decarboxylase (ACMSD), and 2-amino-muconate-6-semialdehyde dehydrogenase (AMSDH), compete with the non-enzymatic auto-cyclization of their substrates and products for further metabolism (Fig. 1). The trio of enzymes is also present in the 2-nitrobenzoic acid biodegradation pathway.
FIGURE 1.
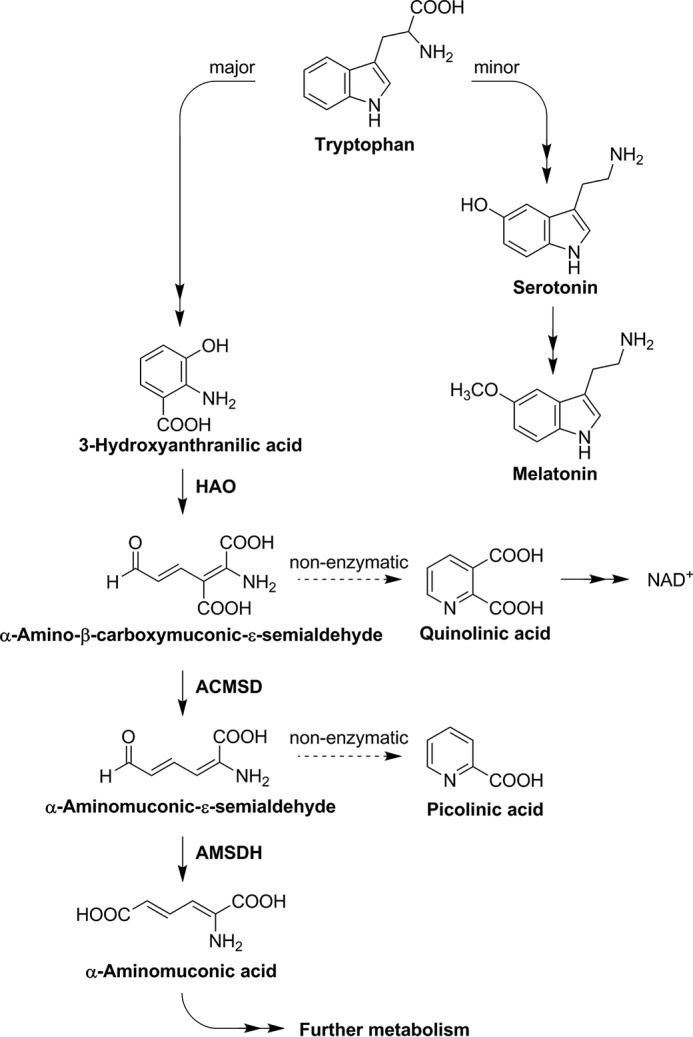
Tryptophan catabolic pathways.
Investigations at the molecular level of the kynurenine pathway were extended to AMSDH in our recent work (5). AMSDH is a 216-kDa homotetrameric protein (500 amino acid residues in each subunit) that belongs to the aldehyde dehydrogenase superfamily. It competes with a spontaneous, non-enzymatic cyclization of 2-aminomuconate semialdehyde (2-AMS) to prevent overproduction of picolinic acid. The off-pathway product, picolinic acid, is a metal chelator in human milk that is barely detectable in blood serum and below the detection limit in other tissues (6). AMSDH oxidizes 2-AMS to 2-aminomuconate and directs the metabolic flux to enzyme-controlled reactions. We have shown the anticipated enzymatic activity of AMSDH using isolated protein and determined its first crystal structure (5). Furthermore, the binary and ternary complexes as well as two catalytic intermediates, thiohemiacetal and thioacyl, were characterized by soaking single crystals of the binary enzyme-NAD+ complex under varied time periods with substrates, widening our knowledge of the catalytic mechanism of this semialdehyde dehydrogenase.
In the active site of AMSDH, as in ACMSD (7), two arginine residues appear to stabilize the carboxyl group of their respective substrates (5). In AMSDH, Cys-302 and Glu-268 are reported to have critical roles in the reaction catalyzed by AMSDH (5). Cys-302 serves as a catalytic nucleophile and Glu-268 acts as a general base based on our recent findings and literature reports of enzymes in the same family (5, 8, 9). In addition to their catalytic nucleophile and general base, members of the aldehyde dehydrogenase superfamily also possess a strictly conserved asparagine in their active site (supplemental Fig. S1), i.e. residue 169 in AMSDH. Such an active-site asparagine residue is hypothesized to provide catalytic contribution as an oxyanion hole residue in other aldehyde dehydrogenases (10, 11). Substitution of the asparagine by alanine eliminated the dehydrogenase activity (12, 13). However, the precise role of the asparagine residue remains unexplored in any member of the aldehyde dehydrogenase superfamily, including AMSDH.
Asn-169 is within H-bonding distance with the substrate-derived intermediates of the catalytic cycle (5). As shown in the thioacyl intermediate structure of AMSDH (Protein Data Bank (PDB) entry: 4NPI), the amide moiety of Asn-169 forms a hydrogen bond with the oxo moiety of the thioacyl, substrate-enzyme adduct. Thus, Asn-169 is expected to stabilize the bound substrate, 2-AMS, as well as to stabilize the thiohemiacetal and thioacyl intermediates through hydrogen-bonding interactions during the oxidation of 2-AMS by AMSDH.
In this work, we constructed and expressed several mutants to illuminate the precise role of Asn-169 in the reaction catalyzed by AMSDH. Kinetic analysis and crystallography were employed to study those mutants. Unexpectedly, we captured an isomerization reaction intermediate in addition to the previously trapped intermediates in the native protein.
Results
Steady-state Kinetics
Asn-169 was mutated to alanine, glutamine, aspartate, and serine, respectively, to explore the function of the active-site asparagine residue. The steady-state kinetic parameters were determined for each of the active mutants. As compared with the kinetic parameters of the wild-type enzyme (wtAMSDH), mutants of N169S, N169Q, and N169D have similar Km but a much lower kcat. The observation of a greater than 100-fold reduction in reaction rate with a less than 2-fold change in the Km indicates that before the first irreversible step of the reaction, Asn-169 is not heavily involved in catalysis. Therefore, the hydrogen-bonding interaction between Asn-169 and the C6 oxygen of the alternate substrate, 2-hydroxymuconate-6-semialdehyde (2-HMS) (5), appears to be essential to the rate-limiting steps of the reaction. Substitution of Asn-169 with alanine creates a mutant protein that does not contain any side-chain functional group capable of forming H-bonds, eliminating a possible stabilizing interaction at this position. As indicated in Table 1, there is no detectable steady-state activity with the N169A mutant.
TABLE 1.
