


Abstract
The small nuclear RNA (snRNA) components of the spliceosome undergo many conformational rearrangements during its assembly, catalytic activation and disassembly. The U4 and U6 snRNAs are incorporated into the spliceosome as a base-paired complex within the U4/U6.U5 small nuclear ribonucleoprotein (tri-snRNP). U4 and U6 are then unwound in order for U6 to pair with U2 to form the spliceosome's active site. After splicing, U2/U6 is unwound and U6 annealed to U4 to reassemble the tri-snRNP. U6 rearrangements are crucial for spliceosome formation but are poorly understood. We have used single-molecule Förster resonance energy transfer and unwinding assays to identify interactions that promote U4/U6 unwinding and have studied their impact in yeast. We find that U4/U6 is efficiently unwound using DNA oligonucleotides by coupling unwinding of U4/U6 stem II with strand invasion of stem I. Unwinding is stimulated by the U6 telestem, which transiently forms in the intact U4/U6 RNA complex. Stabilization of the telestem in vivo results in accumulation of U4/U6 di-snRNP and impairs yeast growth. Our data reveal conserved mechanisms for U4/U6 unwinding and indicate telestem dynamics are critical for tri-snRNP assembly and stability.
INTRODUCTION
In all eukaryotes, precursors to mRNAs (pre-mRNAs) are transcribed with noncoding intronic regions that must be excised and flanking exonic regions that must be ligated together through splicing. Splicing is carried out by the spliceosome, an essential macromolecular machine composed of more than 80 proteins and five small nuclear RNAs (snRNAs) (1). Each of the snRNAs (U1, U2, U4, U5 and U6) associates with multiple proteins forming small nuclear ribonucleoprotein particles (snRNPs). Assembly of the spliceosome proceeds by association of the U1 snRNP at the 5′ splice site (SS) and U2 at the branchpoint sequence to form the spliceosomal A complex (1,2). The U4, U5 and U6 snRNAs join the A complex together as a pre-assembled particle called the U4/U6.U5 tri-snRNP and form the spliceosomal B complex. Following integration of the tri-snRNP, the spliceosome is activated for catalysis through a series of conformational and compositional rearrangements (3). These rearrangements result in expulsion of the U1 and U4 snRNPs, recruitment of the Prp19-associated complex and other changes in protein composition and RNA and protein conformation that ultimately result in formation of the spliceosomal active site and 5′ SS cleavage (1,3,4).
The U6 snRNA undergoes major structural rearrangements during tri-snRNP assembly and spliceosomal activation. Within both the Saccharomyces cerevisiae (yeast) U6 snRNP and free U6 snRNA, U6 forms multiple intramolecular base-pairing interactions that create the internal stem loop (ISL) and the telestem (Figure 1A) (5,6). Within the spliceosome, the ISL ultimately functions to help position catalytically important metal ions for catalysis (1,7,8). The ISL is not always present in U6: it forms in the U6 snRNP and in the spliceosomal active site but is unwound and base-paired to U4 (Figure 1B) in the U4/U6 di-snRNP, tri-snRNP and spliceosomal B complex (1,2,9). Even though both the ISL and telestem are present in the U6 snRNP, the telestem is unlikely to be involved in catalysis since it must be unwound to permit U6 base-pairing with U2 (3,10–13). Instead, the telestem may prove most important at other stages in splicing. It has recently been demonstrated that the telestem assists in U4/U6 annealing by the assembly factor Prp24 (14). Thus, while the U6 ISL is an essential component of spliceosomal catalysis, the U6 telestem likely functions in tri-snRNP assembly.
Figure 1.

