

Abstract
Huntingtin-associated protein 1 (Hap1) was originally identified as a protein that binds to the Huntington disease protein, huntingtin. Growing evidence has shown that Hap1 participates in intracellular trafficking via its association with various microtubule-dependent transporters and organelles. Recent studies also revealed that Hap1 is involved in exocytosis such as insulin release from pancreatic β-cells. However, the mechanism underlying the action of Hap1 on insulin release remains to be investigated. We found that Hap1 knock-out mice had a lower plasma basal insulin level than control mice. Using cultured pancreatic β-cell lines, INS-1 cells, we confirmed that decreasing Hap1 reduces the number of secreted vesicles and inhibits vesicle exocytosis. Electrophysiology and imaging of intracellular Ca2+ measurements demonstrated that Hap1 depletion significantly reduces the influx of Ca2+ mediated by L-type Ca2+ channels (Cav). This decrease is not due to reduced expression of Cav1.2 channel mRNA but results from the decreased distribution of Cav1.2 on the plasma membrane of INS-1 cells. Fluorescence recovery after photobleaching showed a defective movement of Cav1.2 in Hap1 silencing INS-1 cells. Our findings suggest that Hap1 is important for insulin secretion of pancreatic β-cells via regulating the intracellular trafficking and plasma membrane localization of Cav1.2, providing new insight into the mechanisms that regulate insulin release from pancreatic β-cells.
Keywords: calcium channel, Huntington disease, insulin secretion, pancreas, transport
Introduction
Huntingtin-associated protein 1 (Hap1) was the first huntingtin-interacting protein identified in yeast two-hybrid screens (1). Hap1 is expressed abundantly in the brain and is also present in some of the peripheral endocrine systems, such as the pituitary, thyroid, adrenal glands, and pancreas (2, 3). In the pancreas, Hap1 is selectively expressed in β-cells that release insulin (4). A recent study showed that Hap1 is important for insulin release from β-cells, and when Hap1 expression is reduced, glucose-mediated insulin release is inhibited (5). Mutant mice with the selective Hap1 deficiency in pancreatic β-cells had impaired glucose tolerance and decreased insulin secretion in response to glucose stimulation (5). Because Hap1 binds the Huntington disease (HD)4 protein huntingtin, Hap1 dysfunction may also be involved in metabolic abnormalities in HD, which is evident in HD mouse models and patients. For example, the R6/2 HD mice develop diabetes due to reduced β-cell mass and deficient insulin exocytosis (6). Mutant huntingtin forms aggregates in the cytoplasm and reduces insulin secretion from huntingtin-transfected pancreatic β-cell lines, NIT-1 cells, which could be reversed by overexpression of heat shock protein 40 (HSP40) (7). Patients with HD exhibit increased incidence of diabetes mellitus (8). Also, mutant huntingtin was found to interact with β-tubulin and disrupt vesicular transport and insulin secretion (9). Given these findings, it would be interesting to investigate how Hap1 is involved in insulin release in pancreatic β-cells.
Many proteins known to bind Hap1 are associated with endocytosis and vesicular trafficking. The binding of Hap1 with p150Glued subunit of dynactin and huntingtin could enhance vesicular transport of brain-derived neurotrophic factor (10). Reducing Hap1 protein has been shown to disrupt kinesin light chain-dependent neurite outgrowth and transport of amyloid precursor protein vesicles (11). Phosphorylation at the C terminus of Hap1A, the A isoform of Hap1, plays an important role in the association of Hap1A with kinesin light chain and p150Glued (12). Phosphorylation of Hap1A also enhances the binding of 14-3-3 protein involved in the assembly of protein complexes and the regulation of protein trafficking (13). In cultured primary neurons, lack of Hap1 decreased intracellular TrkA amounts and impaired neurite outgrowth (12). Reducing Hap1 level resulted in decrease of TrkB internalization and signaling transduction (14). Hap1 also associates with the inositol 1,4,5-triphosphate receptor (InsP3R) (15), forming a complex required for proper Ca2+ signaling (16). Consistently, Ca2+ mobilization defects in β cells are involved in the diabetes reported in HD patients (17).
Endocytosis and exocytosis function in many common ways in neurons and endocrine cells. Pancreatic β-cells, a major cellular component of the islets, are electrically excitable and exquisitely sensitive to glucose. In response to blood glucose, β-cells secrete insulin and play a unique role in glucose homeostasis. Glucose homeostasis is critical to the health and survival of mammals. L-type Ca2+ channels take center stage in the process of insulin release (18–21). Neurotransmitter release is attributed to Ca2+ influx through P/Q- and N-type Ca2+ channels, and the L-type (Cav1.2 and Cav1.3) channels are considered as responsible for hormone secretion (22). Electrophysiological and molecular biological studies indicate that pancreatic β-cells express several subtypes of Cav channels. In particular, L-type Ca2+ channels are considered crucial for β-cell function, given that dihydropyridines can potently suppress insulin secretion (23). Both Cav1.2 and Cav1.3 have been found in human and rodent islets, as well as in various β-cell lines (23, 24). Up-regulation of β-cells' Ca2+ channel activity and/or density results in enhanced insulin exocytosis and more efficient glucose homeostasis (25). Moreover, down-regulation of Cav channel activity and/or density causes less insulin secretion and glucose intolerance, which is associated with a group of type 2 diabetic patients (26–29). Studies in mutant mice with Cav1.2 or Cav1.3 deficiency have confirmed that L-type Ca2+ channels are crucial for the physiology of β-cells (30, 31). Proper trafficking and distribution of L-type Ca2+ channels are essential for insulin release.
Growing evidence indicates that Hap1 is likely involved in microtubule-dependent trafficking in neurons, such as vesicular transport, membrane receptor trafficking, and Ca2+ release (32–34). Our previous study has also demonstrated that Hap1 may help to regulate transport of insulin-containing secretory granules along cortical actin filaments in pancreatic β-cells (35). Whether and how Hap1 regulates the trafficking or surface expression of L-type Ca2+ channel in endocrine cells have not been reported. In this study, we provide both in vitro and in vivo data that Hap1 is required for insulin secretion from β-cells. Using patch clamp recordings, we demonstrated that Hap1 depletion significantly reduces the L-type Ca2+ currents. Using biochemical and molecular biology techniques, we also found that Hap1 regulates the surface expression level and intracellular trafficking of L-type Ca2+ channels Cav1.2 in INS-1 cells. These data suggest that Hap1 plays an important role in regulation of insulin secretion in β-cells and offer a new therapeutic target for ameliorating metabolic disorders due to defective insulin release from pancreatic β-cells.
Results
Reducing Hap1 Expression Decreases the Release of Insulin
To provide further evidence for the idea that Hap1 regulates insulin release from β-cells, we measured the plasma insulin level of Hap1 knock-out (KO) or Hap1−/− mice and investigated the effect of Hap1 deficiency on secretion of cultured β-cell lines INS-1. Because Hap1−/− mice have retarded growth and die 3–4 days after birth, and Hap1+/− heterozygous mice showed no obvious behavioral and body weight abnormalities to wild-type mice and lived as long as WT mice (36), we focused our studies on Hap1−/− 3–4-day-old mice to investigate the role of Hap1 in insulin secretion. We collected the blood of Hap1 KO and WT pups and used the plasma to analyze insulin levels via radioimmunoassay (RIA). The results showed that the insulin level of KO mice was significantly lower than WT mice (Fig. 1A, upper panel), indicating that lack of Hap1 may impair insulin release in mice.