Kinetic assays of N169 mutants with 2-HMS at pH 7.5
ND, not detected.
| Km | kcat | kcat/Km | |
|---|---|---|---|
| μm | s−1 | m−1 s−1 | |
| Native | 10.4 ± 0.2 | 1.30 ± 0.01 | 1.25 × 105 |
| N169Q | 5.7 ± 0.7 | 0.0034 ± 0.0001 | 5.9 × 102 |
| N169S | 8.7 ± 0.8 | 0.0077 ± 0.0003 | 8.8 × 102 |
| N169D | 7.7 ± 0.6 | 0.0110 ± 0.0004 | 1.4 × 103 |
| N169A | ND | ND | ND |
In the thioacyl intermediate of wtAMSDH, the amide side chain of Asp-169 forms an H-bond with the C6 oxo of the 2-HMS-enzyme adduct (2.8 Å distance). In this arrangement, the -NH group of Asn-169 is expected to be the H-bond donor, and the adduct oxo group is expected to be the H-bond acceptor. With this understanding, the N169D mutant should only be able to donate a hydrogen for the formation of an intermediate-stabilizing H-bond at lower pH values. To test this hypothesis, pH profiles were obtained for N169D using wtAMSDH as the control. As shown in Fig. 2, the kcat value of N169D increases with decreasing pH such that at pH 4.5, the kcat value is an order of magnitude larger than at pH 8.0. Meanwhile, the Km values present less change with no consistent trend. By comparison, the kcat value of wtAMSDH actually decreases with decreasing pH, opposite to what was observed with N169D, but their Km profiles are similar. The increase in the catalytic rate of N169D with decreasing pH is consistent with the hypothesis that Asn-169 acts to stabilize intermediates and transition states by donating a hydrogen for H-bonding interactions to the C6 oxygen of the substrate. At lower pH conditions, Asp-169 becomes increasingly protonated, enhancing its ability to function as an H-bond donor, fulfilling the same role as Asn-169 in the wild-type enzyme.
FIGURE 2.
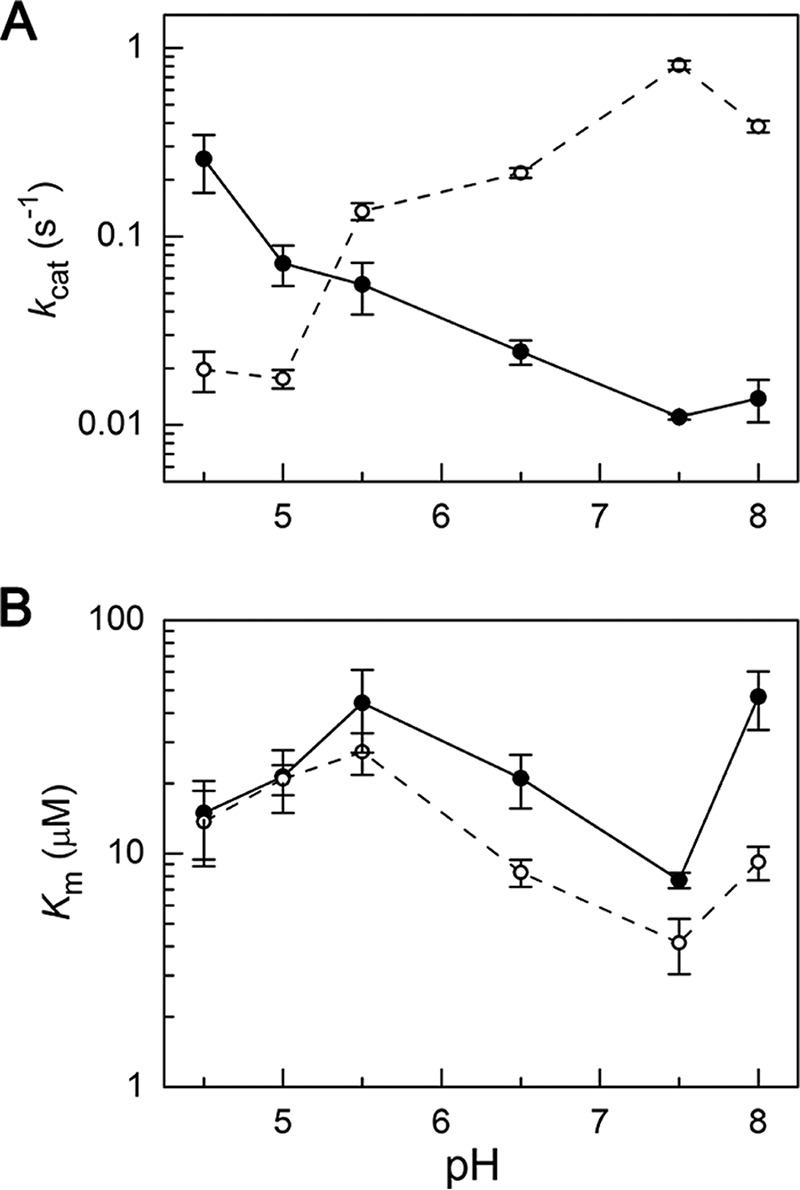
pH profile of Michaelis-Menten parameters of wild-type AMSDH (dashed line) and N169D mutant (solid line). A, kcat and B, Km values for N169D and wtAMSDH at varying pH values. Error bars indicate means ± S.E.
Crystal Structure of N169A and a Thioacyl Intermediate
To rule out aberrant structural changes as the cause for a lack of steady-state activity in the N169A mutant, the mutant protein was crystallized, and its structure was determined by X-ray diffraction and refined to 1.99 Å resolution. The NAD+-bound N169A mutant structure is a homotetramer and agrees well with that of the binary complex structure of wtAMSDH (Fig. 3A), with a root mean square deviation (RMSD) of 0.197 Å (PDB entry: 5KJ5).
FIGURE 3.
