

Abstract
HIV reverse transcriptase plays a central role in viral replication and requires coordination of both polymerase and RNase H activities. Although this coordination is crucial in viral replication, whether a DNA/RNA hybrid can simultaneously engage both active sites has yet to be determined as structural and kinetic analyses have provided contradictory results. Single nucleotide incorporation and RNase H cleavage were examined using presteady-state kinetics with global data analysis. The results revealed three interconverting reverse transcriptase-DNA/RNA species; 43% were active for both sites simultaneously, 27% showed only polymerase activity, and the remaining 30% were nonproductive. Our data clearly demonstrated that the DNA/RNA hybrid could contact both active sites simultaneously, although the single nucleotide incorporation (105 s−1) was ∼5-fold faster than the cleavage (23 s−1). By using a series of primers with different lengths, we found that a string of at least 4–6 nucleotides downstream of the cleaving site was required for efficient RNA cleavage. This was corroborated by our observations that during processive nucleotide incorporation, sequential rounds of RNA cleavage occurred each time after ∼6 nucleotides were incorporated. More importantly, during processive primer extension, pyrophosphate (PPi) release was rate-limiting so that the average rate of nucleotide incorporation (∼28 s−1) was comparable with that of net RNA cleavage (∼27 nucleotides(s)). Although polymerization is efficient and processive, RNase H is inefficient and periodic. This combination allows the two catalytic centers of HIVRT to work simultaneously at similar speeds without being tightly coupled.
Keywords: cooperativity, DNA polymerase, enzyme kinetics, human immunodeficiency virus (HIV), reverse transcription, RNase H, global data fitting, reverse transcriptase, transient kinetics
Introduction
HIVRT2 is a heterodimer, consisting of two related subunits: the catalytic p66 subunit and a smaller p51 subunit that appears to play only a structural role. The p66 subunit has two active sites, namely the N-terminal polymerase and the C-terminal RNase H, which perform DNA- and RNA-dependent DNApolymerization and RNase H cleavage, respectively. The two active sites are separated by ∼60 Å, spanning 18 base pairs of a DNA/RNA (primer/template) hybrid (1, 2). Despite the long distance, the process of reverse transcription requires a close coordination between the two catalytic centers during minus-strand DNA synthesis. As the polymerase copies the viral RNA genome, the RNase H site degrades the recently copied RNA segments to free the nascent DNA strand so that it may serve as the template for plus-strand DNA synthesis and to facilitate template strand transfer. The fact that three activities of HIVRT evolved to be present in a single protein and that their coupled activities are required during reverse transcription suggested a possible spatial and temporal coordination between the two active sites.
This hypothesis has been supported by several lines of evidence. First, RNase H in homologous enzymes is able to function independently; however, the isolated RNase H domain of HIVRT is inactive, although its nuclease activity could be partially restored by adding various N- or C-terminal extensions (3–8), indicating that other parts of RT were also required for the substrate binding and/or activity of RNase H. Second, the first RNA cleavage event occurs 18 base pairs from the 3′-OH and is 7-fold slower than DNA synthesis but proceeds without a lag phase (2), suggesting that the two reactions can occur simultaneously. Subsequent RNase H cleavage events result in shorter products, but in the presence of a trap to inhibit substrate rebinding only the 18–19-nt products were observed, suggesting that sequential RNase H cleavage events are slow relative to dissociation of the DNA/RNA duplex from the enzyme (9). Finally, a more recent biochemical study suggested that HIVRT can simultaneously engage its DNA/RNA substrate at both active sites based on the observations that small ligands capable of stabilizing the nucleic acid in the polymerase active site only affected the pattern but not the efficiency of RNA cleavage under single-turnover conditions (10).
Despite the abundance of HIVRT structures, only three of them were in complex with a DNA/RNA hybrid, and none of them exhibited an RNase H-competent mode (1, 11, 12). Therefore, the structural basis for the mechanism by which the two catalytic centers might coordinate with each other is still elusive. One study suggested that by positioning the DNA primer strand the “RNase H primer grip” might also help to determine the trajectory of the hybrid substrate around the RNase H active site and thereby play an important role in coupling the two active sites (1). A later biochemical study then revealed that the cleavage specificity by RNase H could be modified by altering the RNase H primer grip (13).
Whether the two active sites of HIVRT are able to act simultaneously is still under debate as structural and biochemical data suggested that primer extension and template degradation may be mutually exclusive and might occur sequentially. An attempt to model a DNA/RNA hybrid into the RNase H active site of HIVRT revealed that even with a severe bending, the substrate was not able to simultaneously occupy the two catalytic centers (14). These structural analyses suggested a “toggling” model, where the bound substrate must toggle between the two active sites of HIVRT, presumably through conformational changes in the enzyme·DNA/RNA complex. The rate of primer extension was determined to be 7-fold faster than that of RNA cleavage monitored simultaneously on a millisecond time scale (2), but the template was not degraded processively in RNA-dependent extension assays (15), both of which are inconsistent with the tight temporal coupling between the two active sites. Moreover, RNA cleavage tended to occur more efficiently when primer extension paused before a hairpin on the RNA template (16–18). NNRTIs (non-nucleoside reverse transcriptase inhibitors) inhibitory to DNA synthesis were able to activate RNA cleavage, presumably through increasing the accessibility of the DNA/RNA hybrid substrate to the RNase H site (19). Based on these observations, it has been proposed that the nucleic acid substrate could only engage one active site at one time, but the data are not compelling.
In the present study, whether a DNA/RNA hybrid can simultaneously engage both active sites was further explored using presteady-state kinetics together with global data analysis to simultaneously examine polymerization and RNase H cleavage during processive polymerization and in single turnover kinetic assays. These studies reveal a novel mechanism of coordinating the two activities by coupling efficient processive polymerization with inefficient and periodic RNase H cleavage.
Results
Kinetics of Simultaneous Nucleotide Incorporation and RNase H Cleavage
To measure nucleotide incorporation and polymerase-dependent RNase H cleavage activities simultaneously, a double-labeled DNA/RNA substrate with a 25-nt primer recessed at the 3′-end on a 45-nt RNA template was designed (d25/r45, Fig. 1A). Because sequence preferences have been observed for DNA 3′-end-directed RNase H cleavage (20), a string of Gs covering the cleavage window was placed in the template to avoid effects due to nucleotide preferences. Primer extension and template degradation were then studied simultaneously by rapid quench-flow methods under single turnover conditions, and both activities were monitored within a single time frame (Fig. 1B). During TTP incorporation, RNA cleavage products mainly accumulated at positions −18 and −17, with minor bands formed at positions −16 and −15. It has been reported that the high resolution site-specific footprinting was indicative of the translocation status of HIVRT (21), so we initially thought that the cleavage pattern observed here may have also reflected translocation stages of HIVRT on the DNA/RNA hybrid before and after nucleotide incorporation. Note that our nomenclature for naming the cleavage site is static (Fig. 1A), so that after primer extension, cleavage 18 base pairs from the new primer terminus resulted in a −17 product, numbered based upon the starting material. This nomenclature was chosen so that a given band is consistently named even when we do not know the primer length during a given cleavage event when measuring both reactions simultaneously (as in Fig. 1). Accordingly, one might expect to see cleavage at −18 before polymerization and −17 after a single TTP incorporation. However, control experiments performed in the absence of nucleotides revealed that the DNA 3′-end-directed RNA cleavage failed to strictly follow the translocation status of RT (Fig. 2A). For substrate d25/r45, cleavage occurred predominately at position −18 but with minor products at −17 and −16. However, when 26-nt primers were used, cleavage products accumulated at 4 distinct positions instead of the two that were predicted for the pre- and post-translocation stages. To further analyze our data, polymerization and cleavage were plotted versus time (Fig. 1, C and D, respectively). Both the incorporation and cleavage data were biphasic, so rates of TTP incorporation and concomitant RNA degradation were estimated by fitting each trace to a double-exponential equation. The results showed that TTP was incorporated at a rate of 107 ± 11 s−1, which was 6∼7-fold faster than the template degradation at 17 ± 2 s−1, derived from the fast phase of each reaction. This difference in rates of incorporation versus cleavage was consistent with our previous data (2). Cleavage rates measured in the absence of nucleotides were determined as 7.7 ± 1.7 s−1 and 10.2 ± 1.9 s−1 for the 25-nt and 26-nt primers, which were a bit slower than the cleavage during TTP incorporation (Figs. 1D and 2B). Rates for the slower phase were ∼0.5 s−1, which could be attributed to DNA/RNA dissociation and rebinding to the enzyme, discussed further below.
FIGURE 1.

Kinetics of nucleotide incorporation and RNase H cleavage. A, the “static” nomenclature for naming the cleavage sites are shown in the schematic; numbers refer to the distance from the primer terminus before polymerization. The sequence of the double-labeled DNA/RNA hybrid is shown. The blue oval represents HIVRT, where POL and RNH denote the polymerase and RNase H active sites, respectively. Positions of cleavage sites are counting from the 3′-terminus of the DNA primer, e.g. −18 means the cleavage site is 18 nucleotides upstream of the 3′-end of the DNA primer at the start of the reaction. B, to explore the potential coordination between the two active sites, polymerase (lower panel) and RNase H (upper panel) activities were measured simultaneously by preincubating 175 nm HIVRT with 75 nm double-labeled DNA/RNA hybrid and then mixing with 100 μm TTP or ddTTP in the presence of 10 mm Mg2+. Reactions were stopped by adding 0.5 m EDTA at various time points up to 10 s. The 45-nt RNA template and RNase H cleavages at positions −18 to −15 are marked as are positions of the 25-nt primer and extended products. Incorporation (C) and cleavage (D) data were analyzed by conventional fitting. The insets better display data points within 1 s. Nucleotide incorporation was fit to the double-exponential equation to derive rates of TTP or ddTTP incorporation as 107 ± 11 s−1 and 5 ± 0.3 s−1 for the fast phase and 2 ± 0.8 s−1 and 0.5 ± 0.1 s−1 for the slower phase, respectively. Similarly, rates of RNA cleavage during TTP or ddTTP incorporation were determined to be 17 ± 2 s−1 and 12 ± 0.7 s−1 for the fast phase and 0.5 ± 0.1 s−1 and 0.4 ± 0.04 s−1 for the slower phase.
FIGURE 2.

RNA cleavage of substrates before or after primer extension. A, the 45-nt RNA template (r45) was annealed to 25-nt, 26-nt, or 26ddT (26-nt terminated with ddT) primers to mimic substrates before or after nucleotides (TTP or ddTTP) incorporation. An RNA cleavage assay for each substrate was performed as mentioned in Figs. 3 and 4. The 45-nt RNA template and RNase H cleavages at positions −18 to −15 are marked. B, the data showing the time dependence of RNA cleavage of various DNA/RNA hybrids were analyzed by conventional fitting; the inset better displays data points <1 s. Cleavage data were fit to the double-exponential equation to derive rates measured with d25/r45, d26/r45, and 26ddT/r45 as 7.7 ± 1.7 s−1, 10.2 ± 1.9 s−1, and 12 ± 3 s−1 for the fast phase and 0.3 ± 0.1 s−1, 0.4 ± 0.1 s−1, and 0.5 ± 0.1 s−1 for the slower phase, respectively.
Incorporation and cleavage activities were observed simultaneously, but this observation did not exclude a sequential polymerization-hydrolysis model because incorporation was faster and, therefore, could have been completed before the RNA cleavage. To test this scenario we explored RNA cleavage activity during ddTTP incorporation, which was much slower than TTP incorporation. A similar cleavage pattern was observed, with the major or minor products accumulating at positions −18 and −17 or at positions −16 and −15, respectively (Fig. 1B, right panel). The rate of ddTTP incorporation (5 ± 0.3 s−1) was determined to be much slower than TTP incorporation (107 ± 11 s−1) but was slightly slower than the RNA cleavage (12 ± 0.7 s−1). Concomitant polymerase and RNase H activities were still observed even if they had comparable rates, suggesting that the two active sites were able to function simultaneously and independently. That is, the DNA/RNA duplex can simultaneously engage both active sites, and the two activities occur independently. It is important to note that in the single turnover experiment, if the two reactions had occurred sequentially, one would have expected to see a lag in the second reaction that is a function of the rate of the first reaction, but no lag was observed.