FIGURE 1.

Reducing Hap1 expression decreases the release of insulin. A, insulin levels in blood plasma of KO and WT pups by RIA (upper panel) (**, p < 0.001; KO n = 25,WT n = 30). INS-1 cells treated with Hap1-siRNA displayed an obvious reduction in insulin release compared with control cells (bottom panel) (**, p < 0.001; n = 10). B, immunofluorescent staining of Hap1 in INS-1 cells transfected with Hap1-siRNA or scramble-siRNA. C, Western blotting of INS-1 cells showing that Hap1 protein level was reduced by Hap1-siRNA treatment but not by scramble-siRNA. Scale bars, 20 μm.
INS-1 has been widely used to investigate the mechanisms underlying insulin release. We used small interference RNA (siRNA) to reduce endogenous Hap1 expression in INS-1 cells. RIA detection found that the INS-1 cells transfected with Hap1-siRNA plasmid showed a lower level of insulin release compared with the control cells transfected with scramble plasmid (Fig. 1A, bottom panel). Immunofluorescent staining of the Hap1 signal in Hap1-siRNA-treated cells was markedly reduced compared with cells treated with scramble-siRNA (Fig. 1B). Western blotting also demonstrated that Hap1 expression is inhibited via Hap1-siRNA (Fig. 1C). We then performed membrane capacitance measurement experiments to examine the exocytosis of insulin-containing vesicles in INS-1 cells. To do so, we designed a voltage clamp depolarization protocol to distinguish vesicle release from the immediately releasable pool (IRP) and readily releasable pool (RRP). The stimulation protocol consisted of a train of six 30-ms depolarization (ΔCm6) to deplete the IRP, followed by 10 100-ms depolarization (ΔCm10) to elicit secretion from the RRP, and the holding potential from −80 to 0 mV (Fig. 2A). In response to this stimulation protocol, there was a small capacitance increase after ΔCm6 and ΔCm10 corresponding to release from the IRP and RRP in Hap1-siRNA-treated cells. In contrast, the stimulated exocytosis in control cells was significantly greater for both IRP and RRP. The quantification results suggested that there was a decrease in the number of releasing vesicles in Hap1-silenced cells versus control cells (Fig. 2B). Taken together, these findings indicate that Hap1 deficiency affects insulin release from β-cells.
FIGURE 2.

Reducing Hap1 expression decreases the exocytosis activities of INS-1 cell. A, stimulation protocol consisted of a train of six 30-ms and ten 100-ms pulses (upper panel) as well as exocytotic events (bottom panel) in INS-1 cells transfected with Hap1-siRNA or scramble-siRNA. The initial six exocytotic events represent exocytosis from IRP, and the following 10 events are secretions from RRP. B, averaged capacitance increases after ΔCm6 (IRP) or ΔCm10 (RRP) from INS-1 cells (**, p < 0.001; scramble n = 30, siRNA n = 35).
Dynamics of Insulin Granules Are Inhibited in Hap1-deficient β-Cells
Release of neurotransmitters and peptide hormones involves exocytotic fusion of secretory vesicles with the plasma membrane (37). Furthermore, we examined the dynamic parameters of exocytosis in INS-1 cells by imaging vesicle membrane-targeted fluorescent probes (VAMP2-pHluorin). Live cells were examined in a confocal microscope at the cell footprint to better visualize near PM fluorescent spots. We used fusion constructs of vesicle membrane protein synaptobrevin-2 (VAMP2) with a pH-sensitive green fluorescent protein PHluorin (VAMP2-pHluorin) to measure the vesicle release (38) by taking advantage of the drastically greater pH sensitivity of the probe. Because the lumen of the insulin vesicle is acidic (pH5.5–6.0), we captured a fluorescence image of VAMP2-pHluorin under the conditions in which pHluorin fluorescence is expected to be close to zero. Stimulation of Ca2+ influx with 30 mm K+ caused insulin-containing spots to brighten and spread suddenly, which is consistent with release of the fluorescent peptide. As shown in Fig. 3, A and B, fluorescence intensity increased first in a central region, then spread into a surrounding annulus, and finally declined as dye diffused away. To determine whether there are any effects of loss of Hap1 on insulin release, we co-transfected both constructs siRNA and PH-VAMP2 simultaneously into the same cells and compared those transfected with Hap1-scramble. When cells were stimulated with 30 mm K+ for 300 s, the number of secreted vesicles in Hap1 knockdown cells was decreased compared with that from the control cells (Fig. 3D). Comparing the fluorescence intensity of each sample revealed that there is a significant reduction of insulin release events from the Hap1 deficiency cells (Fig. 3, C and E). Inhibiting Hap1 also diminished the duration of insulin release from secreted vesicles (Fig. 3F). Taken together, these findings indicate that Hap1 deficiency affects the dynamics of insulin release from β-cells.
FIGURE 3.

Inhibiting Hap1 expression decreases the formation of secreted vesicles. A and B, visualization of secretory activities in INS-1 cells co-expressed PH-VAMP2 with mCh-Hap1-scramble or mCh-Hap1-siRNA. Confocal imaging at the cell footprint shows a number of fluorescent spots at PM that spread and are diffuse, which represent the secreting events. Sequential images of insulin-secreting events were observed in control cells after high K+ stimulation, whereas reduced releasing events were seen in Hap1-siRNA. Scale bar, 10 μm. C, representative time course of the fluorescence changes measured at the fluorescent spots (arrow) in scramble-siRNA and Hap1-siRNA cells. D, INS-1 cells treated with Hap1-siRNA displayed a significant reduction in insulin release number after high K+ stimulation for 300 s, compared with control cells. E, fluorescent intensity for insulin decreases in INS-1 cells treated with Hap1-siRNA. F, inhibiting Hap1 diminished the duration of insulin release from secreted vesicles (*, p < 0.05; **, p < 0.001; scramble n = 56, siRNA n = 48).
Hap1 Deficiency Reduces L-type Ca2+ Currents in INS-1 Cells
Cav channels play a critical role in insulin secretion as well as the survival of β-cells. Cav channels are ubiquitously expressed in various cell types throughout the body. There is considerable consensus that L-type Ca2+ current is the major Ca2+ current subtype in the β-cells from all the examined species (25, 39, 40). The β-cell Cav channel-mediated Ca2+ entry directly stimulates secretory granule trafficking and triggers insulin exocytosis. This is the paramount way whereby Cav channels regulate insulin secretion (25, 41). To determine the role of Hap1 in the Ca2+-dependent secretory pathway, we next compared the L-type Ca2+ current in Hap1-siRNA-treated and control INS-1 cells using whole cell patch clamp measurements. TEA-containing extracellular solution and Cs+ were used to block the voltage-gated K+ channels and tetradotoxin to block the voltage-gated Na+ channels. From recording of whole cells with a holding potential of −70 mV, β-cells exhibited inward currents typical for Ca2+ channels current in response to depolarizing voltage from a holding potential of −70 to +50 mV in 10-mV increments (Fig. 4A, upper panel). Hap1-siRNA-treated cells showed distinctly small L-type Ca2+ currents evoked by depolarizing step pulses (Fig. 4A, bottom panel). Under the same condition, control cells exhibited larger Ca2+ currents (Fig. 4A, middle panel). The mean current-voltage relationships (I-V) of L-type Ca2+ channels were then obtained (Fig. 4B). As shown in Fig. 4C, the averaged Ba2+ current density (pA/pF) at 10 mV in control INS-1 cells expressing scramble-siRNA was much higher than that of Hap1-siRNA cells.