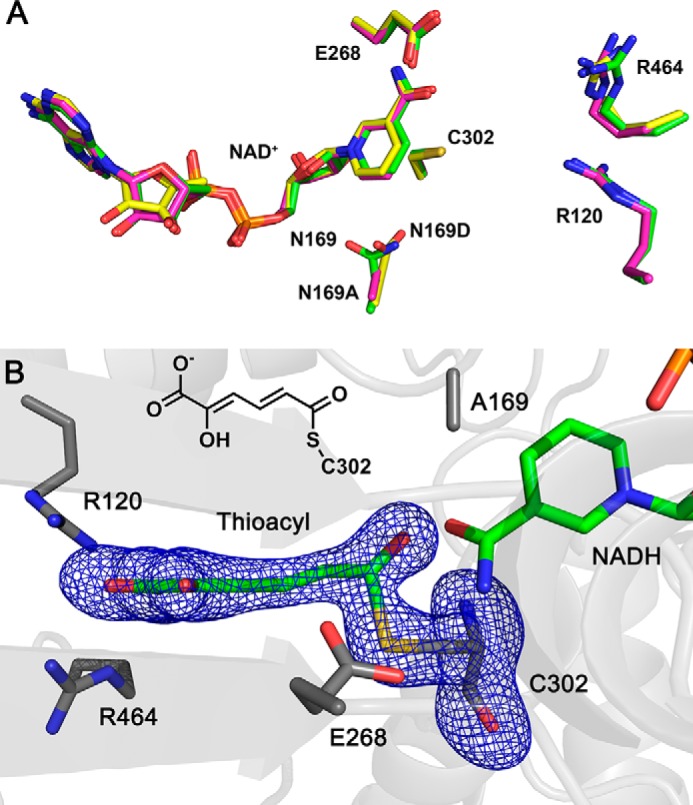
Mutation of Asn-169 does not affect the active-site geometry of AMSDH binary complex or thioacyl intermediate. A, superimposition of N169A (magenta, 5KLN) and N169D (yellow, 5KJ5) active site with wtAMSDH (green, 4I1W) in the binary complex form. B, electron density map of the thioacyl intermediate obtained from N169A-NAD+ crystal reacted with 2-HMS for 20 h. Cys-302, Glu-268, Arg-464, Arg-120, Asn/Asp-169, and NAD+ are present as sticks. The omit maps for ligands and Cys-302 are contoured to 3.0 σ and shown as a blue mesh.
Crystals of N169A were then soaked with the stable alternate substrate 2-HMS for 5 min to 20 h. Of all the datasets collected, those with shorter soaking times (less than 20 h) yielded diffraction maps with poor density for substrate or NAD+, whereas the ones with the longest soaking times (20 h) show clear and continuous electron density for 2-HMS- and NAD+-derived intermediate and product in the active site (Fig. 3B).
The N169A intermediate structure from the 20-h in crystallo reaction was refined to 1.79 Å resolution. As compared with the NAD+-bound N169A structure, Glu-268 rotates more than 70° from its “passive” to its “active” conformation upon formation of the thioacyl intermediate to activate a water molecule for hydrolysis, and the nicotinamide head of NAD+ moves 6 Å away from the active site as it has been reduced to NADH as seen in the crystal structure of wtAMSDH (5). Therefore, the crystal structure shows clear evidence of the in crystallo formation of a substrate-N169A adduct corresponding to a thioacyl intermediate (Fig. 3B). However, under the same conditions, this intermediate was formed by wtAMSDH in 30–40 min. The lack of steady-state activity measured for this mutant may be accounted for by a combination of inefficient formation of the newly observed substrate-derived intermediate and an inability to prepare the thioacyl adduct for hydrolysis through the formation of a second tetrahedral intermediate.
Time-lapse in Crystallo Reaction of N169D
The NAD+-bound N169D binary complex structure was determined and refined to 2.11 Å resolution. The mutant structure exhibits high similarity with the wtAMSDH binary complex with an RMSD of 0.173 Å (Fig. 3A). Moreover, to ascertain whether there is any further information about the influence of the Asn-to-Asp alteration, the crystals of the N169D-NAD+ mutant were mixed with 2-HMS for varying times. As a result, 11 crystal structures were solved with reaction times of 1 min to 23 h.
Among all structures solved, most of the structures showed incomplete or low occupancies of a specific reaction intermediate at the active site. However, three distinct reactive intermediates were captured with reaction times of 1, 5, and 30 min. These intermediates were reproducible, and their structures were refined to resolutions of 2.01, 2.17, and 2.10 Å, respectively (Table 2). The N169D ternary complex with 1-min reaction time (PDB entry: 5KLK) dataset exhibits the same tertiary and quaternary structural characteristics as compared with the wtAMSDH ternary complex structure. 2-HMS is H-bonded with Arg-120 and Arg-464 as its keto tautomer form, and the enol end is pointed to the opposite direction toward Cys-302 (supplemental Fig. S2A). The N169D ternary complex with 5-min reaction time also has the same overall structural features. Interestingly, significant differences in the details of the active site of the N169D ternary complex with 5-min reaction time are found as compared with the wtAMSDH enzyme, where a thioacyl intermediate was observed (5).
TABLE 2.
Crystallization data collection and refinement statistics
| NAD+-N169D | 2-HMS-NAD+-N169D (1 min) | 2-HMS-NAD+-N169D (5 min) | 2-HMS-NAD+-N169D (30 min) | NAD+-N169A | N169A-thioacyl intermediate (20 h) | |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions a, b, c (Å) | 88.6, 142.4, 173.4 | 88.2, 142.9, 174.2 | 88.4, 143.1, 174.1 | 88.4, 142.0, 171.6 | 88.7, 141.0, 173.3 | 88.1, 141.7, 171.5 |
| Resolution | 31.5 − 2.11 (2.19 − 2.11)a | 35.1 − 2.01 (2.08 − 2.01) | 35.2 − 2.17 (2.25 − 2.17) | 45.6 − 2.10 (2.18 − 2.10) | 34.4 − 1.99 (2.06 − 1.99) | 34.9 − 1.79 (1.85 − 1.79) |
| No. of observed reflections | 125557 (12419) | 145427 (14114) | 115719 (11331) | 124700 (12305) | 148478 (14680) | 193366 (17447) |
| Redundancy | 14.2 (14.1) | 12.0 (11.4) | 14.3 (14.0) | 7.9 (8.0) | 12.1 (9.7) | 11.7 (11.8) |
| Completeness (%) | 99.8 (100.0) | 98.7 (96.9) | 99.6 (98.9) | 99.8 (100.0) | 99.8 (99.6) | 95.3 (86.8) |
| I/σ(I) | 27.8 (2.9) | 23.5 (2.0) | 26.5 (3.5) | 20.2 (1.8) | 29.3 (2.9) | 34.6 (2.7) |
| Rmerge (%)b | 11.6 (92.5) | 11.6 (89.6) | 13.1 (92.9) | 11.7 (96.2) | 10.1 (69.2) | 7.6 (81.3) |
| Refinementc | ||||||
| Rwork | 19.0 | 18.8 | 17.0 | 18.6 | 18.7 | 17.9 |
| Rfree | 23.7 | 23.1 | 21.8 | 23.0 | 22.2 | 20.3 |
| RMSD bond length (Å)d | 0.008 | 0.007 | 0.008 | 0.007 | 0.008 | 0.007 |
| RMSD bond angles (°) | 1.10 | 1.12 | 1.07 | 1.10 | 1.14 | 1.11 |
| Ramachandran statisticse | ||||||
| Preferred (%) | 97.2 | 97.1 | 97.1 | 97.2 | 97.2 | 97.7 |
| Allowed (%) | 2.6 | 2.8 | 2.7 | 2.6 | 2.7 | 2.1 |
| Outliers (%) | 0.2 | 0.1 | 0.3 | 0.2 | 0.2 | 0.2 |
| All-atoms Clash score/Percentilee | 3.64/99 | 2.78/99 | 2.46/100 | 3.17/99 | 3.50/99 | 2.68/99 |
| MolProbity score/percentilee | 1.72/94 | 1.56/95 | 1.56/98 | 1.60/96 | 1.76/88 | 1.51/93 |
| Average B-factor (Å2) | ||||||
| Protein/atoms | 44.6/14691 | 39.0/14705 | 36.1/14726 | 45.8/14685 | 37.6/14701 | 33.2/14679 |
| NAD+/atoms | 47.6/176 | 42.9/176 | N/A | 51.4/176 | 34.0/176 | 39.5/176 |
| Na+/atoms | N/A | 51.6/3 | 35.7/4 | 48.5/4 | 37.7/4 | 32.2/4 |
| 2-HMS or intermediates/atoms | N/A | 51.8/30 | 38.2/40 | N/A | N/A | 33.7/40 |
| Solvent/atoms | 48.3/843 | 44.0/1092 | 41.9/1179 | 51.3/864 | 46.2/1179 | 42.0/1475 |
| PDB code | 5KJ5 | 5KLK | 5KLL | 5KLM | 5KLN | 5KLO |
a Values in parentheses are for the highest resolution shell.