Global Data Fitting of Nucleotide Incorporation and RNase H Cleavage
Conventional fitting of the above data provided some evidence that the two catalytic centers of HIVRT work independently. However, conventional analyses were unable to rule out the possibility that the simultaneous activities observed previously were due to mixed enzyme states where one enzyme catalyzes polymerization, whereas another catalyzes RNase H hydrolysis. Conventional fitting largely ignores the amplitude of the reaction, whereas information to distinguish whether a single enzyme species catalyzes both reactions simultaneously lies in analysis of the relative amplitudes of the reactions. The inherent relationships between the rates and amplitudes of the incorporation and cleavage reactions provide more information to determine whether both reactions occur simultaneously. This can be investigated using global data fitting. Global data analysis was first performed for the four experiments measuring TTP incorporation (Fig. 1B, bottom left), concomitant RNA cleavage (Fig. 1B, upper left), and RNase H activities of RT against substrates before or after (Fig. 2A) primer extension.
Data were fit using a minimal model shown in Scheme 1, as described under “Experimental Procedures.” Distinct from the conventional data fitting where the concentration of products of various lengths was summed to define the net reaction rate, for global fitting each band was quantified and analyzed individually. Fig. 3A shows the time course of primer extension during TTP incorporation, whereas Fig. 3B shows the simultaneous accumulation of RNase H cleavage products. Fig. 3, C and D, show the time course of RNase H cleavage of substrates that represent the state before and after primer extension, respectively (25- and 26-nt primers). The smooth lines represent the global fit to the entire data set according to Scheme 1.
SCHEME 1.

Simultaneous TTP incorporation and RNA cleavage. A minimal model for simultaneous TTP incorporation and RNA cleavage during a single turnover is shown. Three different RT·DNA/RNA states were identified and included in the model: XD25R45 (inactive), YD25R45 (polymerase (Pol) active only), and ED25R45 (polymerase and RNase H both active). These three species underwent slow but reversible inter-conversion. Both YD25R45 and ED25R45 could extend the 25-nt substrate to the 27-nt product (after one correct base and one mismatch incorporation). RNA cleavage was able to occur before (ED25R45) or after (ED26R45) primer extension, yielding products with various lengths ranging from 35-nt to 38-nt. To describe the complicated process as completely as possible, secondary cuts (e.g. ED25R38 to ED25R37) and nucleotide incorporation after template degradation (e.g. ED25R38 to ED26R38) were also included in the model. Rate constants for each step are shown in the model and labeled by kn (for n = 1–13).
FIGURE 3.

Global data fitting of TTP incorporation and RNase H cleavage. Data for TTP incorporation (A) and concomitant RNase H cleavage (B) were analyzed by global data fitting. Two control experiments were performed to measure the RNase H activities of HIVRT against substrates before (C) or after (D) primer extension. Briefly, preformed RT·DNA/RNA complexes (175 nm HIVRT and 75 nm d25/r45 (C) or d26/r45 (D)) were rapidly mixed with 10 mm Mg2+ in the absence of any nucleotides. Each band was quantified individually so more information could be derived from the global fitting. Color codes are as follows. A, 25-nt primer, red; 26-nt product, green; 27-nt product, blue. B–D, 45-nt RNA template, red; cleavage at position −18 (38-nt), green; cleavage at position −17 (37-nt), blue; cleavage at position −16 (36-nt), yellow; cleavage at position −15 (35-nt), cyan. Note that because the data are displayed on a logarithmic timescale, the zero time point, which was included in the data fitting, is not shown. All the data were fit to a single model using KinTek Explorer to derive the rate constants summarized in Scheme 1 and Table 1. Smooth lines in each panel represent the best global fit of all four experiments.
The comprehensive mechanism showed in Scheme 1 was derived as a minimal model to account for the data shown in Fig. 3. In this model, we allow for RNase H cleavage before and after polymerization to account for simultaneous reactions from the productive ED25R45 complex. We include an inactive state (XD25R45) and a state that only catalyzes polymerization (YD25R45) to account for the lower reaction amplitudes. Although the model shown in Scheme 1 may appear to be complex, rates of the same reactions from different states were linked to be identical, thereby reducing the complexity and number of unknowns so that the parameters are all well constrained based on confidence contour analysis (22). Our data clearly showed that the majority of species represent the active form of RT·DNA/RNA complexes that could perform both activities simultaneously (ED25R45).
It is evident from the data that neither the 25-nt primer (Fig. 3A, red curve) nor the 45-nt RNA template (Fig. 3B, red curve) decayed with a single fast rate, suggesting the existence of a nonproductive form of RT·DNA/RNA complexes (XD25R45) that slowly converts to the active state accounting for the slow phase. The amplitude of the fast phase of decay of the 25-nt species (due to polymerization) was approximately the same as the sum of amplitudes of the fast and slower phases of 45-nt depletion (RNase H), suggesting that a fraction of polymerase-competent RT·DNA/RNA complexes was not active for RNase H cleavage (species YD25R45) and the slower phase of 45-nt RNA consumption was due to the delayed switch from the inactive form (YD25R45) to the active form (ED25R45).
Apparent equilibria between the three species were determined by the ratios of the amplitudes; the ratio of amplitudes of fast and slower phases of TTP incorporation determined the equilibrium between the polymerase active and inactive forms (YD25R45 + ED25R45, 70%; XD25R45, 30%), whereas the ratio of amplitudes of fast and slower phases of RNA cleavage determined the equilibrium between the bifunctional and polymerase-only forms (ED25R45, 43%; YD25R45, 27%). Rates of conversions among those species were listed in Scheme 1 and Table 1. The results showed that conversions between the active and inactive forms were very slow, with the forward and reverse rates of ∼0.014 and ∼0.012 s−1, respectively. The polymerase-only form (YD25R45) switched to the bi-functional form (ED25R45) at a rate of 0.5 s−1, which was in good agreement with the rate of slower phase (0.5 ± 0.1 s−1) obtained from the conventional fitting of the cleavage data (Fig. 1D). Incorporation rates were determined as 105 s−1 for correct base pairing and 1.4 s−1 for formation of a mismatch (27-mer). Rates of RNA cleavage before primer extension were 20 s−1 at position −18 and 3 s−1 at position −17, consistent with the strong bias toward −18 cleavage observed in the control experiment (Fig. 2). Rates of RNA cleavage after primer extension were 5, 4, 2.5, and 1.5 s−1 for cleavages at positions −18 to −15, respectively. The sum of all rates in a branched pathway yielded net rates of RNA cleavage before and after primer extension as 23 and 13 s−1, respectively. Therefore, the observed rate of 17 ± 2 s−1 determined from the conventional fitting (Fig. 1D) appears to be an average rate combining these two reactions. Secondary cuts also occurred, as cleavage products corresponding to position −18 increased with time at the beginning but eventually decreased (Fig. 3, B and D, green curves), the rate of which, however, was much slower (∼0.05 s−1), suggesting that it was inefficient.
TABLE 1.
Kinetic parameters governing nucleotide incorporation and RNase H cleavage
The lower and upper limits of each rate constant derived from confidence contour analysis of the simulation are included in the parentheses. The nomenclature of rate constants was illustrated in Schemes 1 and 2.
| Nucleotides | k1 | k2 | k3 | k4 | k5 | k6 | k7 | k8 | k9 | k10 | k11 | k12 | k13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | s−1 | |
| TTP | 0.014 (0.009∼0.05)a | 0.012 (0.008∼0.04)a | 0.3 (0.18∼0.36) | 0.5 (0.30∼0.63) | 105 (67∼152) | 1.4 (0.4∼2.5)b | 20 (8∼24) | 3 (1.3∼4.9) | 0.05 (0.007∼0.09)b | 5 (3.6∼7.4) | 4 (3.4∼5.6) | 2.5 (1.9∼3.1) | 1.5 (0.9∼2.2) |
| ddTTP | 0.07 (0.04∼0.09) | 0.04 (0.02∼0.05) | 0.23 (0.18∼0.30) | 0.43 (0.33∼0.56) | 5 (4.9∼6.2) | 9.6 (8.6∼10.9) | 2.2 (1.6∼2.7) | 0.01 (≤0.03)b | 0.05 (0.03∼0.06) | 1.8 (0.9∼2.8) | 2.4 (1.6∼3.8) | 2 (1.3∼3.1) | 2.2 (1.4∼3.5) |
a Individual rate constants k1 and k2 were not well defined by the data, as illustrated by the relatively wide range of values. The product to define K1k2, however, was well constrained.
b Rate constants governing those slow reactions were not well defined by the data. Therefore, either a wide range or limit at one side is specified.
A similar data analysis was performed for RNA cleavage during ddTTP incorporation (Fig. 4) to explore the effect of a slower polymerization rate on the RNase H activity. Rate constants governing the equilibrium between various RT·DNA/RNA complexes, nucleotide incorporation, and RNA cleavage were derived from the global data fitting and summarized in Scheme 2 and Table 1. Similar to our analysis of TTP incorporation, the nonproductive (37%), polymerase-competent only (22%), and bi-functional species (41%) of RT·DNA/RNA complexes were also identified. The rate of ddTTP incorporation was determined as 5 s−1, which was consistent with the number obtained from conventional fitting (5 ± 0.3 s−1). The rate of RNA cleavage before ddTTP incorporation was 11.8 s−1, equal to the sum of 9.6 and 2.2 s−1 to form −18 and −17 cleavage products, respectively. A net rate of 8.4 s−1 was determined for RNA cleavage after primer extension, with comparable rates derived for cleavages at various positions (−18, 1.8 s−1; −17, 2.4 s−1; −16, 2 s−1; −15, 2.2 s−1). The cleavage rates were also consistent with the value (12 ± 0.7 s−1) derived from the conventional fitting (Fig. 1D). Again, the secondary cuts (those acting on the products of the first cleavage) occurred at very slow rates of ∼0.01 and 0.05 s−1, respectively.
FIGURE 4.

Global data fitting of ddTTP incorporation and RNase H cleavage. Data showing ddTTP incorporation (A) and concomitant RNase H cleavage (B) were analyzed by global data fitting. Two control experiments were performed to measure the RNase H activities of HIVRT against substrates before (C) or after (D) primer extension. Briefly, preformed RT·DNA/RNA complexes (175 nm HIVRT and 75 nm d25/r45 (C) or d26ddT/r45 (D) were rapidly mixed with 10 mm Mg2+ in the absence of any nucleotides. All the data were fit to a single model using KinTek Explorer to derive rate constants summarized in Scheme 2 and Table 1. Smooth lines in each panel represent the best fit to the model. Each band was quantified individually so more information can be derived from the global fitting. The same color codes were used here as in Fig. 3. Note that because the data are displayed on a logarithmic timescale, the zero time point, which was included in the data fitting, is not shown.
SCHEME 2.

Simultaneous ddTTP incorporation and RNA cleavage. The minimal model for simultaneous ddTTP incorporation and RNA cleavage is shown. Three different RT·DNA/RNA complexes were identified and included in the model: XD25R45 (inactive), YD25R45 (polymerase (Pol) active only), and ED25R45 (polymerase and RNase H both active), which underwent slow but reversible inter-conversion. Both YD25R45 and ED25R45 could extend the 25-nt substrate to the dideoxy-terminated 26-nt product (mismatch extension was blocked). RNA cleavage was able to occur before (ED25R45) or after primer extension (ED26ddR45), yielding products with various lengths ranging from 35-nt to 38-nt. To describe the complicated process as completely as possible, secondary cuts (e.g. ED25R38 to ED25R37) and nucleotide incorporation after template degradation (e.g. ED25R38 to ED26ddR38) were also included in the model. Rate constants for each step are shown in the model and labeled by kn (for n = 1–13).