FIGURE 4.

Knockdown of Hap1 reduces L-type Ca2+ currents in INS-1 cells. INS-1 cells were transfected with Hap1-siRNA or scramble-siRNA plasmids. Whole cell L-type Ca2+ currents were measured using Ba2+ as the charge carrier. A, representative L-type Ca2+ current traces from scramble-siRNA (middle traces) or Hap1-siRNA (bottom traces) groups. Cells were held at a holding potential of −70 mV and stepped from −60 to +50 mV in 10-mV increments. B, mean current-voltage relations of L-type Ca2+ currents recorded from INS-1 cells transfected with Hap1-siRNA (▴) or scramble-siRNA (●). C, averaged peak L-type current density recorded from INS-1 cells transfected with Hap1-siRNA or scramble-siRNA plasmids at +10 mV. Ba2+ currents were elicited by 200-ms pulses to 0 mV from a V-h of −70 mV (**, p < 0.001, scramble n = 21, siRNA n = 21).
Inhibiting Hap1 Expression Lowers the Ca2+ Influx of INS-1 Cells
The low levels of L-type Ca2+ current and current density in Hap1 deficiency cells led us to speculate that the knockdown of Hap1 expression may decrease the Ca2+ influx. To verify this hypothesis, we used Ca2+ microfluorimetry to measure Ca2+ entry evoked by extracellular high K+ stimulation in INS-1 cells, which expressed either Hap1-siRNA or scramble-siRNA and were loaded with Fura-2-AM. We found the increase in [Ca2+]i in Hap1-siRNA and control cells in response to 30 mm K+ stimulation. Deficiency of Hap1 led to a lower level of high K+-stimulated [Ca2+]i rise than control cells (Fig. 5, A and B). The averaged changes in 340/380 intensity ratio are plotted in Fig. 5C. We found that INS-1 cells expressed endogenous L-type voltage Ca2+ channels predominantly, as the intracellular Ca2+ level was rapidly and significantly inhibited by nifedipine, a typical L-type Ca2+ channel blocker (Fig. 5D). These findings indicate that Hap1 is likely to functionally regulate the activity of L-type Ca2+ channels so that loss of Hap1 reduces the L-type Ca2+ currents and membrane depolarization-induced Ca2+ influx.
FIGURE 5.

Hap1 deficiency decreases the Ca2+ influx. Fura-2 Ca2+ microfluorimetry of INS-1 cells transfected with Hap1-siRNA or scramble-siRNA plasmids. The cells were incubated in Fura-2 for 20 min prior to 30 mm K+ stimulation, which induced a transient increase of cytosolic Ca2+ concentration. A, representative trace of depolarization-induced increase in [Ca2+]i in INS-1 cells expressing scramble-siRNA vector. B, depolarization-induced increase in [Ca2+]i in INS-1 cells expressing Hap1-siRNA vector. C, averaged peak 340/380 intensity ratio recorded from INS-1 cells transfected with Hap1-siRNA or scramble-siRNA vector (**, p < 0.001; scramble n = 26, siRNA n = 26). D, Ca2+ entry through L-type Ca2+ channels was activated by a depolarizing stimulation of extracellular 30 mm K+, and the elevation level of intracellular Ca2+ was completely inhibited by 10 μm L-type Ca2+ channel blocker nifedipine.
Loss of Hap1 Down-regulates the Surface Expression of Cav1.2 in INS-1 Cells
To rule out the possibility that Hap1 may regulate L-type Ca2+ channel transcription, we performed RT-PCR using INS-1 cells treated with Hap1-siRNA or scramble-siRNA for 48 h. Our data show that Hap1 deficiency has no effect on Cav1.2 mRNA expression. Hap1 is involved in the intracellular transport of several membrane receptors (12, 16). To reveal whether the reduction of L-type Ca2+ currents and decrease of Ca2+ influx in Hap1 deficiency INS-1 cells resulted from diminishment of transportation of Cav channel to the cell surface, we checked the effect of Hap1 loss on the cell surface expression of Cav1.2 in INS-1 cells. The plasmid of YFP-Cav1.2-HA was constructed to express YFP at the intracellular N terminus and a HA epitope in the extracellular S5-H5 loop of domain II (YH-Cav1.2). The YFP and HA tags had no effect on the channel but can help us to measure the total number of Cav1.2 channels expressed on the cell surface (42). To verify the effect of Hap1 on Cav1.2 channel localization on the plasma membrane, YH-Cav1.2, β1b, α2δ, and Hap1-siRNA or scramble-siRNA were co-transfected in INS-1 cells, and 48 h later we measured the average YFP intensity expressed on the PM. The YFP fluorescence at the edge of the cell membrane in control cells was much stronger than Hap1-silencing cells (Fig. 6A). As a result, there was a significant decrease of Cav1.2 expression level on membrane in Hap1-deficient cells compared with control cells (Fig. 6, A and B). We then analyzed the ratio of Cav1.2 mean intensity in the PM versus the cytosol in cells by fixing them and staining with anti-Cav1.2 antibodies (Fig. 6C). There was a small decrease of this ratio in the Hap1-deficient cells, compared with control cells (Fig. 6, C and D). We also investigated the membrane localization of YH-Cav1.2 channels by fixing the cells and staining with anti-HA antibodies without membrane permeabilization. The non-permeabilized cells had YFP fluorescence throughout the whole cell, but HA staining was seen only at the edge of the cell membrane. There was a significant decrease of Cav1.2 expression levels on the membrane in Hap1 deficiency cells compared with control cells (Fig. 6, E and F). Evidence for the effect of Hap1 on Cav1.2 surface expression was also obtained by immunoblotting of subcellular fractions samples (Fig. 6G). Furthermore, using immunoprecipitation with antibodies to Cav1.2 or Hap1, we found that Cav1.2 interacts with Hap1 (Fig. 7, A and B), also supporting the idea that Hap1 is important for the localization of Cav1.2 to the plasma membrane in INS-1 cells.
FIGURE 6.