U6 exhibits dynamics once annealed to U4. (A) Proposed base-pairing structure of the U6 RNA construct (U625-112) used in smFRET assays. FRET donor (Cy3) and acceptor (Cy5) fluorophores are denoted by green and red stars, respectively. The 3′ extension represents additional RNA nucleotides (nt) appended to U6 to facilitate surface immobilization with a 3′ terminal biotin (B). The U6 ISL and telestem are noted on the structure with the telestem strands colored in blue. (B) The U4WT/U625-112 di-RNA used in smFRET assays. Telestem forming regions of U6 are colored in blue. Predicted secondary structure of telestem formation in the U4/U6 di-snRNA (inset) (C) Histogram of EFRET values obtained from single molecules of U625-112 (N = 143). The distribution could be fit to a single Gaussian function centered at 0.95 ± 0.01 (red line). No dynamics were observed in single molecule EFRET time trajectories (inset). (D) Histogram of EFRET values obtained from single molecules of U4WT/U625-112 (N = 121). The distribution could be fit to a sum of two Gaussian functions centered at 0.24 ± 0.01 and 0.68 ± 0.01 (red line). Dynamics consistent with a reversible two-state conformational transition were apparent in single molecule EFRET time trajectories (inset). Data in (C and D) were collected in imaging buffer with 400 mM NaCl (see ‘Materials and Methods’ section). The small number of molecules showing a constant high EFRET of ∼0.95 consistent with the absence of U4 were not included in the histogram of U4WT/U625-112 shown in (D).
How U6 snRNA interconverts between different conformational states remains an outstanding question in understanding spliceosome assembly and activation. During the activation process, U6 must be unwound from U4 by disruption of U4/U6 stems I and II (Figure 1B). Unwinding requires an adenosine triphosphate (ATP)-dependent RNA helicase, Brr2, that loads onto U4 and translocates 3′→5′ to promote U4 release (15). How exactly this occurs is not known; however, Brr2 alleles [e.g. brr2-1 (16,17)] can decrease U4/U6 unwinding in vitro and splicing in vivo. In catalytic spliceosomes, U6 is extensively base-paired with U2 and this base-pairing is presumably established at some point during spliceosomal activation prior to 5′ SS cleavage (1,3). The mechanism of U4 release and the structures of U6 intermediates formed during Brr2 unwinding are still unclear.
To investigate the mechanisms of U4/U6 unwinding, we have used single molecule Förster resonance energy transfer (smFRET) in combination with a novel oligonucleotide-dependent U4/U6 unwinding assay and yeast molecular genetics to study how regions of U6 not base-paired to U4 can nonetheless influence U4/U6 stability. The data reveal that the U6 telestem can form transiently while U6 is basepaired to U4 and that telestem formation promotes U4/U6 unwinding. Telestem stabilization in vivo leads to defects in yeast growth and a substantially altered steady-state distribution of U6, U4/U6 and U4/U6.U5 snRNPs. This work suggests a multi-step pathway for yeast U4/U6 unwinding in which telestem formation precedes U4/U6 stem I unwinding, followed by stem II unwinding and U6 ISL formation.
MATERIALS AND METHODS
Preparation of fluorescent RNAs for smFRET
RNA fragments for smFRET experiments were purchased from IDT containing C6-aminoallyl modifications at the positions indicated (Supplementary Table S1). RNAs were fluorescently labeled with N-hydoxysuccinimidylester derivatives of Cy3 or Cy5 fluorophores (GE Healthcare) by incubation of the RNA (5 nmol) with the fluorophore (40 nmol) overnight at room temperature in labeling buffer (33% v/v dimethylsulfoxide, 100 mM sodium bicarbonate pH 8.5). Excess free dye was removed using an Illustra microspin G-25 column (GE Healthcare). Labeled RNA fragments were then purified by 12% denaturing polyacrylamide gel electrophoresis (PAGE).
The U625-112 fragment was prepared by splinted ligation of two labeled RNAs. The 3′ fragment was first phosphorylated at its 5′ end by incubation of the RNA (60 pmol) with T4 polynucleotide kinase (20 units (U), New England Biolabs) in the provided buffer with 3 mM ATP for 30 min at 37°C. The 5′ fragment (120 pmol) and DNA splint (100 pmol) were then added and annealed together with the 3′ fragment by heating to 95°C for 5 min followed by cooling to 25°C over 30 min. T4 RNA Ligase II (10 U, New England Biolabs) was then added, and RNAs ligated for 30 min at 37°C. Ligated products were purified by 12% denaturing PAGE and quantified by measurement of UV-Visible absorbance and use of calculated extinction coefficients for the RNAs.