b Rmerge = ΣhklΣi |Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl), in which the sum is over all the i measured reflections with equivalent Miller indices hkl; 〈I(hkl)〉 is the averaged intensity of these i reflections, and the grand sum is over all measured reflections in the data set.
c All positive reflections were used in the refinement.
d According to Engh and Huber (32).
e Calculated by using MolProbity (33).
As expected, 2-HMS is bound to the enzyme active site by two arginine residues in the active site of N169D in the same manner as seen in the wtAMSDH ternary complex structure (PDB entry: 4I2R). After soaking the co-crystalized NAD+-N169D with 2-HMS for 5 min (PDB entry: 5KLL), 2-HMS adopts an extended conformation reminiscent of the previously captured thioacyl intermediate, but the electron density of the nicotinamide head of NAD+ is not well defined. At longer soaking times (30 min, PDB entry: 5KLM), the electron density of the aldehyde portion of 2-HMS is not well defined, presumably due to increased conformational heterogeneity. The position of Glu-268 and the nicotinamide head of NAD+, however, are both well defined and indicate that even after 30 min of soaking, hydride transfer from the substrate to NAD+ has not yet occurred (supplemental Fig. S2B).
Crystallographic Capture of a New Tautomerized Intermediate
In the 5-min N169D intermediate structure, Glu-268 surprisingly remains in its passive state (i.e. pointing away from the substrate-binding pocket). In all previous intermediate structures (5) obtained with the same in crystallo reaction procedure from wild-type AMSDH, Glu-268 is in an active position, rotated 73° toward the bound substrate from its resting passive position. The electron density of the nicotinamide moiety of NAD+ is not well defined. Careful inspection of the electron density of 2-HMS in the N169D active site reveals that the carbon backbone of the substrate is distorted as compared with all previous structures (5). Specifically, 2-HMS shows significant out-of-plane rotation about its C3–C4–C5–C6 dihedral. Another key distinction between the 5-min N169D intermediate and previously solved structures is the interatomic distances around the aldehydic carbon of the substrate and nearby residues. The two values of interest are the distances between C6 of 2-HMS and the sulfur of Cys-302 and between the oxo of 2-HMS and the terminal atom of Asn/Asp-169, respectively.
In the N169D structure, the thiol moiety of Cys-302 has two alternative conformations. In the first conformation, the thiol is pointed toward the nicotinamide ring of NAD+, as seen previously in the wtAMSDH structure (PDB entry: 4I26) (5). In the other confirmation, the thiol is in position for nucleophilic attack on the substrate, similar to the scenario found in the ternary complex structures of the wtAMSDH (5). In wtAMSDH,the C6-sulfur distance between the substrate and Cys-302 is 1.8 Å, indicating a formal covalent bond, whereas in the N169D mutant, the shortest distance is 2.4 Å (Fig. 4A), at the shorter end of hydrogen-bonding range and too long to indicate a formal covalent carbon-sulfur bond. The other conformation of Cys-302 is farther away from the substrate, 4.4 Å from C6 of the substrate. Thus, the substrate-derived intermediate is not covalently bound to the enzyme in this intermediate.
FIGURE 4.

Snapshot of a new tautomerized intermediate. A, the stereographic view of omit map of a substrate-based intermediate in the co-crystalized crystals of N169D-NAD+ soaked with 2-HMS for 5 min. The ligand density is fit with the enol tautomer of 2-HMS. B, this is compared with the thioacyl intermediate trapped in the native enzyme (PDB entry: 4NPI) under identical conditions. The omit maps of intermediates in the active site are contoured to 3.0 σ and shown as a blue mesh. The active-site residues interacting with the intermediate are included in the presentation. C, the isomerization reaction in AMSDH N169D. D, optimized geometry of 2-AMS in its imine/enol tautomer with Cys-302 and Asp-169. Carbon, hydrogen, oxygen, nitrogen, and sulfur atoms are shown in gray, white, red, blue, and yellow, respectively.
Additionally, the substrate oxo-Asp distance in the N169D mutant is 2.2 Å (Fig. 4A), whereas the substrate oxo-Asn distance in the wild-type thioacyl intermediate is 2.8 Å (Fig. 4B). The finding of what appears to be a very strong H-bond between the C6 oxygen of 2-HMS and the carboxylate moiety of Asp-169 is unexpected, as the crystallization conditions (pH 7.9) should ensure that the side chain of Asp-169 is deprotonated, and an aldehydic oxygen is expected to carry a significant partial negative charge. With such a close observed distance, either Asp-169 is protonated at pH 7.9, or 2-HMS can act as the donor in the formation of this hydrogen bond. The positions of NAD+ and Glu-268 in the structure of the 5-min reaction intermediate of N169D are also consistent with the structure of a later (30-min reaction time) structure (PDB entry: 5KLM) as shown in supplemental Fig. S2, which has increased conformational heterogeneity for the bound substrate.
Taken together, the details of the N169D active site after reacting with 2-HMS for 5 min point to the capture of a keto/enol intermediate, which corresponds to an imine/enol intermediate, 1, in the AMSDH mechanism (Fig. 4C). The findings of an extended substrate conformation in the active site with a C6-sulfur distance outside the covalent bond range, a glutamate in the passive position, an unreduced NAD+, and a very short substrate oxo-Asp distance all suggest that N169D stabilizes an enol tautomer of the substrate that was not previously seen but was anticipated to facilitate isomerization prior to the dehydrogenation reaction.