Conventional fitting and global analysis largely agreed. The only exception was that the rate constant governing the conversion of XD25R45 to YD25R45 (∼0.012 s−1, k2 in Scheme 1) was much slower than the rate of slower phase (2 ± 0.8 s−1) derived from the conventional fitting of the incorporation data (Fig. 1C). We propose that the slower phase of TTP incorporation might mainly reflect the dissociation and rebinding of the DNA/RNA hybrid substrate (∼0.3s−1); this was supported by the fact that the second slower phase almost disappeared when heparin trap was added to prevent rebinding (data not shown). This rationale also explained why individual parameters k1 and k2 (Scheme 1) were not well constrained in the confidence contour analysis, although their ratio was constant and accurately determined by the simulation-based data fitting (Table 1). During ddTTP incorporation, the corresponding rate of conversion (XD25R45 to YD25R45) was ∼0.04 s−1 (k2 in Scheme 2) with a shorter half-time (∼15 s) compared with TTP, and therefore, the rate constant was better defined by the data and was well constrained. On the other hand, the slower phase of RNA cleavages (Fig. 1D) still existed even in the presence of heparin albeit with a slightly reduced amplitude (data not shown), suggesting that the second phase probably involved substrate dissociation and rebinding as well as the conversion to the RNase H competent species (YD25R45 to ED25R45). Because the former (∼0.3 s−1) was defined by the burst experiment (data not shown) and locked in the simulation, parameters governing this conversion were well constrained in the confidence contour analysis. Actually, most of the parameters were well defined in our models, as illustrated by the narrow ranges of rate constants listed in Table 1. A few exceptions were observed for slow reactions, including the mismatched extension and secondary RNase H cleavages.
Our global data analysis demonstrated that nucleotide incorporation and RNA cleavage occur simultaneously under single-turnover conditions, and we have defined the largest fraction of RT·DNA/RNA complexes had the substrate engaged at both active sites. Also, RNA cleavage could take place at the same time as nucleotide incorporation even if rates of the two activities were comparable, which disproved the sequential model where one active center acted faster and, therefore, accomplished its tasks before the second catalytic center. Moreover, we show the coincident rather than sequential appearance of multiple cleavage bands, providing evidence for some plasticity in the location of the cleavage site. For example, during incorporation of either TTP or ddTTP, we see the nearly equal formation of −18 and −17 cleavage products but see no evidence for a sequential cleavage at position −18 before and at −17 after nucleotide incorporation (Fig. 1B). Note that our numbering system is fixed relative to the starting material before polymerization so that one might expect to see cleavage at −18 before polymerization and −17 after a single incorporation.
Effects of Small Ligands on Polymerase-dependent RNase H Cleavage
Previous structural analyses have suggested a toggling model, where the bound substrate must toggle between two active sites of HIVRT, possibly through conformational changes in the enzyme (1, 11, 12, 14). However, a recent biochemical study provided data to argue against this mechanism based on observations that small molecule ligands capable of stabilizing the nucleic acid in the polymerase active site only affected the pattern but not the efficiency of RNA cleavage under single-turnover conditions (10). This issue was revisited here as a quantitative analysis of the previously published cleavage data was missing. In addition, our study performed with the substrate without sequence bias may be more definitive.
The pyrophosphate analogue phosphonoformic acid (PFA) and next correct nucleotide (with dideoxy-terminated primers) were previously reported to be able to stabilize the pre- or post-translocated RT-DNA complexes (21, 23) to lock the nucleic acid substrate at the DNA polymerase active site. Effects of these ligands on polymerase-dependent RNase H activities were explored with various DNA/RNA hybrids containing 25-nt or 26-nt primers in the absence or presence of ligands (Fig. 5, A and B). When 25-nt primers were examined, predominant cleavage products accumulated at position −18 in the absence of a ligand. PFA (a PPi analog) shifted more than half of the cleavage events toward position −19, whereas a saturating concentration of TTP (with a ddAMP-terminated primer) biased the cleavage toward position −18, with minor products formed at position −17 (Fig. 5A). Despite different cleavage patterns, the cleavage efficiencies remained almost unchanged by the ligands (Fig. 5C). Rates of cleavage in the absence of ligands in the presence of PFA or using ddAMP-terminated primer with TTP were determined as 11.3 ± 1.4 s−1, 9.4 ± 0.8 s−1, and 13.4 ± 3.5 s−1 for the fast phase and 0.4 ± 0.07 s−1, 0.4 ± 0.06 s−1, and 0.6 ± 0.2 s−1 for the slower phase. The data clearly disagreed with the toggling mechanism postulating that the engagement of the substrate at the polymerase active site will lead to the exclusive cleavage at a single position in the RNase H site. Rather, multiple cleavage bands appeared that differed in comparing 25- and 26-nt primers. We also noted slower cleavage events, which we attributed to the conversion of YD25R45 to ED25R45 (Scheme 1 and 2), the rate of which also remained the same, suggesting that ligands binding to the polymerase site did not affect the rates of conversion from nonproductive to productive states.
FIGURE 5.

Effects of small polymerase-site ligands on RNase H activity. RNA cleavage assays were performed by mixing preformed RT·DNA/RNA complexes (175 nm HIVRT and 75 nm d25(ddA)/r45 (A) or d26(ddT)/r45 (B)) with 10 mm Mg2+ with or without small ligands. A, RNase H activities were studied in the absence of any ligands, in the presence of 1 mm PFA, or using ddAMP-terminated primer with the next correct nucleotide (0.5 mm TTP). Reactions were stopped by adding of 0.5 m EDTA at various time points up to 10 s. The 45-nt RNA template and RNase H cleavages at positions −19 to −17 are indicated. B, similar experiments using 26-nt primers were performed in the absence of any ligands, in the presence of 1 mm PFA, or using ddTMP-terminated primer with the next correct nucleotide (0.5 mm dCTP). The 45-nt RNA template and RNase H cleavages at positions −18 to −15 are indicated as well. Cleavage data obtained with d25(ddA)/r45 (C) or d26(ddT)/r45 (D) hybrids were analyzed by conventional data fitting. The insets better display data points up to 1 s. C, cleavage data were fit to the double-exponential equation to derive cutting rates measured in the absence of ligands, in the presence of PFA, or using 25ddA with TTP as 11.3 ± 1.4 s−1, 9.4 ± 0.8 s−1, and 13.4 ± 3.5 s−1 for the fast phase as well as 0.4 ± 0.07s−1, 0.4 ± 0.06 s−1, and 0.6 ± 0.2 s−1 for the slower phase. D, similarly, rates of RNA cleavage using 26-nt primers were determined as 10.2 ± 1.9 s−1, 8.8 ± 0.7 s−1, and 10.3 ± 1.9 s−1 for the fast phase as well as 0.4 ± 0.13 s−1, 0.3 ± 0.08 s−1, and 0.5 ± 0.13 s−1 for the slower phase.
Similar conclusions can be drawn from the data obtained with DNA/RNA hybrids containing 26-nt primers, although different cleavage patterns were observed (Fig. 5, B and D). The DNA 3′-end-directed RNA cleavage failed to strictly follow the translocation status of RT when 26-nt primers were used; inthe absence of ligands, cleavage products accumulated at four distinct positions (from −18 to −15) instead of two that are characteristic of the pre- and post-translocation stages. In the presence of PFA, positions of cuts reduced to −18 and −17, whereas the next cognate nucleotide (dCTP) was unable to lock RT·DNA/RNA complexes at the post-translocation stage. Rather, the RNase H yielded two major bands at positions −17 and −16. Consequently, RNA cleavage assays are not an ideal method to study translocation dynamics of RT on DNA/RNA hybrids even if sequence biases are avoided as in this study. Similarly, rates of RNA cleavage were not affected by the ligands, which were 10.2 ± 1.9 s−1, 8.8 ± 0.7 s−1, and 10.3 ± 1.9 s−1 for the fast phase as well as 0.4 ± 0.13 s−1, 0.3 ± 0.08 s−1, and 0.5 ± 0.13 s−1 for the slower phase (Fig. 5D).
Effects of Mutations Causing Impaired TTP Induced Conformational Change on RNase H Activity
Previous studies of HIVRT revealed that the binding of the correct nucleotide could induce large conformational changes in structure from an open to a closed state (24, 25), which was later shown to govern the specificity by HIVRT (26). Whether this crucial conformational change at the polymerase domain affects RNase H activity and/or specificity remains unknown. Here, the activity and specificity of RNA cleavage were examined using three RT mutants (W71A, W71D, and R72A) that we show impair the nucleotide induced isomerization step. The kinetic characterization of the R72A mutant is shown in Fig. 6 as an example. Briefly, quench-flow and stopped-flow experiments measuring nucleotide binding, incorporation, and PPi release were performed and analyzed globally to define the kinetic pathway of nucleotide incorporation by each mutant (26). The extent to which each mutation affected the conformational change step was determined by evaluating the change of the rate and equilibrium constants governing the isomerization step (K2 = k2/k−2). The results revealed that the R72A mutation had the most adverse effect and impaired the isomerization step by 170-fold. Similarly, W71A and W71D mutations were revealed to disfavor the conformational change step by 7- and 14-fold, respectively (Table 2).
FIGURE 6.
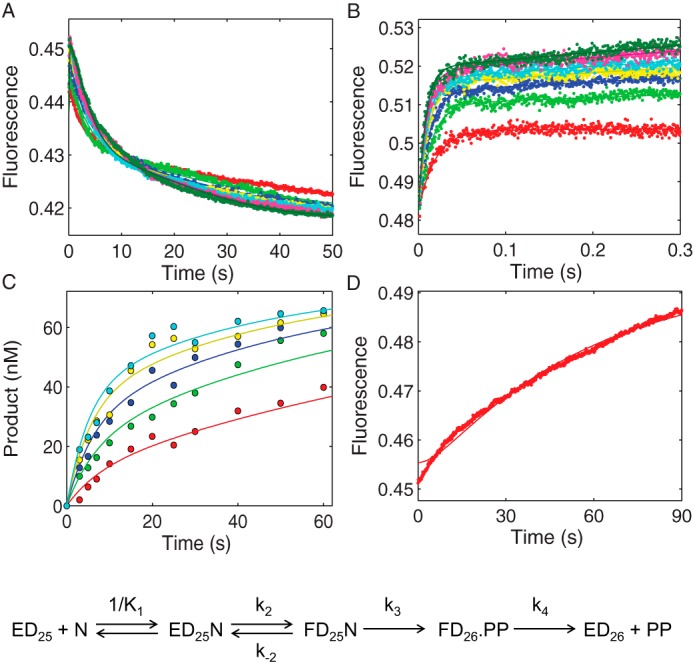
Kinetic characterization of MDCC-labeled HIVRT R72A mutant by global analysis of TTP binding, incorporation, and PPi release. A, the time dependence of the fluorescence change during TTP incorporation was monitored after mixing various concentrations of TTP (5, 10, 20, 30, 40, 60, 80, 100, and 150 μm) with a preformed enzyme·DNA/RNA complex (100 nm MDCC-labeled HIVRT_R72A and 150 nm d25/r45). B, TTP-induced conformational change was measured with dideoxy-terminated primer to prohibit the chemistry step. Various concentrations of TTP (20, 30, 40, 60, 80, 100, 150, and 200 μm) were mixed with a preformed enzyme·DNAdd/RNA complex (100 nm MDCC-labeled HIVRT_R72A and 150 nm d25ddA/r45). C, the concentration dependence of TTP incorporation was measured using the manual quench method. Various concentrations of TTP (5, 12.5, 25, 50, and 100 μm) were mixed with a preformed enzyme·DNA/RNA complex (175 nm MDCC-labeled HIVRT_R72A and 75 nm d25/r45). D, rate of PPi release during TTP incorporation was monitored by rapidly mixing 25 μm TTP with a preformed enzyme·DNA/RNA complex (150 nm MDCC-labeled HIVRT_R72A and 100 nm d25/r45) in the presence of 1.5 μm MDCC-PBP and 0.6 μm pyrophosphatase. Fluorescence was observed by excitation of MDCC at 425 nm and monitoring emission with a 475-nm band-pass filter with a 50-nm bandwidth. Global data analysis of all four experiments was performed to derive rate constants governing TTP binding and incorporation. The TTP ground-state binding constant (1/K1) was determined as 92 ± 3 μm. The forward (k2) and reverse (k−2) rates for the conformational change were 127 ± 2 and 32 ± 1 s−1, which defined a K2 value of 4 ± 0.2 that was 170-fold smaller than that of WT HIVRT-MDCC (690 ± 67) (35). Rates of chemistry and PPi release were determined as 0.18 ± 0.003 and 0.04 ± 0.008 s−1, respectively.