Preparation of non-fluorescent RNAs
Non-fluorescent U4 and U6 RNAs (Supplementary Table S2) were prepared by in vitro transcription from a double stranded DNA template using T7 RNA polymerase and purified by denaturing PAGE. When appropriate, RNAs were labeled with radioactive [32P] by inclusion of α-[32P]-UTP (Perkin Elmer) during transcription or by end-labeling with γ-[32P]-ATP (Perkin Elmer). U649-88 was purchased from IDT with an aminoallyl modification at U54 that was left unlabeled in these experiments, and instead was 5′ end labeled with [32P].
Annealing of U4 and U6 RNAs and analysis of complex formation
For single molecule experiments, U4/U6 di-RNAs were prepared by heat annealing of biotinylated, fluorescent U6 RNAs (200 nM) with U4 (3 μM) in annealing buffer (50 mM Tris pH 7.4, 400 mM NaCl) at 95°C for 5 min followed by cooling to room temperature over 30 min. For unwinding and other assays, U4/U6 di-RNAs were prepared using a similar procedure to that described for single molecule experiments. In experiments containing radioactive RNAs, the non-radioactive RNA was always in excess over the radioactive RNA (200 nM [32P]-U6 and 3 μM U4, unless otherwise noted). Complex formation was confirmed by electrophoresis of annealed complexes on a native 6% 29:1 acrylamide:bisacrylamide gel and run in 1 × Tris-borate-ethylenediaminetetraacetic acid (EDTA) buffer (TBE) at 300V for ∼2 h at 4°C.
Analysis of U6 RNA conformation and dynamics by smFRET
smFRET experiments were carried out at room temperature on quartz microscope slides derivatized with a mixture of polyethylene glycol (PEG) and biotin-derivatized PEG and assembled into microfluidic flow chambers as previously described (18). Before use, slides were washed with 200 μl of 1× phosphate buffered saline (PBS) to remove unreacted PEG. Streptavidin (2 μM, Prozyme) was added, incubated for 2 min and then removed by repeated washing steps with 1 × PBS. RNAs were immobilized on the streptavidin-coated surface by incubation with a dilute solution of the RNAs (25 pM) in annealing buffer. Slides were then washed and imaging buffer was added (50 mM Tris–HCl pH 7.5, 400 mM NaCl, 4.5 mg/ml glucose, 150 U/ml catalase, 40 U/ml glucose oxidase, 3 mM Trolox). Single-molecule data were collected on a home-built prism-based total internal reflection fluorescence microscope using 532 and 640 nm lasers for excitation and a cooled electron multiplying-charge coupled device camera (Andor) for image recording after passage of the emitted light through a Dual View apparatus (DV2, Photometrics) to spatially separate Cy3 and Cy5 emission on the detector. Data were collected continuously at 5 frames/s with most experiments lasting ∼3 min. Molecules were typically excited with the 640 nm laser at the beginning and end of each experiment to confirm presence of the Cy5 fluorophore, while the 532 nm laser was used to excite Cy3 and for smFRET.
For data analysis, a mapping function to translate locations between the short (<640 nm) and long wavelength (>640 nM) channels produced by the Dual View apparatus was created using fluorescent beads that fluorescence in both channels. Well-resolved single spots appearing in the long wavelength channel upon excitation with the 640 nm laser were selected and translated to the corresponding location in the short wavelength channel. Translated spot locations in the short wavelength channel showing Cy3 fluorescence or smFRET upon excitation with the 532 nm laser likely contained molecules with both Cy3 and Cy5 fluorophores. FRET donor (Cy3) and acceptor (Cy5) intensities were obtained for each frame by integrating the measured fluorescence intensity from boxes drawn around each spot in each channel. FRET efficiency values (EFRET) for each molecule were then calculated as IA/(IA+ ID), where IA is the intensity of the acceptor at a particular frame and ID is the intensity of the donor at that same frame using vbFRET (19). Histograms were generated by binning the EFRET values for many molecules obtained from replicate experiments.