Quantum Chemical Investigation of a Crystallographically Captured Intermediate
The crystal structure of the 2-HMS-derived intermediate structure of N169D is distinct from all previous structures of this enzyme or its mutants (5). The straightforward explanation for the disparities between the N169D intermediate and the thiohemiacetal or thioacyl structures is that the introduction of a negative charge to the active site in the mutant protein leads to the preferential stabilization of the imine/enol tautomer. Such an intermediate is necessary for the isomerization of the substrate before the nucleophilic attack by Cys-302. With such an isomerization reaction, the next dehydrogenase step is less sterically hindered (Fig. 5).
FIGURE 5.
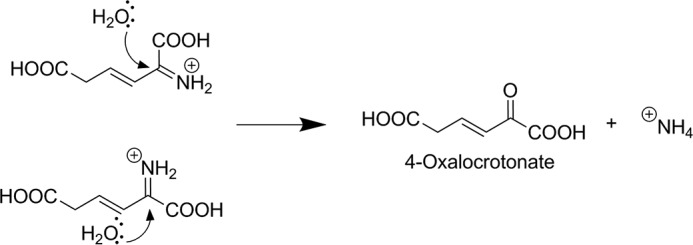
Backside attack on the immonium ion of 2-AM is less sterically hindered after isomerization. Shown is the expected chemical step for the enzyme downstream of AMSDH if there had been no isomerization (top left), and with isomerization (bottom left).
Density functional theory calculations were carried out to assess the feasibility of this explanation. The starting models were generated from the 5-min 2-HMS-NAD+-derived intermediate of N169D crystal structure (PDB entry: 5KLL) and optimized at the B3LYP/6-31G*+ level of theory. For calculations, the native substrate, 2-AMS, was used. The results are summarized in Table 3, where values are shown as the difference in free energy between the aldehyde tautomer as compared with the enol tautomer optimized under the same restraints (negative values indicate lower energy for the enol tautomer). Numbering convention and a representative optimization can be found in Fig. 4D.
TABLE 3.
Geometry optimization of 2-AMS under various conditions: relative energy of the enol minus aldehyde tautomers
| ΔG | |
|---|---|
| kcal mol−1 | |
| 2-AMSa only | −1.15 |
| 2-AMSb, Asp169 | −11.1 |
| 2-AMSa,c, Asp169 | −6.87 |
| 2-AMSd, Asp169, Cys302 | −5.81 |
a The C3-C4-C5-C6 dihedral is frozen.
b All heavy atoms except nitrogen are frozen.
c C1, C2, and all oxygens are frozen.
d C1, C2, O1, and O2 are frozen.
The first row of Table 3 shows the results of optimizing each of the tautomers with the C3–C4–C5–C6 dihedral angle fixed to what is observed in the crystal structure. Although the enol form is lower in energy, the difference between the two tautomers is small. The second row shows that inclusion of the carboxylate group of Asp-169 drastically increases the difference in free energy between the aldehyde and the enol tautomers. Allowing more flexibility during the geometry optimization reduces the difference, as seen in the third row; however, the enol is still significantly lower in energy than the aldehyde. The inclusion of the side chain of Cys-302 does not qualitatively change the results of optimization. Interestingly, if the optimization is performed with the carboxylate group of Asp-169 protonated, as would be expected at lower pH values, the aldehyde tautomer optimizes to a structure with geometry nearly identical to the previously published thiohemiacetal intermediate trapped in the E268A mutant crystal (PDB entry: 4OU2). This observation suggests that when protonated, Asp-169 can play the same role as Asn-169 does in the wild-type enzyme.
Discussion
An Update to the Mechanism of AMSDH Action
In a previous report, insight was gained into the dehydrogenation mechanism of AMSDH by the capture of two important catalytic intermediates, thiohemiacetal 2 and thioacyl adducts 3 (5). The primary substrate and the catalytic intermediates are in distinct E/Z configurations at the C2–C3 position. Following our recent success, here, we have captured a new intermediate, which was not previously seen by reacting single crystals of the N169D mutant with a substrate analog. The new intermediate was trapped prior to the NAD+-dependent oxidation reaction. A perusal of the new intermediate structure and computational analysis point to an isomerization intermediate in AMSDH before its expected dehydrogenase activity. A hidden isomerase-like catalytic mechanism is revealed for the dehydrogenase. Thus, an updated, and more complete, catalytic cycle of AMSDH is proposed (Fig. 6). As compared with our first mechanistic model (5), the new catalytic mechanism removes the puzzle of the E-to-Z conformation difference previously observed in the catalysis and defines an unprecedented isomerization reaction mechanism mediated by a dehydrogenase.
FIGURE 6.
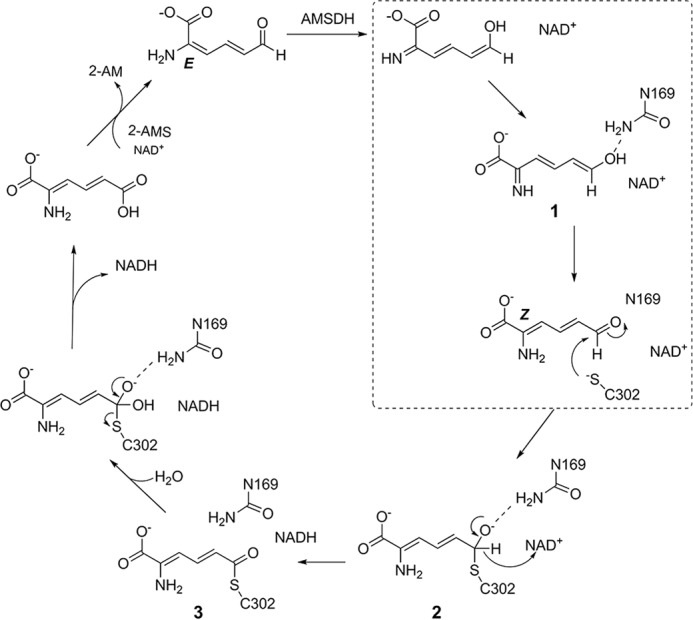
Proposed catalytic mechanism of AMSDH. The isomerase reaction is highlighted in a dotted box.
The new tautomerization intermediate was captured from the N169D mutant of AMSDH. The substitution of asparagine to aspartic acid at the 169 position provides enhanced stabilization of the enol intermediate as compared with the native enzyme, so that it accumulates in the mutant. It is likely that a similar intermediate also occurs in the native enzyme, but presumably it decays faster than it forms. At physiological pH, aspartic acid is typically deprotonated. The side chain of N169D is only able to donate an H-bond when protonated. While in the deprotonated state, the carboxylate moiety is restricted to receiving H-bonds. N169D should only be able to donate a hydrogen for the formation of an H-bond at lower pH values to stabilize intermediates. With this understanding, the pH profile of the mutant was determined, and the results indicate a critical role of an H-bonding stabilization of the catalytic intermediates by Asn-169. In contrast to aspartic acid, asparagine can both donate and receive hydrogen for forming H-bonding interactions and thus possesses a different pH profile.