TABLE 2.
Kinetic parameters governing TTP binding and incorporation by three HIVRT mutants
| HIVRT mutants | 1/K1 | k2 | k−2 | K2a | Fold changeb | k3 | k4 |
|---|---|---|---|---|---|---|---|
| μm | s−1 | s−1 | s−1 | s−1 | |||
| W71A | 280 ± 20 | 1330 ± 80 | 14 ± 1 | 95 ± 7 | 7 ± 1 | 21 ± 0.2 | 500c |
| W71D | 200 ± 10 | 570 ± 20 | 12 ± 1 | 48 ± 3 | 14 ± 2 | 21 ± 0.3 | 500c |
| R72A | 92 ± 3 | 127 ± 2 | 32 ± 1 | 4 ± 0.2 | 170 ± 19 | 0.18 ± 0.003 | 0.04 ± 0.008 |
a For the data reported here, confidence contours were sufficiently symmetrical so we report only the standard error derived from the nonlinear regression.
b Fold change is defined by the K2 value of WT HIVRT-MDCC (690 ± 67) (35) divided by the K2 value of each mutant.
c Rate of pyrophosphate release was fixed at a nominal value of 500 s−1 for simplicity in the global fitting to derive the remaining rate constants.
Despite the notable influences on the nucleotide-induced conformational change, these mutations had modest or no effect on the activity and specificity of RNA cleavage (Fig. 7). Similar to the WT protein, all the mutants yielded one major band corresponding to the cutting at position −18 (Fig. 7A). However, observable cleavage products also accumulated at various positions, including −19, −17, −16, and −15. This was more evident for W71A and W71D mutants, implying these two RT mutants were probably less rigidly locked on the DNA/RNA hybrid. In our quantitative analysis, the W71A and W71D mutants showed nearly unchanged cleavage activities compared with the WT protein, with cleavage rates determined as 7.7 ± 1.3 s−1 and 8.1 ± 0.9 s−1 for the faster phase as well as 0.2 ± 0.07 s−1 and 0.3 ± 0.04 s−1 for the slower phase (Fig. 7B). The rate of the faster phase for R72A mutant was slightly reduced at 6.3 ± 1.2 s−1. Although the rate of the slower phase remained the same (0.3 ± 0.1 s−1), its amplitude decreased significantly. As analyzed above (Scheme 1 and 2), the second slower phase corresponded to a conversion from an RNase H inactive form (YD25R45) to the active form (ED25R45), so R72A mutant might impair this process but did not affect the RNase H activity directly. Consequently, the nucleotide-induced conformational change might play a role in structural rearrangements involved in conversions between the “pol-only” and bifunctional species of RT·DNA/RNA complexes but did not directly affect the RNase H activity. These results further supported our “working simultaneously” model and denied the competing model, in which the two active sites compete with each other to engage substrates, and therefore, impaired polymerization activity should have led to enhanced RNase H activity.
FIGURE 7.

Effects of mutations causing impaired TTP induced conformational change on RNase H activity. A, RNA cleavage assays were performed by rapidly mixing preformed RT·DNA/RNA complexes (175 nm HIVRT_W71A, W71D, or R72A mutants and 75 nm d25/r45 hybrid) with 10 mm Mg2+. Reactions were stopped by the addition of 0.5 m EDTA at various time points in 10 s. The 45-nt RNA template and RNase H cleavages at positions −18 to −15 were marked. B, the data of RNA cleavage by various RT mutants were analyzed by conventional fitting. The insets better display data points within 1 s. Cleavage data were fit to the double-exponential equation to derive rates of RNA cleavage by W71A, W71D, or R72A mutants as 7.7 ± 1.3 s−1, 8.1 ± 0.9 s−1, and 6.3 ± 1.2 s−1 for the fast phase as well as 0.2 ± 0.07 s−1, 0.3 ± 0.04 s−1, and 0.3 ± 0.1 s−1 for the slower phase.
Efficient RNase H Cleavage Requires at least Four Nucleotides Downstream of the Cutting Site
We next examined how the two active sites of HIVRT work simultaneously but at different speeds. Previous studies have suggested that the RNase H activity of HIVRT acted as an endonuclease with partial 3′-5′ processivity (27, 28), which was consistent with our observations that the secondary cuts (38–37-mer, ∼0.05 s−1) were 30–380-fold slower than the primary cuts (1.5 to 20 s−1, Scheme 1). Based on these results, we postulated that a minimum length of RNA strand extending from the 3′-end to the cutting site was required for efficient DNA 3′-end-directed RNA cleavages by ensuring a proper alignment of the hybrid substrate near the RNase H active site. Under this scenario, the processive RNA cleavage was largely prohibited and a next round of cleavage would occur only when the cutting site shifted toward the 5′-end of the RNA strand after multiple nucleotide incorporation events that could regenerate a downstream RNA strand long enough for the proper binding of the DNA/RNA duplex at the RNase H site.
To test this hypothesis, a series of DNA/RNA hybrids was made by annealing primers with various lengths onto a radioactive-labeled 45-nt RNA template with a predefined semi-random sequence (Fig. 8B). RNA cleavage assays were then performed with these hybrids. When the d25rd/r45rd hybrid was examined, two major cleavage products were noticed at position +7 and +8. Note that a new nomenclature of cutting positions was applied here to better illustrate the number of nucleotides extending from the 3′-end of the RNA strand to the cleavage site. Theoretically, shortening the primer by one nucleotide will shift the cleavage site accordingly by one nucleotide toward the 3′-end of the RNA strand. The real data, however, did not follow this rule strictly (Fig. 8, A and B). Shortening the primer by 1–3 nucleotides resulted in cleavages at positions +4 and +5 but with different biases. For the d24rd/r45rd hybrid, cleavages products accumulated almost exclusively at position +5, whereas experiments performed with d23rd/r45rd and d22rd/r45rd hybrids showed a strong bias toward +4 cleavages. Further shortening of the primer by 1 or 2 nucleotides (21- or 20-mer) nearly eliminated the cleavage activity, yielding faint bands at positions +4 and +5 as well as almost negligible bands at position +2. These data supported the idea that at least four nucleotides downstream of the cutting site (toward the 3′-end of the RNA strand) were necessary for efficient RNA cleavage to occur. As shown in Fig. 8C, cleavage data were fit to the single-exponential equation to derive cleavage rates as 5 ± 0.4 s−1, 8.8 ± 1.7 s−1, 22.8 ± 3.8 s−1, 26.7 ± 2.5 s−1, 13.4 ± 0.7 s−1, and15.3 ± 2.4 s−1 for DNA/RNA hybrids with lengthening primers. Corresponding reaction amplitudes were determined as 16.2 ± 0.4, 18.5 ± 1, 33.5 ± 1.1, 40 ± 1, 33 ± 0.4, and 31.7 ± 1.2, respectively. The quantitative analysis of the cleavage data also revealed that both cleavage rates and amplitudes reduced significantly when the number of nucleotides extending from the 3′-end of the RNA strand to the cutting site was <4. Consequently, a next round of RNA cleavage would not occur until the incorporation of at least four nucleotides, which might account for the fact that the processive template degradation was not observed in previously performed RNA dependent extension assays (15).
FIGURE 8.

Efficient RNase H cleavage required at least four ribonucleotides downstream of the cutting site. A, RNA cleavage assays were performed by rapidly mixing preformed RT·DNA/RNA complexes (175 nm HIVRT and 75 nm DNA/RNA hybrids (the 45-nt RNA template was annealed with primers of lengths ranging from 20–25 nucleotides) with 10 mm Mg2+. Reactions were stopped by adding 0.5 m EDTA at various time points up to 1 s. The position of the uncleaved45-nt RNA template was marked as 0. Different from the nomenclature previously used in this manuscript, positions of RNA cleavage products were marked as positive numbers here by counting the number of ribonucleotides from the 3′-end the RNA template to the cutting site. As shown in panels A and B, cleavage sites shifted from +8/+7 to +5/+4 when lengths of primers decreased from 25 to 20 nucleotides. Negligible bands were observed at position +2 in cases of 20 and 21mer. B, the cleavage sites were illustrated in a schematic, where thick or light arrows represented cutting sites for major or minor cleavage products, respectively. Short and light arrows indicated the site where negligible cleavage occurred. C, cleavage data were fit to the single-exponential equation to derive cleavage rates as 5 ± 0.4 s−1, 8.8 ± 1.7 s−1, 22.8 ± 3.8 s−1, 26.7 ± 2.5 s−1, 13.4 ± 0.7 s−1, and 15.3 ± 2.4 s−1 for DNA/RNA hybrids with lengthening primers. Corresponding reaction amplitudes were determined as 16.2 ± 0.4, 18.5 ± 1, 33.5 ± 1.1, 40 ± 1, 33 ± 0.4, and 31.7 ± 1.2 nm, respectively.
RNase H Cleavage during Processive Nucleotide Incorporation
RNase H activities during processive nucleotide incorporation were also explored to further examine the mechanism by which the two catalytic sites of HIVRT were able to work simultaneously but at different rates. As shown in the left panel of Fig. 9A, the 25-nt primer was extended continuously to the full-length 45-nt product, with markers to indicate intermediates in one-nucleotide increments. Whereas polymerization is a continuous process at a nearly constant rate, the RNA cleavage activity appears to be discontinuous, supporting our previous observation that at least four nucleotides downstream of the cutting site were necessary for efficient RNA cleavage. As we expected, the first round of template degradation started at position +7 with minor bands spreading to position +10 (corresponding to positions −18 to −15 in our previous nomenclature). Incorporation of 9 nucleotides shifted the cutting site accordingly by 9 nucleotides toward the 5′-end of the RNA strand, which triggered the second round of RNA cleavage at position +16, with only negligible bands formed between the two rounds of cleavage. Similarly, after the incorporation of 4 and 5 nucleotides, the third and fourth rounds of RNA cleavage started at positions +20 and +25, respectively. On average, the RNA cleavage occurred repeatedly each time after ∼6 nucleotides were incorporated.
FIGURE 9.