Knockdown of Hap1 reduces the surface level of Cav1.2. A, INS-1 cell overexpression of YH-Cav1.2 together with scramble-siRNA (upper) or Hap1-siRNA (bottom). Profiles of YFP intensity along the yellow lines through the cell bodies are shown at bottom left of the 1st image. The signal of YFP in control cells shows sharp peaks at the edges of the cell membrane, whereas the other traces remain elevated throughout the cell compared with Hap1-siRNA cells. Scale bar, 10 μm. B, INS-1 cells treated with Hap1-siRNA displayed a significant reduction in the level of YH-Cav1.2 surface YFP intensity and average ratios (PM/C) compared with scramble-siRNA control (**, p < 0.001; scramble n = 45, siRNA n = 47). au is arbitrary unit. C, immunofluorescent Cav1.2 staining of INS-1 cells transfected with Hap1-siRNA (bottom) or scramble-siRNA (upper). Line scans of the anti-Cav1.2 fluorescence intensity through the cell bodies are shown in the insets. The signal of YFP in scramble-siRNA cells shows sharp peaks at the edges of the cell membrane, whereas no significant peaks were seen in Hap1-siRNA cells. Scale bar, 10 μm. D, surface Cav1.2 intensity and average ratios (PM/C) of endogenous Cav1.2 surface expression in Hap1-siRNA or scramble-siRNA cells (*, p < 0.01; scramble n = 15, siRNA n = 18). E, immunofluorescent of INS-1 cells with an anti-HA antibody without permeabilization. F, cells treated with Hap1-siRNA displayed a significant reduction in the level of YH-Cav1.2 surface HA intensity compared with scramble-siRNA (upper panel) (**, p < 0.001; scramble n = 195, siRNA n = 147). G, immunoblotting of Cav1.2 in plasma membrane (PM), cytosol (C), and homogenate (H) fractions.
FIGURE 7.
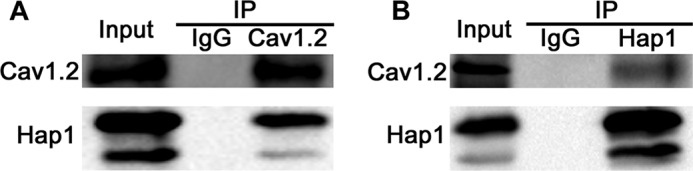
Interaction of Hap1 with Cav1.2. A, endogenous Cav1.2 was immunoprecipitated with Hap1 in INS-1 cells. Immunoblots of immunoprecipitations (IP) were from INS-1 cell lysates using antibodies against Cav1.2; an immunoprecipitation with an IgG antibody was used as a control. B, endogenous Hap1 was immunoprecipitated, and Cav1.2 was found to be co-precipitated with Hap1 in INS-1 cells. Immunoblots of immunoprecipitations (IP) were from INS-1 cell lysates using antibodies against Hap1; an immunoprecipitation with an IgG antibody was used as a control.
Trafficking of Cav1.2 Is Deficient in Hap1-silencing β-Cells
Because the surface level of Cav1.2 was significantly affected by Hap1 deficiency, we then investigated whether Hap1 is involved in the regulation of Cav1.2 intracellular trafficking. To detect the intracellular movement of Cav1.2, we examined Cav1.2 trafficking in INS-1 cells using fluorescence recovery after photobleaching (FRAP). We transiently co-transfected INS-1 cells with Hap1-siRNA + Cav1.2-YFP or scramble-siRNA + Cav1.2-YFP vectors, and we measured the movement of Cav1.2-YFP in these groups. We obtained a series of images for Cav1.2-YFP recovery during 0 to 130 s after photobleaching. Hap1-siRNA cells showed obviously slower recovery after bleaching Cav1.2-YFP (Fig. 8A, bottom panel). Under the same conditions, scramble-siRNA cells exhibited a faster recovery (Fig. 8A, upper panel). Fig. 8B shows the fluorescence recovery data were fit by single exponential curve equation to obtain recovery time constants (τ) (see under “Experimental Procedures”). When measuring the recovery kinetics, Hap1-siRNA-treated cells took a longer time (31.2 ± 5.8 s) than control cells (20.2 ± 7.3 s) with a significant difference (Fig. 8C). It was evident that Hap1 silencing decreases the speed of Cav1.2 intracellular trafficking and also prolongs the recovery time of Cav1.2, implying that Hap1 is able to assist Cav1.2 transport in β-cells.
FIGURE 8.

Lack of Hap1 impacts on the intracellular trafficking of Cav1.2-YFP in INS-1 cells. INS-1 cells were transfected with Hap1-siRNA or scramble-siRNA plasmids, together with Cav1.2-YFP. A, FRAP of Cav1.2-YFP trafficking in living INS-1 cells. The images show Cav1.2-YFP fluorescence before and immediately after photobleaching, as well as after 40, 80, and 120 s of recovery in scramble-siRNA- (upper) and Hap1-siRNA (bottom)-treated cells. The initial fluorescence is indicated as 100%. Scale bar, 5 μm. B, FRAP time course from scramble-siRNA (○)- and Hap1-siRNA (●)-treated cells in A. Fits indicate a τ of 20.1 s (scramble) and 34.6 s (siRNA). C, mean time constant τ of scramble-siRNA-treated cells (scramble) or Hap1-siRNA-treated cells (siRNA). Less time is needed for scramble-siRNA cells to get a fluorescence recovery of Cav1.2-YFP compared with Hap1-siRNA-treated cells (**, p < 0.001; scramble n = 25, siRNA n = 27).
Discussion
Although mainly expressed in the brain, Hap1 is also expressed in endocrine systems that were involved in hormone release (1, 3, 43, 44). Immunofluorescent double staining of mouse pancreas showed that Hap1 is selectively expressed in the insulin-releasing β-cells but not in α-cells and δ-cells that produce glucagon and somatostatin (4). This specific expression in pancreatic β-cells implies that Hap1 is involved in the function of insulin release. Recent studies discovered that mutant mice with selective Hap1 elimination in pancreatic β-cells displayed a significant inhibition of insulin release and glucose tolerance. The same phenomenon was also found in cultured pancreatic β-cell lines Min6 and NIT cells (5). In our studies, we provided new evidence that loss of Hap1 decreased the plasma insulin level in Hap1 knock-out mice and INS-1 cells. Moreover, our mechanistic studies revealed that Hap1 deficiency inhibited the kinetics of insulin exocytosis by reducing the translocation of Cav1.2 to the plasma membrane.
Pancreatic β-cells are electrically excitable and exquisitely sensitive to glucose. The secretory machinery of insulin release from islet β-cells shares many properties with the release of neurotransmitters in neurons, including synaptic vesicle docking, fusion, and release (45, 46). It is known that insulin is produced in pancreatic β-cells, transported by the secretory vesicle, and released from the β-cells by exocytosis. This process requires the fusion of the secretory vesicle membrane with the plasma membrane, followed by release of insulin into the extracellular space to reach the blood circulation (47). In contrast to the exocytosis of neurotransmitters in neurons, insulin release is triggered by Ca2+ influx through L-type Ca2+ channels in pancreatic β-cells (48, 49). L-type Ca2+ channels have been reported to co-localize with insulin-containing vesicles (50). L-type Ca2+ current is the major Ca2+ current subtype in the β-cells (25, 39, 40). All these results indicate that L-type Ca2+ channels play an important role in insulin secretion from pancreatic β-cells. Our studies suggest that Hap1 regulates insulin release through the intracellular trafficking of L-type Ca2+ channels.