For molecules showing transitions between two or more EFRET states, the number of fluorescence states were fit by hidden Markov modeling (HMM) using HaMMy (20,21). Dwell times for each state were then collected, combined with those obtained from other molecules and the unbinned collection fit using MatLab (The Mathworks) to determine the transition lifetimes, τ, using a single or double exponential maximum likelihood function (Equations 1 or 2, respectively) where τm and τmax represent the frame duration and length of the experiment, respectively, and A represents the amplitude. Errors in the fit were determined by bootstrapping.
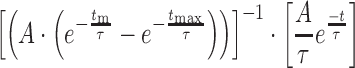 |
(1) |
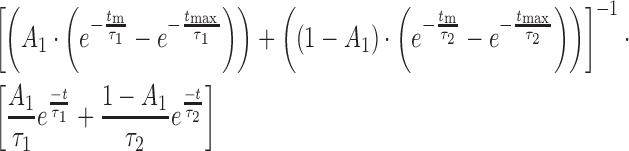 |
(2) |
U4/U6 unwinding assays
U4/U6 unwinding was monitored by 6% acrylamide (29:1) native PAGE using [32P]-labeled U6 annealed to U4. When included, unwinding oligonucleotides (A, B or B2; Supplementary Table S3) were present at 30 μM. Unwinding was carried out at 40°C in annealing buffer (50 mM Tris pH 7.4, 400 mM NaCl), unless otherwise stated. Time points were collected and loaded onto the native gel before electrophoresis at 300V for 2.5 h at 4°C. A phosphorimager screen was then exposed to the gel, imaged on a Typhoon Imager (GE Life Sciences) and data quantified using ImageQuant software (GE Healthcare). The unwinding rate was determined by fitting the fraction of U4/U6 unwound to Equation (3), where A is the amplitude of the reaction, ku is the apparent first-order rate constant for unwinding and t is time.
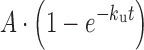 |
(3) |
Plasmids and yeast strains
Mutant U6 snRNA genes were generated by site-directed mutagenesis of the pRS314-U6 plasmid followed by DNA sequencing to confirm the presence of the mutation in the resulting plasmids (Supplementary Table S4). Wild-type (WT) U4 and WT or mutant U6 plasmids were co-transformed into yeast strain CJM000 (Supplementary Table S5) using the lithium acetate method and transformants selected on –trp, –his dropout plates. Selection for loss of the pRS316-U4wt-U6wtmini plasmid was carried out by streaking individual colonies onto –trp, –his dropout plates also containing 5-fluoroorotic acid (5-FOA, 1 mg/ml).
Yeast growth assays
Overnight cultures of each strain were diluted to an OD600 of 0.5 in sterile 10% glycerol and then serially diluted 10-fold before plating onto YPD/agar. Plates were incubated at the indicated temperatures (16, 23, 30, or 37°C) for 2–10 days and monitored for the appearance of colonies.
Solution hybridization assays of U4/U6 di-snRNA abundance
Cellular RNAs were isolated from strains grown at 30°C by phenol extraction at 4°C to maintain U4/U6 base-pairing as previously described (22). Isolated RNA (1 μg) was then hybridized to 5′ [32P] end-labeled oligonucleotides (0.5 pmol each, Supplementary Table S6) in 50 mM Tris pH 7.5, 1 mM EDTA, 150 mM NaCl for 15 min at 37°C (22). Hybrids were resolved by native PAGE (9% acrylamide, 29:1, in 1 × TBE) followed by electrophoresis at 300V for 5 h at 4°C. A phosphorimager screen was then exposed to the gel, imaged, and data analyzed using ImageJ software (23).