RNase H cleavage during processive nucleotide incorporation. A, RNase H activities during processive nucleotide incorporation were explored by performing RNA cleavage assays in the presence of all 4 nucleotides (100 μm). The procedures were the same as described previously except that data were collected for 20 s to allow full extension and multiple rounds of cleavage to occur. To avoid overlapping bands for extension and cleavage products, the two activities were assayed in parallel as described above. Positions for the primer (d25), primer extended by one nucleotide (d26), and fully extended product (d45) were marked. The same nomenclature as used in Fig. 8 was applied here to label positions of full-length RNA template (0) and cleaved products. Positions for the longest cleavage products formed during each round of degradation were marked as +7, +16, +20, and +25. The numbers of nucleotides incorporated before each round of RNA degradation were also labeled as 9, 4, and 5, respectively. B, time courses of processive nucleotide incorporation were simulated according to Scheme 3, with smooth lines indicating the best fit. Data within the first 1 s were shown to better display the main content of the data and avoid the long non-zero tails of fitting curves. The fitted rates for sequential polymerization steps were 94 ± 6, 19 ± 1, 33 ± 3, 30 ± 4, 19 ± 2, 35 ± 8, 36 ± 10, 43 ± 15, 27 ± 6, 29 ± 8, 20 ± 5, 23 ± 7, 36 ± 18, 21 ± 7, 26 ± 12, 21 ± 10, 29 ± 20, 36 ± 34, 20 ± 13, and 10 ± 5 s−1, respectively. The apparent incorporation rates for +N8, +N4, and +N5 were then determined as 3.4 ± 0.3, 6.3 ± 1.3, and 5 ± 1.7 s−1, respectively. C, the apparent incorporation rates obtained from panel B were used in the global analysis of time courses of RNA cleavage according to Scheme 4. Apparent incorporation rates and fitted rates for each round of RNA degradation are summarized in Scheme 4 and Table 3. Color codes: red, 45-nt RNA template; green, 1st cleavage (38 nt); blue, 2nd cleavage (29 nt); yellow, 3rd cleavage (25 nt); cyan, fourth cleavage (20-nt).
The incorporation and cleavage data were then fit globally, but in an alternative way due to the complexity of the data, as described under “Experimental Procedures.” Briefly, the incorporation data were first analyzed according to Scheme 3 (Fig. 9B) to derive the rate constants governing the incorporation of each nucleotide: 94 ± 6, 19 ± 1, 33 ± 3, 30 ± 4, 19 ± 2, 35 ± 8, 36 ± 10, 43 ± 15, 27 ± 6, 29 ± 8, 20 ± 5, 23 ± 7, 36 ± 18, 21 ± 7, 26 ± 12, 21 ± 10, 29 ± 20, 36 ± 34, 20 ± 13, and 10 ± 5 s−1, respectively. The average incorporation rate of the 2nd to 9th nucleotides was 27.2 ± 2.7 s−1. When the eight-step incorporation was simplified kinetically to a one-step process as we did in Scheme 4 (+N8), its apparent net rate was approximately determined by (27.2 ± 2.7)/8 = 3.4 ± 0.3 s−1, representing the time required to form the final (+N8) product. Similarly, apparent incorporation rates for +N4 and +N5 were determined as 6.3 ± 1.3 and 5 ± 1.7 s−1, respectively.
SCHEME 3.
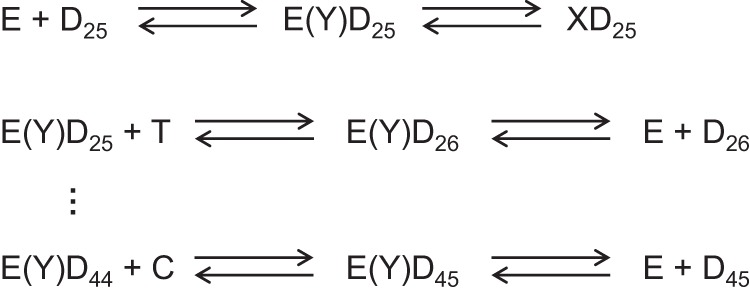
Pathway for processive synthesis. The minimal pathway for processive polymerization is given, starting with the binding of the enzyme (E) to the DNA (D25 primer/template) and formation of a nonproductive complex (XD25). E(Y)Dn designates the productive complex. At each step, the polymerization reaction can continue or the DNA can dissociate; the kinetic partitioning between the two reactions defines the processivity of the enzyme.
SCHEME 4.

Minimal model for processive polymerization and simultaneous RNase H cleavage. A minimal, abbreviated model describing processive polymerization and simultaneous RNase H cleavage is shown starting with the ED25R45 complex, where the subscript defines the length of each oligonucleotide. We also include the nonproductive complex: X(Y)D25R45 to quantitatively account for the amplitudes of the reactions. In this diagram polymerization steps go from left to right, whereas RNase H cleavage events go from top to bottom. Rate constants are given adjacent to the arrows for each reaction. The reaction starts with kinetic partitioning between polymerization to form D26 and RNase H to form R38; to describe the two activities as independent, we explicitly allow for either order of the two reactions. The next 8 nucleotide incorporations (+N8) are modeled as 1 step kinetically at 3.4 s−1 (8 nucleotides at 27 s−1 each). Subsequently, either the ED34R45 or the ED34R38 can be cleaved to form ED34R29 with the rate constants shown. Alternatively, either intermediate can be extended by 4 polymerization events (+N4) at a net rate of 6.3 s−1 (4 nucleotides at 25 s−1 each). Again, each elongation product can then either be extended by +N5 (at 5 s−1) or the RNA can be cleaved to form R25 (with different DNA states). Note that identical polymerization reactions are given identical rate constants independent of the state of the RNA, and identical RNase H cleavage events are given identical rate constants independent of the state of the DNA. This is an essential element of our model showing that the two reactions are independent. To simplify the model, we described several polymerization steps as occurring kinetically as a single step. Modeling the individual steps of polymerization gave the same results but required a much more complex model and computation.
These rates were then applied to constrain the simulation of RNA degradation data according to Scheme 4 (Fig. 9C). The comprehensive model shown in Scheme 4 was derived based on the data and our rule of deriving a minimal model. At the beginning, RT·DNA/RNA complexes equilibrated between the RNase H active form (ED25R45) and inactive forms (X(Y)D25R45), with the forward and reverse rates as 0.05 and 0.03 s−1. Then, the ED25R45 complex underwent RNA cleavage before or after TTP incorporation to yield the 38-nt products, whereas the cleaved substrates were still extended. Rates of TTP incorporation (105 s−1) and RNA cleavage before primer extension (23 s−1) were determined previously (Fig. 3) and fixed in the simulation to reduce the number of variable parameters. After TTP incorporation, the ED26R45 species underwent a kinetic partitioning between the RNA cleavage (ED26R38) and multiple nucleotide incorporations (ED34R45).
To simplify the model, the incorporation of 8 nucleotides was expressed as only one kinetic step (+N8), with a net rate of incorporation as 3.4 ± 0.3 s−1. Consequently, the rate of RNA cleavage after TTP incorporation was also slower, 4.4 s−1, compared with the 13 s−1 we determined previously (Fig. 3). After the incorporation of 8 nucleotides, the second round of RNA cleavage occurred, yielding 29-nt cleavage products. The cleavage reactions could start from ED34R45 (0.06 s−1) or ED34R38 (4.8 s−1) complexes, but the latter was the kinetically dominant pathway, as indicated by the rapid decrease of 38-nt products after an initial accumulation (Fig. 9C, green curve). Similarly, the cleavage rate determined here (4.8 s−1) was constrained by the kinetic partitioning between RNA cleavage and the next round of multiple nucleotide incorporation events (+N4) at an apparent rate of 6.3 ± 1.3 s−1. Then, the third round of RNA cleavage could start from three substrates, namely, ED38R45 (4.4 s−1), ED38R38 (0.06 s−1), and ED38R29 (0.06s−1). Because the amount of 29-nt cleavage products remained almost constant after its peak value (Fig. 9C, blue curve), the observed 25-nt products were not due to a secondary cut of the 29-nt products. Therefore, cleavage starting from ED38R45 became the main pathway at this time. The final round of RNA cleavage occurred after the incorporation of another 5 nucleotide (+N5) at a rate of 5 ± 1.7 s−1. Cleavage reactions started from four precursors, including ED43R45 (4.4 s−1), ED43R38 (0.4 s−1), ED43R29 (0.06 s−1), and ED43R25 (0.06 s−1). Cleavages of the 45-nt RNA precursor (ED43R45) remained the dominant kinetic pathway as the amounts of both 29-nt and 25-nt cleavage products were almost unchanged after their initial formation (Fig. 9C, blue and yellow curves). Cleavage of the ED43R38 precursor corresponded to the slower phase of the disappearance of the 38-nt products after the initial rise (Fig. 9C, green curve) at a rate of 0.4 s−1.
The average rate of nucleotide incorporation was ∼28 s−1 (except for the first TTP). To maintain the same kinetic partitioning between RNA cleavage and nucleotide incorporation activities in a complete model, the average rate of RNA cleavage after TTP incorporation could be approximately determined by 4.5 × 6 = 27 s−1. Accordingly, the discontinuous RNase H activities are due to the fact that 4–6 nucleotides downstream of the cutting site are required for efficient RNA cleavage to occur. Under processive nucleotide incorporation conditions, the processive polymerization and discontinuous RNase H can work simultaneously at the same net rate.
Discussion
Reverse transcription of HIV is a complex process that converts the single-stranded RNA genome of the virus into double-stranded DNA (29) and requires a close coordination between the RNA-dependent polymerase and the RNase H, which degrades the RNA during minus-strand DNA synthesis to allow template switching and efficient plus-strand DNA synthesis. Despite the abundance of structural and biochemical data, little is known about how the polymerase and RNase H catalytic centers work together, whether they act simultaneously or sequentially, and whether there are long range interactions to coordinate the two active sites. Our quantitative analysis under single turnover conditions demonstrated that the two active sites can engage the two ends of the DNA/RNA duplex to perform primer extension and template degradation activities simultaneously (Scheme 1). Although we performed single turnover kinetic analysis-only during TTP incorporation at a single sequence, we showed the results to be applicable to multiple incorporation sites by analysis of processive synthesis. Analysis of processive polymerization revealed that the net rates of synthesis and RNase H cleavage are comparable if one takes into account that each RNase H cleavage event removes 4–6 nucleotides from the template strand (Scheme 4). Under this scenario, the two catalytic centers can coordinate smoothly and harmoniously without the need for elaborate long-distance interactions to regulate the two activities. A critical part of this analysis is provided in the accompanying paper in which we demonstrate that PPi release limits the rate of processive synthesis during reverse transcription (30) so that the two activities occur a comparable net rates.
The potential coupling between the two active sites of HIVRT was first explored under single-turnover conditions. Nucleotide sequence preferences in the vicinity of the cleavage site are a major concern, as previous studies indicated that the DNA 3′-end-directed RNA cleavages occurred within a window of ∼6 nucleotides centered on the predicted distance 18 residues from the 3′-end of the primer (2, 9, 17, 31–34). To remove the sequence biases, the nucleotides near the predicted cleavage sites were replaced by a string of G residues; A residues could not be used because they appear to contribute resistance to cleavage (1) and cause bends in the duplex structure. The incorporation and cleavage data were analyzed using both conventional and global data analysis methods, which yielded comparable results, supporting the validity of the model chosen to fit the data globally.
Our analysis depends on accurate quantification of the products formed per enzyme active site. Previous studies have provided indirect evidence for a non-productive complex between HIVRT and DNA, defined by the fraction of bound duplex that fails to react in a single turnover at concentrations of excess enzyme sufficient to saturate the binding (26, 35–37). Here we defined three distinct RT·DNA/RNA complexes, including the bifunctional, polymerase active only, and inactive (nonproductive) forms. Although the active enzyme concentration in each experiment was defined by active site titration with a DNA template, we still observed that 30% or 37% of the RT·DNA/RNA complexes were inactive as measured by TTP or ddTTP incorporation. Native gel analysis revealed that the DNA primers were in slight excess over the RNA templates in our DNA/RNA substrate, and we corrected the numbers reported here for the amount of free primers. When a DNA/DNA duplex was used, the percentage of nonproductive complexes reduced to ∼20%, but the 100% polymerase activity has never been achieved no matter whether a DNA or RNA template was used. HIVRT has been reported to adopt dynamic binding orientations on the nucleic acid substrates and could switch between these orientations for different functions (38), so the inactive complexes identified here might reflect the nonproductive intermediates previously inferred in quantitative studies on HIVRT polymerization kinetics (26, 35–37).