The Catalytic Driving Force of Isomerization Reaction and the Role of Asn-169
The results obtained in this work reveal a pitcher-and-catcher mechanism. At one end of the active site, two arginine residues (Arg-120 and Arg-464) function as the “pitcher,” using electrostatic forces to drive an isomerization (Fig. 4C). Because of the two arginine residues, the substrate binds in the 2-imine, 6-enol form. At the other end of the active site, Asn-169 acts as the “catcher” by stabilizing the 2-enamine, 6-aldehyde form of the substrate, the necessary tautomer for dehydrogenation. In the N169D mutant, however, the scenario is somewhat different. In wtAMSDH, the side chain of Asn-169 can act as an H-bond donor with its amide moiety to stabilize the aldehydic oxygen of the substrate and subsequent reactive intermediates. By contrast, in the N169D mutant, the deprotonated carboxylate group of the Asp-169 can only accept H-bonds, giving an unexpected opportunity to capture an imine/enol tautomer during the in crystallo chemical reaction.
During the reaction catalyzed by AMSDH, the role of Asn-169 is to stabilize partial negative charges of intermediates and transition states by acting as an H-bond donor with the C6 oxo group of the substrate-derived intermediates. It was a fortuitous discovery to capture a tautomer of the primary substrate during our quest to determine the precise role of Asn-169. This finding allows for a deeper understanding of AMSDH (Fig. 6), and the enzymatically mediated tautomerization mechanism is fully established.
The Necessity of the Isomerization Reaction
Upon recognizing that AMSDH performs an unexpected isomerization on its substrate, the question of why such an isomerization should take place naturally arises. There is a difference of ∼4.2 kcal mol−1 in free energy between the 2-AMS 2-enamine, 6-aldehyde, 2E isomer and 2Z isomer (5). One reason for the presence of an intrinsic isomerase activity is perhaps to utilize this small but noticeable energy for the dehydrogenation reaction. Moreover, in the tertiary complex, the substrate in the active site was recognized by arginine residues in its original “compact” conformation (Fig. 4). After isomerization, the distance between C6 of the substrate and the sulfur of cysteine decreased from ∼3.5 Å to ∼2 Å, which also facilitates the nucleophilic attack from the cysteine in the subsequent step of the reaction. Thus, the isomerization reaction forces the substrate binding to the active site in a correct configuration for dehydrogenation.
Another consideration is that the enzyme following AMSDH in the kynurenine pathway is a deaminase that presumably works by adding water to the iminium ion of 2-AM, generating ammonia and 4-oxalocrotonate (Fig. 5) (14). The chemistry of deamination has been well studied and proceeds via backside nucleophilic attack of a water molecule or hydroxide ion on the electrophilic imine carbon. Such a reaction would be less sterically hindered on the observed product of AMSDH as compared with the expected product had no isomerization taken place.
A broader look at the kynurenine pathway may provide further insight into both why an isomerization is needed at this point in the pathway as well as why AMSDH is best suited to perform such an activity. The metabolic intermediate two reactions upstream of 2-AMS is 3-hydroxyanthranilic acid (3-HAA) with a substituted benzene ring. The aromaticity of 3-HAA is broken by HAO as molecular oxygen is added across its C3–C4 bond. During the addition of oxygen, 3-HAA bidentately chelates the active-site iron ion of HAO with its hydroxyl and amino groups, ensuring that the resulting product, 2-amino-3-carboxymuconic semialdehyde (ACMS), will be formed with its two carboxylate groups trans to each other and its amine group cis to its 3-carboxyl group. This much of the stereochemistry has been previously verified (15). A crystal structure of ACMSD bound with a competitive inhibitor also agreed with the two carboxylate groups of ACMS being trans to each other (16). After decarboxylation by ACMSD, the kynurenine metabolite, 2-AMS, can rapidly, spontaneously decay to picolinic acid, presumably by an electrocyclization like its upstream metabolite, ACMS, which decays to quinolinic acid. The decay reaction to picolinic acid, a metabolic dead end, is relatively rapid with a half-life of 35 s at room temperature (5).
If one is to accept that an isomerization must be performed before the downstream deamination reaction, HAO, the first enzyme of the pathway available to perform the isomerization, is an untenable choice because it is directly chelated by its substrate and products across the very bond to isomerize. The next candidate, ACMSD, is more promising; however, if it were to catalyze the isomerization of the 2–3 bond of ACMS or 2-AMS, it would increase the rate of an already fast decay process (17) by putting 2-AMS in the correct conformation to form picolinic acid. AMSDH is then the logical choice to perform the isomerization, as it is the last enzyme for which the amine and aldehyde of the metabolite are in full conjugation to allow for facile tautomerization to an imine and enol form that can readily rotate about the C2–C3 bond. After oxidation to 2-aminomuconic acid, the barrier for tautomerization to the imine form to allow for isomerization is expected to be much larger. Hence, from both the metabolic pathway and the chemical reaction standpoints, there is an intrinsic need for isomerization.
A Conserved Substrate Recognition Model in Kynurenine Pathway
In the kynurenine pathway, several intermediates are unstable. The in vitro decay rates of ACMS and 2-AMS are 0.015 and 1.2 min−1 at pH 7.4, respectively (17), which means that the enzymes responsible for them must compete with their non-enzymatic decay under differing metabolic states. Therefore, it is essential to recognize and stabilize those unstable intermediates by H-bonding within a short time during the enzymatic reactions. It was reported that two arginine residues from the adjoining units in ACMSD (7), the upstream neighbor of AMSDH in kynurenine pathway, are located in the binding pocket for H-bonding with the two carboxylate groups of ACMS and possible catalytic intermediates (7, 16). In AMSDH, the substrate, 2-AMS, is even more unstable than ACMS, and thus it needs to be efficiently recognized and stabilized by the two arginine residues in AMSDH at one end and an asparagine residue at the other end to prevent spontaneous autocyclization of the substrate inside the enzyme.
Comparison with the 4-Oxalocrotonate Tautomerase
Based on previously characterized dehydrogenases, there is no precedent for an aldehyde dehydrogenase to isomerize its bound substrate before performing its primary redox reaction. Interestingly, an enzyme in the tautomerase superfamily has been characterized in which 2-hydroxymuconate, the product of AMSDH with its alternate substrate, 2-HMS, is a reaction intermediate in the isomerization of 2-oxo-4-hexenedioate to 2-oxo-3-hexenedioate (18, 19). The enzyme, 4-oxalocrotonate tautomerase, has been well studied (18–24), and it binds its substrate with three arginine residues. This binding model is the same as that in the active site of AMSDH tertiary complex. The implication is that the two arginine residues, Arg-120 and Arg-464, in the active site of AMSDH facilitate a similar tautomerization.