Analysis of RNA levels by primer extension and northern blotting
Yeast cultures (30 ml) from each strain were grown at 30°C to an OD600 0.6-0.8 and 10 OD600 units were collected. Pelleted cells were washed with MilliQ water followed by resuspension in 400 μl disruption buffer (10 mM Tris pH 7.5, 10 mM EDTA pH 8, 0.2% sodium dodecyl sulphate). Acid phenol (400 μl) and disruption beads (∼200 μl, 0.5 mm) were added to the cells and vortexed for 5 min at room temperature using a Disruptor Genie. Samples were centrifuged and the aqueous layer was collected. To the aqueous layer, acid phenol (400 μl) was added and the samples were incubated at 65°C for ∼30 min. Following heating, the aqueous layer was collected and two additional phenol:chloroform:isoamyl alcohol (25:24:1) extractions were performed followed by a chloroform:isoamyl alcohol extraction to get rid of excess phenol. Samples were then ethanol precipitated and quantified by UV spectroscopy.
Primer extension was performed using a qScript Flex cDNA kit (Quanta Biosciences). Briefly, 10 μg of total RNA was incubated at 65°C for 5 min with ∼0.7 μM of each Cy5-labeled primer (Supplementary Table S7) and 2 μl of GSP enhancer (Quanta Biosciences) in a 10 μl reaction. Reaction buffer (3 μl, Quanta Biosciences) and reverse transcriptase (1 μl) were added and the reactions were incubated at 42°C for 1 h. Reactions were inactivated by incubating at 85°C for 5 min. Deionized formamide RNA loading buffer (15 μl) was added, and samples were heat denaturated at 95°C for 2 min and then loaded onto a 7% denaturing PAGE gel. Electrophoresis was carried out at 22W for 1 h in 1 × TBE. Fluorescent products were imaged using a Typhoon Gel Imager (GE).
Northern blotting was performed by loading 5 μg of total RNA on a 5% denaturing PAGE gel followed by electrophoresis at 30W for ∼1 h in 1 × TBE. RNAs were then transferred to a Hybond-XL membrane (Amersham) at 25V for 1 h in 0.5 × TBE using a Trans-Blot SD Semi-Dry Transfer Cell (BioRad). Blots were dried at room temperature for 30 min and then crosslinked at 1200 mJ using a UV-crosslinker (UVP). Blots were then blocked, probed, washed and imaged as previously described (22). Probes used for northern blotting of denaturing gels are outlined in Supplementary Table S6 (U1-Long, U4-Long, U5-Long and U6-Long).
Native gel analysis of snRNP distribution
Native PAGE analysis of spliceosomal snRNPs was carried out by first preparing whole cell extracts from the appropriate yeast strains as previously described (22) but using the following protease inhibitors: 1 mM PMSF, 2 mM benzamidine, 1 μM leupeptin, 2 μM pepstatin A, 4 μM chymostatin and 2.6 μM aprotinin. Lysates were clarified by centrifugation using a Beckman JA-20 rotor at 18 000 rpm for 30 min followed by centrifugation at 36 000 rpm for 1 h in a Beckman SW40-Ti rotor. Extract was collected and dialyzed using a 10 kD cutoff-membrane (Slide-A-Lyzer, ThermoFisher) twice against 1.5 l of 20 mM HEPES pH 7.9, 50 mM KCl, 0.5 mM EDTA, 0.2 mM DTT and 20% glycerol for 1.5–2 h.
Native PAGE analysis of spliceosomal snRNPs was carried out as described (16,22). Diluted extracts (40% v/v; diluted with buffer to a final concentration of 60 mM potassium phosphate pH 7.3, 2.5 mM MgCl2, 1 mM spermidine and 3% w/v PEG-8000) were incubated at the indicated temperatures for 10 min and loaded directly into the wells of a non-denaturing acrylamide gel (4%, 80:1; 20 × 24 × 1.5 cm) and electrophoresis occurred at 240V over 5 h at 4°C in TGM buffer (50 mM Tris, 50 mM glycine, 2 mM MgCl2). When included, ATP was added to a final concentration of 5 mM. Where indicated, proteinase K (2 mg/ml) was added to diluted extracts and incubated at 30°C for 10 min prior to loading.