Growing evidence suggests that Hap1 may maintain neuronal transmission and neurotrophic functions by regulating intracellular trafficking, recycling, and stabilization of receptors. Hap1 may increase the receptors at the plasma surface via inhibition of lysosomal degradation and enhancing endocytic recycling. For example, Hap1 overexpression increases the activity and surface level of GABAA receptor in the hypothalamus (51). Hap1 modulates the normal membrane level of TrkA by preventing the degradation of internalized TrkA (12). Hap1 can also increase the TrkB level by interacting with Ahi1 (52). Overexpression of Hap1 prevents the trafficking of internalized EGF receptor from early endosomes to lysosomes, resulting in the suppression of EGF receptor degradation (36). InsP3R1, an intracellular Ca2+ release channel, is another membrane receptor that also binds to Hap1 (16). The binding of Htt and Hap1 can influence Ca2+ signaling mediated by InsP3R1 in neurons (15). Our studies found that loss of Hap1 could impair L-type Ca2+ currents and Ca2+ influx. Our data from FRAP and immunofluorescence showed that knockdown of Hap1 reduced the intracellular trafficking and surface expression of Cav1.2. We also demonstrated that there was an interaction between Hap1 and Cav1.2 via immunoprecipitation, which suggested that Cav1.2 could be a new target for the future therapeutic regulation of insulin release. INS-1 cells and even mouse islet β-cells express both Cav1.2 and Cav1.3 channels (25, 53, 54). It remains to be determined whether Hap1 protein interacts with Cav1.3 channels.
Ca2+ is a triggering signal for neurotransmitter and hormone release. Alteration in the density and function of Ca2+ channels in β-cells occurs under pathophysiological conditions. The activity of the β-cell Ca2+ channel can be up- or down-regulated in different animal models of diabetes (27, 55). Ca2+ channel mutation results in β-cell dysfunction that is possibly associated with type 2 diabetes (56, 57). How Cav channel dysfunction contributes to the pathological alterations in β-cells remains to be fully investigated. Further analysis of the Ca2+-dependent exocytosis and endocytosis in pancreatic β-cells from a genetically manipulated mouse model will be necessary for elucidating the underlying mechanism. Both HD patients and mouse models showed abnormal glucose tolerance and insulin secretion (6, 9, 58, 59). It would be interesting to investigate whether impaired Hap1 function is involved in HD pancreatic dysfunction and type 2 diabetes and whether the addition of Hap1 could improve the insulin secretion in β-cells. In addition, there is impaired secretion of various hormones in HD patients and hypothalamic and neuroendocrine changes were seen in HD (60–64). Hap1 dysfunction could also be involved in the endocrine alteration and metabolic dysfunction seen in HD. Our finding that Hap1 regulates Cav channel trafficking to influence insulin release from β-cells provides a new direction to investigate the mechanisms underlying impaired insulin release in a variety of pathological conditions.
Experimental Procedures
Animals
Hap1 knock-out mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Animals were kept under a standardized breeding environment with free water and food. Postnatal day 1 Hap1+/+ (wild type, WT) and Hap1−/− (knock-out, KO) pups were obtained from mice crossed between Hap1+/−.
Radioimmunoassay of Insulin Secretion
Mice were sacrificed by decapitation; the blood was collected and centrifuged to collect plasma. All steps are operating on the ice. The plasma was diluted with PBS, and the basal insulin level was measured using RIA with the DFM 96-type multitube radioimmunoassay counter. For measuring insulin release from INS-1 cells, Hap1-siRNA or Hap1-scramble-treated cells were plated in 6-well plates and maintained at 37 °C in 5% CO2, 95% air-humidified atmosphere for 48 h. The medium in 6-well plates was collected for measuring released insulin using RIA.
Cell Culture and Transfection
The rat insulin-producing INS-1 cells (ATCC) were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1 mm sodium pyruvate, 10 mm HEPES, 50 μm β-mercaptoethanol, 50 units/ml penicillin, and 50 mg/ml streptomycin. Cell cultures were maintained at 37 °C in 5% CO2, 95% air-humidified atmosphere. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, cells were plated onto 30-mm round glass coverslips in a 6-well plate. The following day, cells were transfected with various plasmids. Six hours later, the medium was replaced with complete DMEM, and the transfected cells were maintained in culture overnight. After 48 h, the transfected cells were plated onto 30-mm round glass coverslips and mounted in a Teflon chamber. YFP-HA-Cav1.2, β1b, and α2δ1 plasmids were obtained from the laboratory of Ricardo E. Dolmetsch, Stanford University.
For siRNA knockdown experiments, the following 19-mer oligonucleotide sequences were selected as described previously (12), 5′-GAAGTATGTCCTCCAGCAA-3′. Short hairpin oligonucleotides were designed and inserted into Pgenesil-1 vector (Invitrogen) between BamHI and HindIII sites to construct two plasmids, mCherry-Hap1-siRNA and EGFP-Hap1-siRNA. This shRNA was effective at reducing the expression of Hap1.
Electrophysiology
Whole cell voltage clamp recordings were applied to INS-1 cells with standard techniques. For knockdown studies, INS-1 cells were transfected with 2 μg of scramble or the corresponding rat Hap1-siRNA plasmid also expressed GFP. GFP-positive cells were recorded 48 h after transfection. Whole cell currents were recorded using EPC 10 amplifier (HEKA) with the following external solution (in mm): 20 BaCl2, 120 TEA-Cl, 1 MgCl2, 10 glucose, 10 HEPES, and 0.001 tetradotoxin, pH adjusted to 7.4 with TEA-OH. Pipette solution contained (in mm) the following: 135 CsMeSO3, 5 CsCl, 0.5 EGTA, 1 MgCl2, 4 Mg-ATP, 0.5 Na-GTP, and 10 HEPES, and pH was adjusted to 7.4 with CsOH. Data were filtered at 2 kHz and digitized at 5 kHz. All experiments were performed at room temperature.
Intracellular Ca2+ Measurements
INS-1 cells were loaded with 2 μm ratiometric dye fura-2/AM (Invitrogen) for 30 min at 37 °C in Ca2+-free solution (140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES). Cells were then washed with Ca2+-free solution, and fura-2 loaded into cells was allowed to de-esterify for 30 min at 25 °C. Intracellular Ca2+ was measured using a dual-wavelength excitation (340/380 nm) photometry system on an inverted microscope (TE2000, Nikon, Japan) equipped with a polychromatic xenon light source (TILL photonics, Germany). Briefly, fura-2 was excited at 340 and 380 nm alternatively at a rate of 1 Hz, and the fluorescence emission was first passed through a 415-nm long pass dichroic filter and then monitored at 515 nm with a photodiode controlled by the TILL photometry system and X-Chart extension of Pulse software (HEKA, Lambrecht, Germany). Intracellular Ca2+ concentrations are represented by the ratio of fluorescence emission at the two excitation wavelengths.
Immunofluorescent Labeling
INS-1 cells transfected with YFP-Cav1.2-HA were washed three times with PBS and fixed with 4% paraformaldehyde for 20 min (for non-permeabilization, this step was omitted) and then incubated with primary antibodies to HA antibody (Sigma) at 1:1000 dilution overnight. Cy3-conjugated secondary antibodies (Jackson ImmunoResearch) were used at a dilution of 1:500 at room temperature under dark conditions for 2 h. Photographs were taken with a laser scanning confocal microscope (Olympus FV1000; Tokyo, Japan) using a-plan Apochromat ×60/NA1.42 oil immersion objective and analyzed using Fluoview 5 software (Olympus).