Extensive interactions between the nucleic acid substrate and the polymerase domain of RT hold the primer 3′-end at the polymerase active site (24). Such interactions, however, appear to be fewer in number in the RNase H site domain (12), which might allow the substrate to temporarily swing in and out of the RNase H active site without affecting contacts at the polymerase site (10). Supporting this postulate, a fraction of the polymerization competent RT·DNA/RNA complexes did not exhibit any RNase H activity (YD25R45) during the time course of a single turnover. ∼40% of RT·DNA/RNA complexes were able to perform RNase H cleavage, whereas ∼70% extendedthe primer. Thus, the sum of the two activities exceeded 100%, providing evidence for the existence of bifunctional enzyme·nucleic acid complexes capable of simultaneous polymerization and RNase H hydrolysis. If the non-productive forms were excluded from the analysis, we computed that >60% of the active RT·DNA/RNA complexes could perform both activities simultaneously.
A nucleic acid substrate bound to HIVRT is in a dynamic equilibrium between the pre- and post-translocation states (21). The molecular details of translocation remain elusive, but its equilibrium may be adjusted by small-molecule ligands. For example, the PPi analogue, PFA, may be able to trap the pretranslocation state of HIVRT (23), whereas the next cognate nucleotide could stabilize the post-translocation complex. In previous kinetic studies, the observation that the RNase H activity can be inhibited by these two ligands under steady-state conditions (10, 19, 31) was taken to suggest that DNA/RNA hybrids stabilized at the polymerase active site were not able to engage the RNase H site. However, these steady-state results could be explained by the effect of the ligands in slowing the rates of dissociation of the DNA/RNA duplex, which wouldbe rate-limiting in these steady-state assays. Therefore, there are no definitive data to support the notion that binding at the polymerase site inhibits RNase H activity. In contrast, a recent study revealed that the binding of PFA did not affect the efficiency of RNase H cleavages under single-turnover conditions (10). This issue was revisited in this study using various substrates and with a string of G residues to avoid nucleotide sequence preferences. Our results showed that the ligands capable of stabilizing the binding of duplex at the polymerase active site mode did not inhibit the RNase H activity, supporting the conclusion that the DNA/RNA hybrid could engage both active sites simultaneously. Although the cleavage patterns were affected by these ligands, they did not follow the predicted modes, especially when the 26-nt primers were tested (Fig. 5B).
In the absence of any ligands, RNase H cleavage occurred at 4 distinct positions, ranging from −18 to −15. Because previous studies have suggested that the RNase H activity of HIVRT acted as an endonuclease with partial 3′-5′ processivity (27, 28), cleavages at position −16 and −15 might reflect secondary cuts of the 38/37-nt cleavage products (−18/−17). However, the global data analysis revealed that only the 38-nt products (−18) underwent negligible levels of secondary cleavages (∼0.05 s−1). Moreover, there were no detectable lag phases, and comparable rates were derived for cleavages at positions −18 to −15 (1.5∼5 s−1, Scheme 1), so the 36/35-nt cleavage products were not due to processive, sequential cleavage events; rather, they resulted from two independent events. The results were consistent with previous observations that the DNA 3′-end-directed RNA cleavages occurred within a window of ∼6 nucleotides centered on the predicted distance (corresponding to position −20 to −15) (2, 9, 17, 31–34). Removal of the nucleotide sequence preference only narrowed down the cleavage window to 4 nucleotides, and this low level of specificity is probably due to the lack of a well defined structural element around the RNase H active site required to bind the DNA/RNA hybrid. Therefore, the RNase H site can temporarily slide back and forth on the substrate, which may be achieved through conformational changes of the DNA/RNA hybrid (12). Both PFA and the next correct nucleotide failed to lock the enzyme·nucleic acid complex in a fixed translocation state, as measured by the RNase H cleavage site. Consequently, we conclude that the RNase H assay is not a reliable method to measure the equilibrium of translocation stages, as cleavages did not occur at the fixed distances, reflecting the translocation state but, rather, within a “cleavage window” that is four nucleotides long.
The nucleotide-induced conformational change is a crucial step during nucleotide incorporation that was shown to govern the specificity and analog discrimination by HIVRT (26). However, whether this crucial conformational change affects RNase H activity and/or specificity remained unknown. Therefore, we examined how mutants with impaired rate and equilibrium constants governing the conformational change would influence RNase H cleavages. Two mutations of W71 were chosen as MD simulations of the conformational change step (39) suggested that this residue was an important contributor to the rate and free energy of the isomerization step. In addition, Arg-72 interacts with the PPi moiety of the incoming nucleotide, and the arginine to alanine substitution was reported to significantly impede the PPi release step (40). Our results revealed that mutations W71A, W71D, and R72A impeded the nucleotide-dependent conformational change step by 7-, 14,- and 170-fold, respectively. However, they did not significantly affect the rate or amplitude of RNase H activity, although minor effects on the cleavage pattern were observed. R72A reduced the amplitude of the second, slower phase during RNA cleavage presumably by inhibiting substrate dissociation and/or the conversion to the RNase H active form (YD25R45 to ED25R45). These results further supported the working simultaneously model and argued against the competing model in which the two active sites compete with each other to engage substrates such that impaired polymerization activity should have led to enhanced RNase H activity or vice versa.
Our data have suggested that the two catalytic centers of HIVRT work simultaneously and independently from each other, except for the obvious fact that continued polymerization provides new substrates for the RNase H active site. Our model also provides a possible explanation for the observation that mixing mutants deficient in either site could fully restore reverse transcription (19). However, our model does not eliminate possible subtle change in enzyme specificity induced by long range structural changes. For example, substitutions at Trp-266 and Phe-61 in the polymerase domain of HIVRT were able to affect the cleavage specificity and render the RNase H unable to generate/remove polypurine tract primer (41–43), implying that the polymerase domain might allosterically attenuate in the RNase H specificity.
The rate of polymerization was initially shown to be ∼7-fold faster than the RNase H cleavage (2), and it was shown that the template was not degraded processively in RNA-dependent extension assays during a single turnover (15). Here we also explored the coordination between the two active sites under processive synthesis conditions by presteady-state kinetics with global data analysis, and we show quantitatively that a minimal model can account for the observed simultaneous polymerization and RNase H activities (Scheme 4). Due to the complexity of the full model, reactions with comparable rates were linked in the simulation to reduce the number of variable parameters. For instance, rates of RNA cleavage starting from 45-nt precursors (ED26R45, ED38R45, and ED43R45, except ED34R45) were linked to be identical in the simulation. Similarly, minor secondary reactions at each round of RNA cleavage were also linked (except ED43R38) and yielded a rate of 0.06 s−1. The biphasic decrease of 38-nt products after their initial rise defined the rates governing the cleavage of ED34R38 and ED43R38 as 4.8 and 0.4 s−1, respectively. Consequently, the simulation only needed to define 6 parameters despite the complexity of the model, which turned out to be all well constrained (Table 3). Accordingly, we could describe a large number of reactions with a simplified model. Although our model may not be unique, there can be no question that it can quantitatively account for the data, because the data were fit based upon numerical integration of the rate equations without simplifying approximations.
TABLE 3.
Kinetic parameters governing RNase H cleavages during processive nucleotide incorporation
The lower and upper limits of each rate constant derived from confidence contour analysis of the simulation were listed in the parenthesis. The nomenclature of rate constants was illustrated in Scheme 4. All parameters were well constrained in the fitting.
| k1 | k2 | k3 | k4 | k5 | k6 |
|---|---|---|---|---|---|
| s−1 | s−1 | s−1 | s−1 | s−1 | s−1 |
| 0.05 (0.043∼0.055) | 0.03 (0.026∼0.033) | 4.4 (3.5∼4.9) | 0.06 (0.05∼0.08) | 4.8 (3.8∼6.1) | 0.4 (0.3∼0.6) |
Multiple incorporation events between sequential rounds of RNA cleavage were “reduced” to one kinetic step. Note that this simplification resulted in what appears to be a slow rate of nucleotide incorporation, which could be problematic without a proper interpretation of the data. First, the “real” average rate for polymerization approximately equaled the apparent average rate of nucleotide incorporation (∼4.7 s−1) multiplied by the average number of nucleotides incorporated (∼6), which is ∼28 s−1 (rate per nucleotide). Second, all non-dead-end species in the model faced a kinetic partitioning between the polymerization and RNase H cleavage activities (e.g. ED26R45), and the real polymerization rate is 6-fold faster than the apparent rate applied in the simulation. To maintain the same kinetic partitioning in a complete model, the average rate of RNase H cleavage derived from this simplified model (∼4.5 s−1) also needed to be multiplied by 6 to approximately yield the net rate of RNase H cleavage to be ∼27 s−1. Consequently, rates of polymerization and RNase H cleavage were comparable to each other.
Our data now suggest a model in which processive polymerization at ∼28 s−1 is coincident with RNase H activity also occurring at ∼28 s−1 but only after every ∼6 nucleotides. Thus, although the RNase H activity appears to be slower than polymerization, the net rate of hydrolysis (net nucleotides removed per second) at the RNase H site is identical to the rate of polymerization. Accordingly, the RNase H produces products ∼6 nucleotides in length and does not impede the processive motion of the polymerase because it acts periodically.
Previous studies have noted that in single turnover experiments, polymerization with an RNA template is much faster than with a DNA template (2). Here we noted that the incorporation of the first nucleotide (∼105 s−1) was much faster than subsequent incorporation events. These data suggested that the processive nucleotide incorporation was constrained by a rate-limiting step occurring after the chemistry step for incorporation of the first nucleotide but before the binding of the next nucleotide. Further studies demonstrated that this rate-limiting step was the slow PPi release (30). Starting from the second nucleotide, polymerization was no longer faster than RNase H cleavage as shown here. Therefore, these two activities could coordinate spatially and temporally. Under this circumstance, relative levels of these two activities were determined by their kinetic partitioning at each branch point. This model also explains how the RNase H cleavage tended to increase efficiency when the extension reaction was paused (16), not because the two activities were mutually exclusive but because the kinetic partitioning more strongly favored the cleavage activity when polymerization stalled. However, we also observed that template degradation was not processive, although polymerization was as fast as the RNase H cleavage. This is consistent with previous observations that the RNase H activity of HIVRT acted as an endonuclease with only partial 3′-5′ processivity (27, 28).
A recent crystal structure of HIVRT in complex with a DNA/RNA hybrid provided some structural basis for this periodic and inefficient RNase H activity (12). First, there are no structural elements of RT that continuously thread the RNA through the RNase H active site; therefore, structural changes in the RT and/or the DNA/RNA duplex appear to be required to align the RNA strand at the RNase H active site. Second, the chelation environment and charge distribution at the RNase H active site changes after the cleavage reaction, which may force the cleaved RNA products to transiently disengage from the RNase H site (12). Moreover, our results showed that four-six nucleotides downstream of the cutting site were required for efficient RNA cleavage, and therefore, a second round RNase H cleavage would not occur until at least four nucleotides were incorporated. The oligonucleotide with a minimum length of four nucleotides extending from the 3′-end of the RNA strand to the cleavage site may be required for the RNA strand to bind productively at the RNase H active site, probably through interactions between the oligonucleotide segment and HIVRT near the RNase H site. This model also suggested a possible mechanism for HIVRT to prevent uncontrolled template degradation that would otherwise cause premature dissociation of the primer strand and termination of synthesis in advance of replication.