RNAs were transferred from the gel to a Hybond-XL membrane (Amersham) for 16–18 h at 1.5A (20–30V) in 50 mM sodium phosphate pH 6.5 in a blotting unit (Hoefer TE42). RNAs were crosslinked to the membrane with a UVP at 1200 mJ for 1.5 min, repeated for a total of three exposures. Membranes were then blocked, probed, washed, and imaged as previously described (22). Probes used for native northern analysis are described in Supplementary Table S6. Before re-probing, membranes were stripped by incubation in water at 90–100°C for 10–30 min followed by blocking.
RESULTS
U6 is conformationally dynamic when base-paired with U4
While previous data from us and others suggest that the three-helix junction formed between the U4 and U6 snRNAs is remarkably rigid in the absence of protein (24,25), we wondered if the peripheral regions of U6, those not base-paired with U4, are dynamic. These peripheral regions extend both 5′ and 3′ from the U6 domain that is base-paired with U4 in the di-snRNA and were not included in previous smFRET or nuclear magnetic resonance small-angle X-ray scattering experiments (24,25). We therefore created a U6 smFRET-reporter molecule containing the majority of the S. cerevisiae (hereafter ‘yeast’) U6 snRNA sequence (nt 25–112; U625-112) and a 20-nt, 3′ extension that terminates with a biotin for surface immobilization (Figure 1A). FRET donor (Cy3) and acceptor (Cy5) fluorophores were incorporated by derivatization of two RNA fragments (Supplementary Table S1) containing amino-allyl modified deoxyuridine nucleotides incorporated at positions corresponding to U54 and U90 in the U6 snRNA, and the final RNA was prepared by splinted ligation of these fragments. We reasoned that fluorophores incorporated at U54 and U90 would exhibit at least two EFRET (FRET efficiency) values: a low EFRET state when U6 was base-paired to U4 and the fluorophores are predicted to be far apart (Figure 1B), and a higher EFRET state in the absence of U4 due to U6 intramolecular base-pairing within the ISL and telestem regions (Figure 1A).
In the absence of U4, single molecules of the U6 smFRET reporter exhibited a predominant high EFRET of ∼0.9 with little evidence of dynamics (Figure 1C, inset). When EFRET values from many molecules of the reporter were combined, the resulting histogram showed a sharp peak that could be fit to a single Gaussian distribution centered at 0.95. By moving the Cy3 FRET donor molecule from U54 to U46 within the U6 asymmetric bulge region, a lower EFRET was observed (0.75) but the smFRET signals remained static (Supplementary Figure S1A and B). These results are consistent with U6 predominantly folding into a single conformation in which U54 and U90 are proximal and U46 resides a greater distance from U90 than does U54.
We next heat-annealed the U6 smFRET reporter to an in vitro-prepared transcript representing the entire yeast U4 snRNA sequence (U4WT). Complex formation was confirmed by native gel electrophoresis (Supplementary Figure S2A, lane 4), and the U4/U6 duplex RNA was analyzed by smFRET by immobilizing the U4/U6 duplexes and washing away any unbound RNA. Unexpectedly, instead of the static smFRET signal observed with U6 alone (Figure 1C) or the U4/U6 three-helix junction (24,25), 90% of U4/U6 molecules showed reversible transitions between two states with EFRET values of ∼0.2 and ∼0.7 (Figure 1D). The small number of remaining molecules that did not transition either exhibited a constant high EFRET of 0.9 consistent with absence of U4 (∼5%) or showed no transitions from the 0.2 EFRET state (∼5%). Thus, in contrast to previous studies of the U4/U6 three-helix junction, larger U6 RNAs become dynamic once base-paired to full-length U4.