Conclusion
An enzyme-mediated substrate tautomerization mechanism is found in the early stage of the dehydrogenase catalytic cycle of AMSDH. Our previous work suggested an E/Z isomerization of the substrate at the enzyme active site. However, the chemical mechanism of the isomerization was not studied (5). The chemical mechanism of the hidden isomerization reaction was solved unexpectedly during our quest to delineate the precise role of Asn-169. On the basis of the newly captured enol tautomer intermediate shown in our crystal structure and computational analysis, we propose an enzyme-mediated isomerization mechanism that proceeds through tautomerization catalyzed by the dehydrogenase as part of the AMSDH catalytic cycle (Fig. 6). First, the 6-aldehyde form of the substrate, 2-AMS, is tautomerized to its 2-imine, 6-eneol form in the active site. Next, the substrate rotates about its C2–C3 bond to an extended conformation under the assistance of Asn-169. Finally, the extended substrate is tautomerized back to the aldehyde form to allow for nucleophilic attack from the thiolate of Cys-302 to carry out the natural dehydrogenation chemistry. As the electrostatic driving force identified for initiating tautomerization during AMSDH turnover is also found in other members of the aldehyde dehydrogenase superfamily, these findings may have broader implications for these and related enzymes.
Experimental Procedures
Site-directed Mutagenesis and Protein Preparation
The cloning and generation of expression plasmid of Pseudomonas fluorescens KU-7 AMSDH were described in a previous publication (5). N169A, N169S, N169D, and N169Q single mutants were constructed by the PCR overlap extension mutagenesis method. The plasmid pET16b-AMSDH containing amsdh KU-7 was used as a template, and the forward primerswere 5′-GTTATTTCTCCGTGGgcgCTGCCGTTGCTGTTG-3′for N169A, 5′-GTTATTTCTCCGTGGtctCTGCCGTTGCTGTTG-3′ for N169S, 5′-GTTATTTCTCCGTGGgatCTGCCGTTGCTGTTG-3′ for N169D, and 5′-GTTATTTCTCCGTGGcagCTGCCGTTGCTGTTG-3′ for N169Q. Each mutant plasmid was verified by DNA sequencing and transformed to Escherichia coli BL21 (DE3). The isolation strategy of each mutant protein is the same as wild-type AMSDH (5).
Preparation of the Substrate 2-HMS and Kinetic Assay
The native substrate of AMSDH, 2-AMS, is too unstable for routine kinetic work (5). As such, an alternate substrate by which the nitrogen atom of 2-AMS is substituted by oxygen, 2-hydroxymuconate semialdehyde, 2-HMS, was used to measure the dehydrogenase activity as described in our previous study (5). 2-HMS was generated as described previously (25). Briefly, 3-hydroxyanthranilic acid dioxygenase was used to catalyze the addition of molecular oxygen to 3-hydroxyanthranilic acid, generating ACMS (16, 17, 26, 27). As described previously, lowering the pH below 2 caused the chemical conversion of ACMS to 2-HMS (5). The steady-state kinetics analyses were performed in a reaction mixture of 1 mm NAD+ and 25 mm citrate buffer (pH 4.5–5.5) or HEPES buffer (pH 6.5–8.0). All assays were done at room temperature; the consumption of 2-HMS (λmax at 375 nm, ϵ375 is 43,000 m−1 cm−1) (5) was monitored with an Agilent 8453 diode-array spectrophotometer.
Crystallization, Data Collection, Processing, and Refinement
The N169A and N169D mutants were incubated with 10 eq of NAD+ for 10 min and crystallized by the hanging-drop method and using a reservoir solution of 20–25% PEG 3350 and 0.2–0.3 m sodium phosphate dibasic monohydrate, pH 9.1. The cryoprotectant solution containing 13% PEG 3350 and ∼1 mm 2-HMS was employed to react with the NAD+ co-crystalized mutant crystals. After incubation for 1 min to 20 h, the soaked crystals were flash-cooled in liquid nitrogen. X-ray diffraction datasets were collected at the SER-CAT beamline 22-ID of the Advanced Photon Source, Argonne National Laboratory, and were processed and scaled by HKL-2000 (28). Using the wild-type AMSDH structure (PDB entry: 4I26) as the template, the structures of the mutants and catalytic intermediates were solved by molecular replacement and refined by employing the Phenix 1.10.1 (29) and Coot 0.8.3 (30). PyMOL (34) was used in generating structural figures.
Quantum Chemical Calculations
Gaussian 03 Revision-E.01 was used to perform all calculations (31). The crystal structure of N169D ternary complex (PDB entry: 5KLL) was used to build the starting models. Geometry optimizations and density functional theory calculations were done at the B3LYP/6-31G*+ levels.
Accession Codes
Coordinates and structure factors for NAD+-bound N169D AMSDH, NAD+- and 2-HMS-bound N169D AMSDH, N169D 2-HMS tautomeric intermediate, N169D 2-HMS intermediate, NAD+-bound N169A AMSDH, and N169A thioacyl intermediate have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank under accession codes 5KJ5, 5KLK, 5KLL, 5KLM, 5KLN, and 5KLO, respectively.
Author Contributions
A. L., I. D., and Y. Y. conceived the study. Y. Y., U. H., Y. W., and I. S. performed experiments, and I. D. conducted computational analyses. Y. Y. and I. D. wrote the manuscript, with input and editing by A. L. All authors participated in discussions and approved the final manuscript.
Supplementary Material
Acknowledgments
X-ray datasets were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory.
This work was supported by the National Institutes of Health Grant MH107985 and the National Science Foundation Grant CHE-1623856. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.

This article contains supplemental Figs. S1 and S2.
- HAO
- 3-hydroxyanthranilate dioxygenase
- ACMS
- 2-amino-3-carboxymuconate-6-semialdehyde
- ACMSD
- ACMS decarboxylase
- 2-AMS
- 2-aminomuconate semialdehyde
- AMSDH
- 2-aminomuconate-6-semialdehyde dehydrogenase
- 2-HMS
- 2-hydroxymuconate semialdehyde
- 3-HAA
- 3-hydroxyanthranilic acid
- RMSD
- root mean square deviation.