Subcellular Fractionation and Immunoprecipitation
Proteins in cytosol and plasma membrane were prepared using Plasma membrane and cytoplasmic protein extraction kit (Beyotime, Jiangsu, China). Briefly, cells were harvested and then resuspended in 1 ml of buffer A. The nucleus and cell debris were removed from the homogenate by centrifugation at 700 × g for 10 min at 4 °C. The resulting supernatant was centrifuged at 14,000 × g for 30 min at 4 °C. The membrane pellet was solubilized in buffer B for 30 min at 4 °C. Insoluble material was removed by centrifugation at 14,000 × g for 10 min at 4 °C. For immunoblotting, 60 μg of proteins were resolved by 10% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. The membrane was incubated with polyclonal anti-Cav1.2 antibody (1:500, Sigma) overnight at 4 °C, followed by incubation with horseradish peroxidase-conjugated secondary antibody for 2 h. The immunoreactive bands were visualized using an enhanced chemiluminescence kit (Pierce Thermo Scientific; Rockford, IL).
For immunoprecipitation (IP), INS-1 cells were washed twice with PBS and then lysed in chilled Nonidet P-40 buffer (1% Nonidet P-40, 150 mm NaCl, 50 mm Tris-HCl, pH 8.0) containing 100 μm phenylmethylsulfonyl fluoride and Sigma protease inhibitor mixture, followed by incubation for 1 h at 4 °C and subsequent centrifugation (12,000 rpm, 10 min at 4 °C). The supernatant was then subjected to immunoprecipitation with anti-Cav1.2 or anti-Hap1 antibodies overnight at 4 °C, and the immunocomplex was recovered by incubating with 20 μl of 50% protein G-Sepharose beads for 1 h at 4 °C followed by brief centrifugation. The immunoprecipitates were washed three times with lysis buffer. After the final wash, beads were resuspended in gel-loading buffer and boiled for 10 min. The lysates and immunoprecipitates were immunoblotted as described above.
Confocal Microscopy Imaging
Experiments were performed using the Olympus FV1000 laser scanning confocal microscopy system (Olympus, Tokyo, Japan). Coverslips containing INS-1 cells co-expressing VAMP2-pHluorin (PH-VAMP2) and mCherry-Hap1-scramble (mCh-Hap1-scramble) or mCherry-Hap1-siRNA (mCh-Hap1-siRNA) were placed into a perfusion chamber on a stage of an inverted Olympus IX71 microscope. Structural data acquisition was performed in the sequential line mode for the best spatiotemporal reliability. Live cells were examined in a confocal microscope at the cell footprint to better visualize near PM fluorescent spots. The sample area is scanned at resolution of 512 × 512 pixels. pHluorin and mCherry were excited at 488 and 543 nm, respectively. Fluoview imaging software (Olympus, Tokyo) and Matlab software (The MathWorks, Inc.) were used for imaging data analysis. All experiments were performed at 22–25 °C.
FRAP in Live Cells
INS-1 cells were seeded on coverslips and co-transfected with Hap1-siRNA or scramble-siRNA together with Cav1.2-YFP. Forty eight hours later, we mounted coverslips in the experimental chamber at 25 °C. The culture medium was then replaced with standard bathing solution medium. Briefly, the fluorescence of YFP was assessed before and after photobleaching in a timed series. To bleach YFP, the laser was set to 100% for 488 nm, and 5 bleaches were taken. A total of 60 scans was performed at 2-s intervals after bleaching. Photobleaching was performed by full argon laser outputs and focused on the regions of interest. We collected images before and after bleaching and analyzed the recovered fluorescent intensity data. Fluorescence recovery traces were fitted with an exponential curve as shown in Equation 1,
| (Eq. 1) |
where Ai is the amplitude of each component; t is time; and τi is the time constant of each component.
Statistics Analysis
Statistical analyses were carried out using the SigmaPlot 11 software (Systat Software Inc.). All of the data indicate the mean ± S.D., with sample number (n) referring to either coverslip or bouton number. Statistical significance was tested using either Student's t test or analysis of variance. Differences were considered statistically significant with a p value <0.05.
Author Contributions
J. P., J. H., and S. Y. conducted most of the experiments and analyzed the results. C. S. and X. L. conducted immunofluorescence experiments and Western blottings. T. Y. conducted radioimmunoassay for insulin secretion of Hap1 knockout mice. J. H. and H. L. conceived the idea for the project, designed the experiments, analyzed and interpreted the results, and wrote the paper with J. P.
Acknowledgments
We are grateful to Professor Ricardo E. Dolmetsch at Stanford University for the kind gift of YFP-HA-Cav1.2, β1b, α2δ1 plasmids. We thank Z. X. Wu at Huazhong University of Science and Technology for VAMP2-PH constructs. We also acknowledge Professor Xiao-Jiang Li at Emory University for assistance in the preparation of the manuscript.
This work was supported by the National Natural Science Foundation of China Grants 30871311, 31171155, and 31371217 and Research Fund for the Doctoral Program of Higher Education of China Grant 20070487184. The authors declare that they have no conflicts of interest with the contents of this article.
- HD
- Huntington disease
- Cav1.2
- L-type Ca2+ channel 1.2
- InsP3R
- inositol 1,4,5-triphosphate receptor
- IRP
- immediately releasable pool
- RRP
- readily releasable pool
- PM
- plasma membrane
- IP
- immunoprecipitation
- TEA
- tetraethylammonium.