Summary
Our data quantitatively demonstrated the existence of a spatial and temporal coordination between the two active sites, arising only because the polymerase continuously feeds new substrate into the RNase H active site. Our results show that whereas the polymerase is efficient and processive, the RNase H is inefficient at any given site and occurs only after 4–6 nucleotides have been extended. What emerges is a model in which the RNase H active site requires a 4–6 nucleotide overhang and then only grips the DNA/RNA duplex infrequently and transiently to promote cleavage. The inefficient RNase H reaction may, in fact, be an important feature of the enzyme activity so that the RNase H site does not bind tightly to the DNA/RNA duplex at any one site as that may impede the movement of the duplex driven by the polymerase. Moreover, we show in the accompanying paper (30) that PPi release limits the rate of reverse transcription so that the net rates of DNA synthesis and RNase H cleavage are comparable, allowing the two reactions to proceed at comparable rates and appear to act simultaneously even though they are not coupled. The inefficient RNase H activity provides the perfect solution to the problem of coupling the two activities without impeding synthesis or requiring an elaborate mechanism to directly coordinate the two active sites. Like its error-prone replication, HIV has evolved an elegant adaptive advantage from the apparent sloppiness of the enzyme.
Experimental Procedures
Expression and Purification of Proteins Used in This Study
WT and mutant HIVRT proteins were expressed, purified, and labeled following the methods previously described (26, 35). Briefly, single mutations (W71A, W71D, and R72A) were introduced in genes of both subunits using a QuikChange Multi kit (Stratagene). The two subunits of each WT or mutant RT were individually expressed in T7 Express Competent Escherichia coli (New England BioLabs), mixed in a 1:1 ratio, and purified by tandem Q-Sepharose and Bio-Rex 70 columns followed by a single-strand DNA affinity column. Labeled enzymes were passed through a Bio-Rex 70 column to remove excess MDCC. The enzymes were assayed by presteady-state burst experiments to determine the active site concentrations, divided into aliquots, and stored at −80 °C. The method for the preparation of MDCC-labeled PBP used in this study was described previously (44), and its active-site concentration was determined by fluorescent titration with various concentrations of inorganic Pi.
Nucleic Acid Substrates for Kinetic Studies
All DNA/RNA hybrids were designed to have a DNA primer recessed at the 3′-end on a RNA template to monitor the primary polymerase-dependent cleavage events. The 45-nt RNA templates with a predefined semi-random sequence (r45rd, 5′-GGA CGG CAU UGG AUC GAC GAU GAG UUG GUU GGA CGG CUG CGA GGC) or a string of Gs covering the RNase H cleavage site (r45, 5′-GGA CGG CAU UGG AUC GAC GAU GAG UUG GUU GGG GGGGGGGGA GGC) were purchased from Integrated DNA Technologies with RNase free HPLC purification. The 25-nt DNA primers complementary with r45rd (d25rd, 5′-GCC TCG CAG CCG TCC AAC CAA CTC A) or r45 (d25, 5′-GCC TCCCCCCCCCCC AAC CAA CTC A) were ordered from Integrated DNA Technologies and purified by gel extraction.
A series of primers with various lengths (Fig. 8B) was designed to study the minimum number of ribonucleotides downstream of the cutting site that are required for efficient cleavage. Shortening the primer by one nucleotide will shift the cutting site by one nucleotide toward the 3′-end of the RNA template in the DNA 3′-end-directed cleavage mode. DNA primers terminated with ddAMP or ddTMP were made through enzymatic synthesis with RT. After the biosynthetic reactions, terminated 25- or 26-nt primers (25ddA or 26ddT) were purified by gel extraction. Primers and templates used in quench flow assays were 5′-32P- labeled with T4 polynucleotide kinase (New England BioLabs). Annealing of DNA primers and RNA templates was carried out by mixing the two oligonucleotides at a 1:1 molar ratio and incubating at 67 °C for 8 min followed by slow cooling to room temperature.
Magnesium Concentrations
All experiments were performed in a buffer containing 50 mm Tris, pH 7.5, 100 mm KCl, and 0.1 mm EDTA at 37 °C. In quench-flow and stopped-flow experiments, 0.1 mm EDTA was included during the preincubation when forming the enzyme·DNA complex, and 10 mm MgCl2 was added with nucleotides or PPi to start the reaction.
RNA Cleavage Assays
Time courses of RNA cleavage were measured by rapid quench flow techniques using a KinTek RQF-3. The 5′-labeled RNA template was annealed to the DNA primer as described above and diluted to a final concentration of 75 nm, preincubated with 175 nm HIVRT (with 0.1 mm EDTA), and then rapidly mixed with 10 mm Mg2+ to initiate the cleavage. Effects of small-molecule ligands on polymerase-dependent RNase H cleavage were studied by performing RNA cleavage assays in the presence of 1 mm PFA or the next cognate nucleotides (500 μm, with dideoxy-terminated primers). To explore the potential coordination between the two active sites, incorporation and RNase H activities were measured simultaneously by preincubating 175 nm HIVRT with 75 nm concentrations of double-labeled DNA/RNA hybrids (with 0.1 mm EDTA) and mixing with 100 μm TTP or ddTTP (with 10 mm MgCl2). Reactions were stopped by the addition of 0.5 m EDTA at various time points within 10 s. Products were separated by 15% denaturing PAGE and quantified using a Typhoon scanner in Imagine Quant 8.1 (GE Healthcare). The incorporation and cleavage data were biphasic and fit with a double exponential equation to estimate the observed rates (“conventional fitting”).
RNase H activities were also explored under the processive nucleotide incorporation condition, in which all four nucleotides were added at 100 μm so the 25-nt primer would be fully extended to the 45-nt product. To avoid the overlap of bands for extension and cleavage products, the two activities were assayed in parallel under the same conditions with the same enzyme by attaching the radioactivity only to one strand ofthe hybrid at one time, e.g. 32P was only labeled to the 5′-end of the primer when the incorporation activity was assayed and vice versa. Other procedures were the same except that data were collected for 20 s to allow full extension and multiple rounds of cleavage to occur.
Characterization of HIVRT Mutants
Single-nucleotide incorporation was monitored by rapid quench flow and stopped flow methods. Chemical quench flow incorporation experiments were performed to determine the incorporation efficiency of TTP by HIVRT mutants. Various concentrations of TTP were rapidly mixed with a preformed enzyme·DNA/RNA complex (175 nm MDCC-labeled HIVRT mutant and 75 nm d25/r45 with 0.1 mm EDTA) using a RQF-3 Rapid Quench Flow. All reactions were conducted in 50 mm Tris, pH 7.5, 100 mm KCl, and 10 mm magnesium acetate (all concentrations final) at 37 °C and stopped by adding 0.5 m EDTA. Products were separated and quantified as described above.
The time dependence of transient MDCC fluorescence changes corresponding to TTP-induced enzyme conformational change was monitored using an AutoSF-120. A preformed enzyme·DNA/RNA duplex (100 nm MDCC-labeled HIVRT mutant, 150 nm d25ddA/r45, 0.1 mm EDTA) was rapidly mixed with various concentrations of TTP (with 10 mm MgCl2) in the stopped flow for 300 ms. MDCC fluorescence was excited at 425 nm, and fluorescence was monitored at 475 nm with a 50-nm band-pass filter (Semrock). To monitor the fluorescence changes corresponding to the chemistry step, a normal primer (not dideoxy-terminated) was used to form the enzyme·DNA/RNA binary complex (100 nm MDCC-labeled HIVRT mutant and 150 nm d25/r45) and mixed with various concentrations of TTP in the stopped flow for a longer time scale of 50 s.
A coupled fluorescence assay described elsewhere (45) was used to measure the kinetics of PPi release. Briefly, the preformed enzyme·DNA/RNA duplex (150 nm HIVRT mutant and 100 nm d25/r45) was rapidly mixed with 25 μm TTP in the presence of 0.6 μm pyrophosphatase, the Pi “mop” containing 100 μm 7-methylguanosine and 0.02 IU/ml purine nucleoside phosphorylase as well as 1.5 μm concentrations of fluorescently labeled E. coli PBP.
Global Data Analysis
The inherent relationship between the rates and amplitudes of incorporation and cleavage reactions is crucial for the exploration of any potential coordination between the two active sites. However, this critical information can be lost in conventional fitting; therefore, simultaneous fitting of all data within a given set is the preferred method. In the case of RNA cleavage during single nucleotide incorporation (TTP or ddTTP), the data were globally fit with two control experiments calibrating the RNase H activity of HIVRT against substrates before or after primer extension. The whole set of data was fit globally to a comprehensive mechanism showed in Scheme 1 using KinTek Explorer software (22, 46). Three different RT·DNA/RNA complexes were identified and included in the model (XD25R45 (inactive), YD25R45 (polymerase active only), and ED25R45 (polymerase and RNase H both active)), which underwent slow but reversible interconversion. Amplitudes of substrate depletion in primer extension (Fig. 3A, red curve) and template degradation (Fig. 3B, red curve) assays defined the equilibrium constants governing the exchanges between different species. Both YD25R45 and ED25R45 could extend the 25-nt substrate to the 27-nt product (with one mismatch extension), rates of which were derived from the nucleotide incorporation data (Fig. 3A). RNA cleavage was able to occur before (ED25R45) or after (ED26R45) primer extension, yielding products with various lengths ranging from 35 to 38 nt. In globally fitting the data, cleavage rates were defined by the cleavage data obtained with (Fig. 3B) or without (Fig. 3, C and D) the addition of an incoming nucleotide so that the cleavage pattern observed during polymerization was modeled based upon the patterns seen with the DNA/RNA duplex before and after elongation. To describe the complicated process as complete as possible, secondary cuts (e.g. ED25R38 to ED25R37) and nucleotide incorporation after template degradation (e.g. ED25R38 to ED26R38) were also included in the model. The simulation of RNA cleavage during ddTTP incorporation was similar except that the mismatch extension was prohibited and not included in the model (Scheme 2). The smooth curves in each figure panel represented the best simultaneous fit of all the data to the model, and the rate constants were summarized in Scheme 1 (TTP) and 2 (ddTTP). Results of confidence contour analysis of these parameters were listed in Table 1.
Simulation of RNA cleavage during processive nucleotide incorporation was performed in an alternative way due to the complexity of reactions occurred in the process. First, the incorporation data were fit to a minimal model including sequential nucleotide incorporation events followed by possible product dissociation after each round of incorporation to account for the retention of a fraction of intermediates at long reaction times as attributed to their dissociation from the enzyme (Scheme 3 and Fig. 9B). Global fitting of primer extension and RNA cleavage data were achieved by using rate constants obtained from the analysis of polymerization to constrain the simulation of RNA degradation data (Scheme 4 and Fig. 9C). At the beginning, RT·DNA/RNA complexes equilibrated between the RNase H active form (ED25R45) and inactive forms (X(Y)D25R45). Then, the ED25R45 complex underwent RNA cleavage (Scheme 4, vertical arrows) before or after TTP incorporation (horizontal arrows), whereas the cleaved substrates were still extended. After the incorporation of multiple nucleotides, the second round of RNA cleavage occurred, yielding shorter products. Then another two rounds of RNA cleavage would occur until the primer was extended to full-length and no more space was available for the polymerase-dependent RNA cleavage. To simplify the model, multiple incorporation events between different rounds of RNA cleavage were expressed as “one” step kinetically, e.g. +N8 represented the incorporation of eight nucleotides in a row (ED26R45 to ED34R45). This simplification was appropriate and required because of the absence of intermediates. The cleavage products listed for each round (e.g. ED25R38 and ED26R38 for the first round) represented the sum of all cleavage products with various lengths and were expressed in that way for simplification. Rate constants derived from the simulation were summarized in Scheme 4 and Table 3.
In fitting fluorescence data (Fig. 6), three fluorescence scaling factors were used, one for the net scale of the reaction to convert volts to concentration units (f1), one for the change in fluorescence in during the conformational change step (f2), and third for the product state (f3) according to the equation,
| (Eq. 1) |
where Y is the observed fluorescence.