References
- 1.Stone T. W., and Darlington L. G. (2002) Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 1, 609–620 [DOI] [PubMed] [Google Scholar]
- 2.Schwarcz R. (2004) The kynurenine pathway of tryptophan degradation as a drug target. Curr. Opin. Pharmacol. 4, 12–17 [DOI] [PubMed] [Google Scholar]
- 3.Schwarcz R., Bruno J. P., Muchowski P. J., and Wu H.-Q. (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis I., and Liu A. (2015) What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev. Neurother. 15, 719–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huo L., Davis I., Liu F., Andi B., Esaki S., Iwaki H., Hasegawa Y., Orville A. M., and Liu A. (2015) Crystallographic and spectroscopic snapshots reveal a dehydrogenase in action. Nat. Commun. 6, 5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dazzi C., Candiano G., Massazza S., Ponzetto A., and Varesio L. (2001) New high-performance liquid chromatographic method for the detection of picolinic acid in biological fluids. J. Chromatogr. B Biomed. Sci. Appl. 751, 61–68 [DOI] [PubMed] [Google Scholar]
- 7.Huo L., Davis I., Chen L., and Liu A. (2013) The power of two arginine 51 and arginine 239* from a neighboring subunit are essential for catalysis in α-amino-β-carboxymuconate-ϵ-semialdyhyde decarboxylase. J. Biol. Chem. 288, 30862–30871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farrés J., Wang T. T., Cunningham S. J., and Weiner H. (1995) Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry 34, 2592–2598 [DOI] [PubMed] [Google Scholar]
- 9.Wang X., and Weiner H. (1995) Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry 34, 237–243 [DOI] [PubMed] [Google Scholar]
- 10.D'Ambrosio K., Pailot A., Talfournier F., Didierjean C., Benedetti E., Aubry A., Branlant G., and Corbier C. (2006) The first crystal structure of a thioacylenzyme intermediate in the ALDH family: new coenzyme conformation and relevance to catalysis. Biochemistry 45, 2978–2986 [DOI] [PubMed] [Google Scholar]
- 11.Steinmetz C. G., Xie P., Weiner H., and Hurley T. D. (1997) Structure of mitochondrial aldehyde dehydrogenase: the genetic component of ethanol aversion. Structure 5, 701–711 [DOI] [PubMed] [Google Scholar]
- 12.Cobessi D., Tête-Favier F., Marchal S., Branlant G., and Aubry A. (2000) Structural and biochemical investigations of the catalytic mechanism of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J. Mol. Biol. 300, 141–152 [DOI] [PubMed] [Google Scholar]
- 13.Park J., and Rhee S. (2013) Structural basis for a cofactor-dependent oxidation protection and catalysis of cyanobacterial succinic semialdehyde dehydrogenase. J. Biol. Chem. 288, 15760–15770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muraki T., Taki M., Hasegawa Y., Iwaki H., and Lau P. C. (2003) Prokaryotic homologs of the eukaryotic 3-hydroxyanthranilate 3,4-dioxygenase and 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase in the 2-nitrobenzoate degradation pathway of Pseudomonas fluorescens strain KU-7. Appl. Environ. Microbiol. 69, 1564–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colabroy K. L., and Begley T. P. (2005) The pyridine ring of NAD is formed by a nonenzymatic pericyclic reaction. J. Am. Chem. Soc. 127, 840–841 [DOI] [PubMed] [Google Scholar]
- 16.Huo L., Liu F., Iwaki H., Li T., Hasegawa Y., and Liu A. (2015) Human α-amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase (ACMSD): a structural and mechanistic unveiling. Proteins 83, 178–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T., Ma J. K., Hosler J. P., Davidson V. L., and Liu A. (2007) Detection of transient intermediates in the metal-dependent non-oxidative decarboxylation catalyzed by α-amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase. J. Am. Chem. Soc. 129, 9278–9279 [DOI] [PubMed] [Google Scholar]
- 18.Chen L. H., Kenyon G. L., Curtin F., Harayama S., Bembenek M. E., Hajipour G., and Whitman C. P. (1992) 4-Oxalocrotonate tautomerase, an enzyme composed of 62 amino acid residues per monomer. J. Biol. Chem. 267, 17716–17721 [PubMed] [Google Scholar]
- 19.Metanis N., Brik A., Dawson P. E., and Keinan E. (2004) Electrostatic interactions dominate the catalytic contribution of Arg39 in 4-oxalocrotonate tautomerase. J. Am. Chem. Soc. 126, 12726–12727 [DOI] [PubMed] [Google Scholar]
- 20.Taylor A. B., Czerwinski R. M., Johnson W. H. Jr., Whitman C. P., and Hackert M. L. (1998) Crystal structure of 4-oxalocrotonate tautomerase inactivated by 2-oxo-3-pentynoate at 2.4 Å resolution: analysis and implications for the mechanism of inactivation and catalysis. Biochemistry 37, 14692–14700 [DOI] [PubMed] [Google Scholar]
- 21.Burks E. A., Yan W., Johnson W. H. Jr., Li W., Schroeder G. K., Min C., Gerratana B., Zhang Y., and Whitman C. P. (2011) Kinetic, crystallographic, and mechanistic characterization of TomN: elucidation of a function for a 4-oxalocrotonate tautomerase homologue in the tomaymycin biosynthetic pathway. Biochemistry 50, 7600–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terrell C. R., Burks E. A., Whitman C. P., and Hoffman D. W. (2013) Structural and kinetic characterization of two 4-oxalocrotonate tautomerases in Methylibium petroleiphilum strain PM1. Arch. Biochem. Biophys. 537, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huddleston J. P., Burks E. A., and Whitman C. P. (2014) Identification and characterization of new family members in the tautomerase superfamily: analysis and implications. Arch. Biochem. Biophys. 564, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burks E. A., Fleming C. D., Mesecar A. D., Whitman C. P., and Pegan S. D. (2010) Kinetic and structural characterization of a heterohexamer 4-oxalocrotonate tautomerase from Chloroflexus aurantiacus J-10-fl: implications for functional and structural diversity in the tautomerase superfamily. Biochemistry 49, 5016–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichiyama A., Nakamura S., Kawai H., Honjo T., Nishizuka Y., Hayaishi O., and Senoh S. (1965) Studies on the metabolism of the benzene ring of tryptophan in mammalian tissues. II. Enzymatic formation of α-aminomuconic acid from 3-hydroxyanthranlic acid. J. Biol. Chem. 240, 740–749 [PubMed] [Google Scholar]
- 26.Li T., Iwaki H., Fu R., Hasegawa Y., Zhang H., and Liu A. (2006) α-Amino-β-carboxymuconic-ϵ-semialdehyde decarboxylase (ACMSD) is a new member of the amidohydrolase superfamily. Biochemistry 45, 6628–6634 [DOI] [PubMed] [Google Scholar]
- 27.Li T., Walker A. L., Iwaki H., Hasegawa Y., and Liu A. (2005) Kinetic and spectroscopic characterization of ACMSD from Pseudomonas fluorescens reveals a pentacoordinate mononuclear metallocofactor. J. Am. Chem. Soc. 127, 12282–12290 [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 29.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31.Frisch M. J., Trucks G., Schlegel H., Scuseria G., Robb M., Cheeseman J., Montgomery J. A. Jr., Vreven J. T., Kudin K., Burant J., Millam J., Iyengar S. S., Tomasi J., Barone V., Mennucci B., et al. (2004) Gaussian-03, Revision E.01, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 32.Engh R. A., and Huber R. (1991) Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A 47, 392–400 [Google Scholar]
- 33.Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLano W. L. (2014) The PyMOL Molecular Graphics System, version 1.7.0.0, Schrödinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.