References
- 1.Li X. J., Li S. H., Sharp A. H., Nucifora F. C. Jr., Schilling G., Lanahan A., Worley P., Snyder S. H., and Ross C. A. (1995) A huntingtin-associated protein enriched in brain with implications for pathology. Nature 378, 398–402 [DOI] [PubMed] [Google Scholar]
- 2.Dragatsis I., Dietrich P., and Zeitlin S. (2000) Expression of the Huntingtin-associated protein 1 gene in the developing and adult mouse. Neurosci. Lett. 282, 37–40 [DOI] [PubMed] [Google Scholar]
- 3.Liao M., Shen J., Zhang Y., Li S. H., Li X. J., and Li H. (2005) Immunohistochemical localization of huntingtin-associated protein 1 in endocrine system of the rat. J. Histochem. Cytochem. 53, 1517–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liao M., Chen X., Han J., Yang S., Peng T., and Li H. (2010) Selective expression of Huntingtin-associated protein 1 in β-cells of the rat pancreatic islets. J. Histochem. Cytochem. 58, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cape A., Chen X., Wang C. E., O'Neill A., Lin Y. F., He J., Xu X. S., Yi H., Li H., Li S., and Li X. J. (2012) Loss of huntingtin-associated protein 1 impairs insulin secretion from pancreatic β-cells. Cell. Mol. Life Sci. 69, 1305–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Björkqvist M., Fex M., Renström E., Wierup N., Petersén A., Gil J., Bacos K., Popovic N., Li J. Y., Sundler F., Brundin P., and Mulder H. (2005) The R6/2 transgenic mouse model of Huntington's disease develops diabetes due to deficient β-cell mass and exocytosis. Hum. Mol. Genet. 14, 565–574 [DOI] [PubMed] [Google Scholar]
- 7.Ye C. F., and Li H. (2009) HSP40 ameliorates impairment of insulin secretion by inhibiting huntingtin aggregation in a HD pancreatic β-cell model. Biosci. Biotechnol. Biochem. 73, 1787–1792 [DOI] [PubMed] [Google Scholar]
- 8.Farrer L. A. (1985) Diabetes mellitus in Huntington disease. Clin. Genet. 27, 62–67 [DOI] [PubMed] [Google Scholar]
- 9.Smith R., Bacos K., Fedele V., Soulet D., Walz H. A., Obermüller S., Lindqvist A., Björkqvist M., Klein P., Onnerfjord P., Brundin P., Mulder H., and Li J. Y. (2009) Mutant huntingtin interacts with β-tubulin and disrupts vesicular transport and insulin secretion. Hum. Mol. Genet. 18, 3942–3954 [DOI] [PubMed] [Google Scholar]
- 10.Gauthier L. R., Charrin B. C., Borrell-Pagès M., Dompierre J. P., Rangone H., Cordelières F. P., De Mey J., MacDonald M. E., Lessmann V., Humbert S., and Saudou F. (2004) Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138 [DOI] [PubMed] [Google Scholar]
- 11.McGuire J. R., Rong J., Li S. H., and Li X. J. (2006) Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J. Biol. Chem. 281, 3552–3559 [DOI] [PubMed] [Google Scholar]
- 12.Rong J., McGuire J. R., Fang Z. H., Sheng G., Shin J. Y., Li S. H., and Li X. J. (2006) Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J. Neurosci. 26, 6019–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rong J., Li S., Sheng G., Wu M., Coblitz B., Li M., Fu H., and Li X. J. (2007) 14-3-3 protein interacts with Huntingtin-associated protein 1 and regulates its trafficking. J. Biol. Chem. 282, 4748–4756 [DOI] [PubMed] [Google Scholar]
- 14.Sheng G., Xu X., Lin Y. F., Wang C. E., Rong J., Cheng D., Peng J., Jiang X., Li S. H., and Li X. J. (2008) Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J. Clin. Invest. 118, 2785–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang T. S., Tu H., Orban P. C., Chan E. Y., Hayden M. R., and Bezprozvanny I. (2004) HAP1 facilitates effects of mutant huntingtin on inositol 1,4,5-trisphosphate-induced Ca release in primary culture of striatal medium spiny neurons. Eur. J. Neurosci. 20, 1779–1787 [DOI] [PubMed] [Google Scholar]
- 16.Tang T. S., Tu H., Chan E. Y., Maximov A., Wang Z., Wellington C. L., Hayden M. R., and Bezprozvanny I. (2003) Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burcelin R., Knauf C., and Cani P. D. (2008) Pancreatic α-cell dysfunction in diabetes. Diabetes Metab. 34, S49–S55 [DOI] [PubMed] [Google Scholar]
- 18.MacDonald P. E., and Rorsman P. (2006) Oscillations, intercellular coupling, and insulin secretion in pancreatic β-cells. PLoS Biol. 4, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian G., Tepikin A. V., Tengholm A., and Gylfe E. (2012) cAMP induces stromal interaction molecule 1 (STIM1) puncta but neither Orai1 protein clustering nor store-operated Ca2+ entry (SOCE) in islet cells. J. Biol. Chem. 287, 9862–9872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gandini M. A., Sandoval A., González-Ramírez R., Mori Y., de Waard M., and Felix R. (2011) Functional coupling of Rab3-interacting molecule 1 (RIM1) and L-type Ca2+ channels in insulin release. J. Biol. Chem. 286, 15757–15765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arredouani A., Evans A. M., Ma J., Parrington J., Zhu M. X., and Galione A. (2010) An emerging role for NAADP-mediated Ca2+ signaling in the pancreatic β-cell. Islets 2, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Catterall W. A., Perez-Reyes E., Snutch T. P., and Striessnig J. (2005) International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425 [DOI] [PubMed] [Google Scholar]
- 23.Mears D. (2004) Regulation of insulin secretion in islets of Langerhans by Ca2+ channels. J. Membr. Biol. 200, 57–66 [DOI] [PubMed] [Google Scholar]
- 24.Safayhi H., Haase H., Kramer U., Bihlmayer A., Roenfeldt M., Ammon H. P., Froschmayr M., Cassidy T. N., Morano I., Ahlijanian M. K., and Striessnig J. (1997) L-type calcium channels in insulin-secreting cells: biochemical characterization and phosphorylation in RINm5F cells. Mol. Endocrinol. 11, 619–629 [DOI] [PubMed] [Google Scholar]
- 25.Yang S. N., and Berggren P. O. (2005) Beta-cell CaV channel regulation in physiology and pathophysiology. Am. J. Physiol. Endocrinol. Metab. 288, E16–E28 [DOI] [PubMed] [Google Scholar]
- 26.Iwashima Y., Pugh W., Depaoli A. M., Takeda J., Seino S., Bell G. I., and Polonsky K. S. (1993) Expression of calcium channel mRNAs in rat pancreatic islets and down-regulation after glucose infusion. Diabetes 42, 948–955 [DOI] [PubMed] [Google Scholar]
- 27.Roe M. W., Worley J. F. 3rd., Tokuyama Y., Philipson L. H., Sturis J., Tang J., Dukes I. D., Bell G. I., and Polonsky K. S. (1996) NIDDM is associated with loss of pancreatic β-cell L-type Ca2+ channel activity. Am. J. Physiol. 270, E133–E140 [DOI] [PubMed] [Google Scholar]
- 28.Sharp G. W. (1996) Mechanisms of inhibition of insulin release. Am. J. Physiol. 271, C1781–C1799 [DOI] [PubMed] [Google Scholar]
- 29.Bito M., Tomita T., Komori M., Taogoshi T., Kimura Y., and Kihira K. (2013) The mechanisms of insulin secretion and calcium signaling in pancreatic β-cells exposed to fluoroquinolones. Biol. Pharm. Bull. 36, 31–35 [DOI] [PubMed] [Google Scholar]
- 30.Namkung Y., Skrypnyk N., Jeong M. J., Lee T., Lee M. S., Kim H. L., Chin H., Suh P. G., Kim S. S., and Shin H. S. (2001) Requirement for the L-type Ca2+ channel α(1D) subunit in postnatal pancreatic β-cell generation. J. Clin. Invest. 108, 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulla V., Renström E., Feil R., Feil S., Franklin I., Gjinovci A., Jing X. J., Laux D., Lundquist I., Magnuson M. A., Obermüller S., Olofsson C. S., Salehi A., Wendt A., Klugbauer N., et al. (2003) Impaired insulin secretion and glucose tolerance in β cell-selective Ca(v) 1.2 Ca2+ channel null mice. EMBO J. 22, 3844–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rong J., Li S. H., and Li X. J. (2007) Regulation of intracellular HAP1 trafficking. J. Neurosci. Res. 85, 3025–3029 [DOI] [PubMed] [Google Scholar]