Author Contributions
A. L. performed all of the experiments except those on the Trp-71 mutants, which were performed by J. L., A. L., and K. A. J. worked together to design the experiments and interpret the results. A. L. wrote the first draft of the paper, which was then edited by K. A. J.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM084741. This work was also supported by a grant from the Welch Foundation (F-1604). K. A. J. is the president of KinTek Corp., which provided the AutoSF-200x stopped flow and RQF-3 quench-flow instruments and KinTek Explorer software used in this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HIVRT
- human immunodeficiency virus-1 reverse transcriptase (RT)
- MDCC
- 7-diethylamino-3-((((2-maleimidyl)ethyl)amino)carbonyl)coumarin
- PBP
- phosphate-binding protein
- ddTTP
- 2′,3′-dideoxythymidine-5′-triphosphate
- ddAMP
- 2′,3′-dideoxyadenosine-5′-monophosphate
- ddTMP
- 2′,3′-dideoxythymidine-5′-monophosphate
- PFA
- phosphonoformic acid or foscarnet.
References
- 1.Sarafianos S. G., Das K., Tantillo C., Clark A. D. Jr, Ding J., Whitcomb J. M., Boyer P. L., Hughes S. H., and Arnold E. (2001) Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 20, 1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kati W. M., Johnson K. A., Jerva L. F., and Anderson K. S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 [PubMed] [Google Scholar]
- 3.Hostomsky Z., Hostomska Z., Hudson G. O., Moomaw E. W., and Nodes B. R. (1991) Reconstitution in vitro of RNase H activity by using purified N-terminal and C-terminal domains of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 88, 1148–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans D. B., Brawn K., Deibel M. R. Jr, Tarpley W. G., and Sharma S. K. (1991) A recombinant ribonuclease H domain of HIV-1 reverse transcriptase that is enzymatically active. J. Biol. Chem. 266, 20583–20585 [PubMed] [Google Scholar]
- 5.Stammers D. K., Tisdale M., Court S., Parmar V., Bradley C., and Ross C. K. (1991) Rapid purification and characterisation of HIV-1 reverse transcriptase and RNaseH engineered to incorporate a C-terminal tripeptide α-tubulin epitope. FEBS Lett. 283, 298–302 [DOI] [PubMed] [Google Scholar]
- 6.Smith J. S., and Roth M. J. (1993) Purification and characterization of an active human immunodeficiency virus type 1 RNase H domain. J. Virol. 67, 4037–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stahl S. J., Kaufman J. D., Vikić-Topić S., Crouch R. J., and Wingfield P. T. (1994) Construction of an enzymatically active ribonuclease H domain of human immunodeficiency virus type 1 reverse transcriptase. Protein Eng. 7, 1103–1108 [DOI] [PubMed] [Google Scholar]
- 8.Keck J. L., and Marqusee S. (1995) Substitution of a highly basic helix/loop sequence into the RNase H domain of human immunodeficiency virus reverse transcriptase restores its Mn2+-dependent RNase H activity. Proc. Natl. Acad. Sci. U.S.A. 92, 2740–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gopalakrishnan V., Peliska J. A., and Benkovic S. J. (1992) Human immunodeficiency virus type 1 reverse transcriptase: spatial and temporal relationship between the polymerase and RNase H activities. Proc. Natl. Acad. Sci. U.S.A. 89, 10763–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beilhartz G. L., Wendeler M., Baichoo N., Rausch J., Le Grice S., and Götte M. (2009) HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J. Mol. Biol. 388, 462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lapkouski M., Tian L., Miller J. T., Le Grice S. F., and Yang W. (2013) Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nat. Struct. Mol. Biol. 20, 230–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das K., Martinez S. E., Bandwar R. P., and Arnold E. (2014) Structures of HIV-1 RT-RNA/DNA ternary complexes with dATP and nevirapine reveal conformational flexibility of RNA/DNA: insights into requirements for RNase H cleavage. Nucleic Acids Res. 42, 8125–8137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rausch J. W., Lener D., Miller J. T., Julias J. G., Hughes S. H., and Le Grice S. F. (2002) Altering the RNase H primer grip of human immunodeficiency virus reverse transcriptase modifies cleavage specificity. Biochemistry 41, 4856–4865 [DOI] [PubMed] [Google Scholar]
- 14.Nowotny M., Gaidamakov S. A., Ghirlando R., Cerritelli S. M., Crouch R. J., and Yang W. (2007) Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol Cell 28, 264–276 [DOI] [PubMed] [Google Scholar]
- 15.DeStefano J. J., Buiser R. G., Mallaber L. M., Myers T. W., Bambara R. A., and Fay P. J. (1991) Polymerization and RNase H activities of the reverse transcriptases from avian myeloblastosis, human immunodeficiency, and Moloney murine leukemia viruses are functionally uncoupled. J. Biol. Chem. 266, 7423–7431 [PubMed] [Google Scholar]
- 16.Purohit V., Roques B. P., Kim B., and Bambara R. A. (2007) Mechanisms that prevent template inactivation by HIV-1 reverse transcriptase RNase H cleavages. J. Biol. Chem. 282, 12598–12609 [DOI] [PubMed] [Google Scholar]
- 17.Suo Z., and Johnson K. A. (1997) Effect of RNA secondary structure on RNA cleavage catalyzed by HIV-1 reverse transcriptase. Biochemistry 36, 12468–12476 [DOI] [PubMed] [Google Scholar]
- 18.Suo Z., and Johnson K. A. (1997) RNA secondary structure switching during DNA synthesis catalyzed by HIV-1 reverse transcriptase. Biochemistry 36, 14778–14785 [DOI] [PubMed] [Google Scholar]
- 19.Shaw-Reid C. A., Feuston B., Munshi V., Getty K., Krueger J., Hazuda D. J., Parniak M. A., Miller M. D., and Lewis D. (2005) Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry 44, 1595–1606 [DOI] [PubMed] [Google Scholar]
- 20.Schultz S. J., Zhang M., and Champoux J. J. (2010) Multiple nucleotide preferences determine cleavage-site recognition by the HIV-1 and M-MuLV RNases H. J. Mol. Biol. 397, 161–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchand B., and Götte M. (2003) Site-specific footprinting reveals differences in the translocation status of HIV-1 reverse transcriptase: implications for polymerase translocation and drug resistance. J. Biol. Chem. 278, 35362–35372 [DOI] [PubMed] [Google Scholar]
- 22.Johnson K. A., Simpson Z. B., and Blom T. (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 [DOI] [PubMed] [Google Scholar]
- 23.Marchand B., Tchesnokov E. P., and Götte M. (2007) The pyrophosphate analogue foscarnet traps the pre-translocational state of HIV-1 reverse transcriptase in a Brownian ratchet model of polymerase translocation. J. Biol. Chem. 282, 3337–3346 [DOI] [PubMed] [Google Scholar]
- 24.Huang H., Chopra R., Verdine G. L., and Harrison S. C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 25.Ding J., Das K., Hsiou Y., Sarafianos S. G., Clark A. D. Jr, Jacobo-Molina A., Tantillo C., Hughes S. H., and Arnold E. (1998) Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 284, 1095–1111 [DOI] [PubMed] [Google Scholar]
- 26.Kellinger M. W., and Johnson K. A. (2010) Nucleotide-dependent conformational change governs specificity and analog discrimination by HIV reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 107, 7734–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeStefano J. J., Buiser R. G., Mallaber L. M., Bambara R. A., and Fay P. J. (1991) Human immunodeficiency virus reverse transcriptase displays a partially processive 3′ to 5′ endonuclease activity. J. Biol. Chem. 266, 24295–24301 [PubMed] [Google Scholar]
- 28.Krug M. S., and Berger S. L. (1989) Ribonuclease H activities associated with viral reverse transcriptases are endonucleases. Proc. Natl. Acad. Sci. U.S.A. 86, 3539–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beilhartz G. L., and Götte M. (2010) HIV-1 ribonuclease H: structure, catalytic mechanism, and inhibitors. Viruses 2, 900–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li A., Gong S., and Johnson K. A. (2016) Rate-limiting pyrophosphate release by HIV reverse transcriptase improves fidelity. J. Biol. Chem. 26554–26565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furfine E. S., and Reardon J. E. (1991) Reverse transcriptase. RNase H from the human immunodeficiency virus. Relationship of the DNA polymerase and RNA hydrolysis activities. J. Biol. Chem. 266, 406–412 [PubMed] [Google Scholar]
- 32.DeStefano J. J., Mallaber L. M., Fay P. J., and Bambara R. A. (1994) Quantitative analysis of RNA cleavage during RNA-directed DNA synthesis by human immunodeficiency and avian myeloblastosis virus reverse transcriptases. Nucleic Acids Res. 22, 3793–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suo Z., and Johnson K. A. (1997) Effect of RNA secondary structure on the kinetics of DNA synthesis catalyzed by HIV-1 reverse transcriptase. Biochemistry 36, 12459–12467 [DOI] [PubMed] [Google Scholar]
- 34.Champoux J. J., and Schultz S. J. (2009) Ribonuclease H: properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 276, 1506–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kellinger M. W., and Johnson K. A. (2011) Role of induced fit in limiting discrimination against AZT by HIV reverse transcriptase. Biochemistry 50, 5008–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kerr S. G., and Anderson K. S. (1997) RNA dependent DNA replication fidelity of HIV-1 reverse transcriptase: evidence of discrimination between DNA and RNA substrates. Biochemistry 36, 14056–14063 [DOI] [PubMed] [Google Scholar]
- 37.Wöhrl B. M., Krebs R., Goody R. S., and Restle T. (1999) Refined model for primer/template binding by HIV-1 reverse transcriptase: pre-steady-state kinetic analyses of primer/template binding and nucleotide incorporation events distinguish between different binding modes depending on the nature of the nucleic acid substrate. J. Mol. Biol. 292, 333–344 [DOI] [PubMed] [Google Scholar]
- 38.Abbondanzieri E. A., Bokinsky G., Rausch J. W., Zhang J. X., Le Grice S. F., and Zhuang X. (2008) Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature 453, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirmizialtin S., Nguyen V., Johnson K. A., and Elber R. (2012) How conformational dynamics of DNA polymerase select correct substrates: experiments and simulations. Structure 20, 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarafianos S. G., Pandey V. N., Kaushik N., and Modak M. J. (1995) Site-directed mutagenesis of arginine 72 of HIV-1 reverse transcriptase: catalytic role and inhibitor sensitivity. J. Biol. Chem. 270, 19729–19735 [DOI] [PubMed] [Google Scholar]
- 41.Gao H. Q., Boyer P. L., Arnold E., and Hughes S. H. (1998) Effects of mutations in the polymerase domain on the polymerase, RNase H, and strand transfer activities of human immunodeficiency virus type 1 reverse transcriptase. J. Mol. Biol. 277, 559–572 [DOI] [PubMed] [Google Scholar]
- 42.Powell M. D., Beard W. A., Bebenek K., Howard K. J., Le Grice S. F., Darden T. A., Kunkel T. A., Wilson S. H., and Levin J. G. (1999) Residues in the αH and αI helices of the HIV-1 reverse transcriptase thumb subdomain required for the specificity of RNase H-catalyzed removal of the polypurine tract primer. J. Biol. Chem. 274, 19885–19893 [DOI] [PubMed] [Google Scholar]
- 43.Mandal D., Dash C., Le Grice S. F., and Prasad V. R. (2006) Analysis of HIV-1 replication block due to substitutions at F61 residue of reverse transcriptase reveals additional defects involving the RNase H function. Nucleic Acids Res. 34, 2853–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auerbach S. D., and Johnson K. A. (2005) Alternating site ATPase pathway of rat conventional kinesin. J. Biol. Chem. 280, 37048–37060 [DOI] [PubMed] [Google Scholar]
- 45.Hanes J. W., and Johnson K. A. (2008) Real-time measurement of pyrophosphate release kinetics. Anal. Biochem. 372, 125–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson K. A., Simpson Z. B., and Blom T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]