- 33.Li X. J., and Li S. H. (2005) HAP1 and intracellular trafficking. Trends Pharmacol. Sci. 26, 1–3 [DOI] [PubMed] [Google Scholar]
- 34.Wu L. L., and Zhou X. F. (2009) Huntingtin-associated protein 1 and its functions. Cell Adh. Migr. 3, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z., Peng T., Wu H., He J., and Li H. (2015) HAP1 helps to regulate actin-based transport of insulin-containing granules in pancreatic β cells. Histochem. Cell Biol. 144, 39–48 [DOI] [PubMed] [Google Scholar]
- 36.Li S. H., Yu Z. X., Li C. L., Nguyen H. P., Zhou Y. X., Deng C., and Li X. J. (2003) Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J. Neurosci. 23, 6956–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schweizer F. E., Betz H., and Augustine G. J. (1995) From vesicle docking to endocytosis: intermediate reactions of exocytosis. Neuron 14, 689–696 [DOI] [PubMed] [Google Scholar]
- 38.Sankaranarayanan S., and Ryan T. A. (2000) Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat. Cell Biol. 2, 197–204 [DOI] [PubMed] [Google Scholar]
- 39.Sher E., Giovannini F., Codignola A., Passafaro M., Giorgi-Rossi P., Volsen S., Craig P., Davalli A., and Carrera P. (2003) Voltage-operated calcium channel heterogeneity in pancreatic β cells: physiopathological implications. J. Bioenerg. Biomembr. 35, 687–696 [DOI] [PubMed] [Google Scholar]
- 40.Satin L. S. (2000) Localized calcium influx in pancreatic β-cells: its significance for Ca2+-dependent insulin secretion from the islets of Langerhans. Endocrine 13, 251–262 [DOI] [PubMed] [Google Scholar]
- 41.Seino S., Shibasaki T., and Minami K. (2011) Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Invest. 121, 2118–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green E. M., Barrett C. F., Bultynck G., Shamah S. M., and Dolmetsch R. E. (2007) The tumor suppressor eIF3e mediates calcium-dependent internalization of the L-type calcium channel CaV1.2. Neuron 55, 615–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutekunst C. A., Li S. H., Yi H., Ferrante R. J., Li X. J., and Hersch S. M. (1998) The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J. Neurosci. 18, 7674–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin E. J., Kim M., Velier J., Sapp E., Lee H. S., Laforet G., Won L., Chase K., Bhide P. G., Heller A., Aronin N., and Difiglia M. (1999) Analysis of Huntingtin-associated protein 1 in mouse brain and immortalized striatal neurons. J. Comp. Neurol. 403, 421–430 [PubMed] [Google Scholar]
- 45.Wang S., and Hsu S. C. (2006) The molecular mechanisms of the mammalian exocyst complex in exocytosis. Biochem. Soc. Trans. 34, 687–690 [DOI] [PubMed] [Google Scholar]
- 46.Park J. J., and Loh Y. P. (2008) How peptide hormone vesicles are transported to the secretion site for exocytosis. Mol. Endocrinol. 22, 2583–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lang J. (1999) Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur. J. Biochem. 259, 3–17 [DOI] [PubMed] [Google Scholar]
- 48.Zhang H., Kelley W. L., Chamberlain L. H., Burgoyne R. D., Wollheim C. B., and Lang J. (1998) Cysteine-string proteins regulate exocytosis of insulin independent from transmembrane ion fluxes. FEBS Lett. 437, 267–272 [DOI] [PubMed] [Google Scholar]
- 49.Ammälä C., Eliasson L., Bokvist K., Larsson O., Ashcroft F. M., and Rorsman P. (1993) Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J. Physiol. 472, 665–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiser O., Trus M., Hernández A., Renström E., Barg S., Rorsman P., and Atlas D. (1999) The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc. Natl. Acad. Sci. U.S.A. 96, 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheng G., Chang G. Q., Lin J. Y., Yu Z. X., Fang Z. H., Rong J., Lipton S. A., Li S. H., Tong G., Leibowitz S. F., and Li X. J. (2006) Hypothalamic huntingtin-associated protein 1 as a mediator of feeding behavior. Nat. Med. 12, 526–533 [DOI] [PubMed] [Google Scholar]
- 52.Mayer B. J. (2001) SH3 domains: complexity in moderation. J. Cell Sci. 114, 1253–1263 [DOI] [PubMed] [Google Scholar]
- 53.Yang S. N., and Berggren P. O. (2006) The role of voltage-gated calcium channels in pancreatic β-cell physiology and pathophysiology. Endocr. Rev. 27, 621–676 [DOI] [PubMed] [Google Scholar]
- 54.Yang G., Shi Y., Yu J., Li Y., Yu L., Welling A., Hofmann F., Striessnig J., Juntti-Berggren L., Berggren P. O., and Yang S. N. (2015) CaV1.2 and CaV1.3 channel hyperactivation in mouse islet β cells exposed to type 1 diabetic serum. Cell. Mol. Life Sci. 72, 1197–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Splawski I., Timothy K. W., Sharpe L. M., Decher N., Kumar P., Bloise R., Napolitano C., Schwartz P. J., Joseph R. M., Condouris K., Tager-Flusberg H., Priori S. G., Sanguinetti M. C., and Keating M. T. (2004) Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 [DOI] [PubMed] [Google Scholar]
- 56.Yamada Y., Masuda K., Li Q., Ihara Y., Kubota A., Miura T., Nakamura K., Fujii Y., Seino S., and Seino Y. (1995) The structures of the human calcium channel α1 subunit (CACNL1A2) and β subunit (CACNLB3) genes. Genomics 27, 312–319 [DOI] [PubMed] [Google Scholar]
- 57.Yamada Y., Kuroe A., Li Q., Someya Y., Kubota A., Ihara Y., Tsuura Y., and Seino Y. (2001) Genomic variation in pancreatic ion channel genes in Japanese type 2 diabetic patients. Diabetes Metab. Res. Rev. 17, 213–216 [DOI] [PubMed] [Google Scholar]
- 58.Podolsky S., Leopold N. A., and Sax D. S. (1972) Increased frequency of diabetes mellitus in patients with Huntington's chorea. Lancet 1, 1356–1358 [DOI] [PubMed] [Google Scholar]
- 59.Podolsky S., and Leopold N. A. (1977) Abnormal glucose tolerance and arginine tolerance tests in Huntington's disease. Gerontology 23, 55–63 [DOI] [PubMed] [Google Scholar]
- 60.Keogh H. J., Johnson R. H., Nanda R. N., and Sulaiman W. R. (1976) Altered growth hormone release in Huntington's chorea. J. Neurol. Neurosurg. Psychiatry 39, 244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hult S., Soylu R., Björklund T., Belgardt B. F., Mauer J., Brüning J. C., Kirik D., and Petersén Å. (2011) Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab. 13, 428–439 [DOI] [PubMed] [Google Scholar]
- 62.Lavin P. J., Bone I., and Sheridan P. (1981) Studies of hypothalamic function in Huntington's chorea. J. Neurol. Neurosurg. Psychiatry 44, 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saleh N., Moutereau S., Durr A., Krystkowiak P., Azulay J. P., Tranchant C., Broussolle E., Morin F., Bachoud-Lévi A. C., and Maison P. (2009) Neuroendocrine disturbances in Huntington's disease. PLoS One 4, e4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petersén A., and Björkqvist M. (2006) Hypothalamic-endocrine aspects in Huntington's disease. Eur. J. Neurosci. 24, 961–967 [DOI] [PubMed] [Google Scholar]