Visual inspection and HMM of individual smFRET trajectories as well as analysis of histograms of EFRET revealed that the dynamic transitions observed in the U4/U6 duplex occurred almost exclusively between two states. Transitions occurred well within the timescale of our smFRET experiments with the lifetime of the low 0.23 EFRET state (7.2 ± 0.3 s) being longer than that of the higher 0.68 EFRET state, whose distribution of lifetimes was best fit to an equation containing two exponential terms (τ1 = 0.7 ± 0.3 s, amplitude = 0.6 ± 0.1; τ2 = 5.2 ± 3.4 s; amplitude = 0.4 ± 0.1; Supplementary Figure S3). The equilibrium constant (Keq) for molecules undergoing reversible transitions from the low to the high EFRET state was measured to be 0.40. This corresponds to a small increase in stability upon transitioning to the low EFRET state (ΔG = −0.53 kcal/mol). The high EFRET state was dependent on salt concentration, with divalent metal ions (10 mM Mg2+) and high monovalent metal ion concentrations (400 mM Na+, consistent with annealing conditions) each favoring its formation (Supplementary Figure S4). These results suggest that cellular ionic conditions may also influence U4/U6 dynamics. Together, the data demonstrate that when U6 is base-paired to U4, the regions of U6 flanking stems I and II undergo spontaneous transitions between conformations with distinct EFRET states and stabilities. A transient long-range intramolecular interaction in U6 that brings these regions into closer proximity could explain these data.
U6 telestem mutations disrupt U4/U6 dynamics
The human U6 RNA contains widely-separated sequences that can form an intramolecular duplex even while U6 is base-paired to U4 (26). We wondered if the dynamics we observe with our yeast U4/U6 model arise from transient formation of a similar intramolecular duplex. In yeast U6, the corresponding duplex is called the telestem, where the 5′-strand of the telestem flanks U4/U6 stem I and the 3′-strand flanks U4/U6 stem II (Figure 1B, inset). We tested this hypothesis by introducing mutations in the U6 smFRET reporter that are predicted to disrupt or stabilize telestem formation.
We generated mutant U6 smFRET reporter RNAs that disrupted two specific regions of the telestem (U6MUT1 and U6MUT2; Figure 2B and C). In the absence of U4, these U6 mutants behaved identically to U625-112 and showed a single, static high EFRET state of ∼0.95 (Supplementary Figure S5). These data indicate that the intramolecular interactions involved in formation of the static EFRET state are not significantly perturbed by these telestem mutations.
Figure 2.

Alteration of telestem base-pairing also alters U6 smFRET dynamics in U4/U6. Each panel has at left a cartoon of the U41-64/U625-112 di-RNA used for the experiment, including any mutations in the U6 telestem (in red) and a histogram of EFRET values obtained from single molecules at right. (A) U41-64/U625-112 di-RNA without any mutations (N = 213). The distribution could be fit to a sum of two Gaussian functions centered at 0.24 ± 0.01 and 0.73 ± 0.01 (red line). Dynamics consistent with a reversible two-state conformational transition were apparent in single molecule EFRET time trajectories (inset). (B) U41-64/U6MUT1 (N = 102). The distribution could be fit to a sum of two Gaussian functions centered at 0.22 ± 0.01 and 0.71 ± 0.04 (red line). Single molecule EFRET time trajectories showed transient excursions to the short-lived 0.71 EFRET state (inset). (C) U41-64/U6MUT2 (N = 104). The distribution could be fit to a single Gaussian function centered at 0.22 ± 0.01 (red line). No dynamics were observed in single molecule EFRET time trajectories (inset). (D) U41-64/U6BASE (N = 114). The distribution could be fit to a sum of two Gaussian functions centered at 0.23 ± 0.01 and 0.69 ± 0.01 (red line). Dynamics consistent with a reversible two-state conformational transition were apparent in single molecule EFRET time trajectories (inset). (E) U41-64/U6ΔBULGE (N = 126). The distribution could be fit to a sum of two Gaussian functions centered at 0.22 ± 0.01 and 0.69 ± 0.01 (red line). Static ∼0.2 and ∼0.7 states were observed (inset). (F) U41-64/U6BASE + ΔBULGE (N = 114). The distribution could be fit to a sum of two Gaussian functions centered at 0.23 ± 0.05 and 0.73 ± 0.01 (red line). Example of a single molecule EFRET time trajectories showing no excursions from the 0.7 EFRET state (inset). Data in (A–F) were collected in imaging buffer with 400 mM NaCl (see ‘Materials and Methods’ section), and molecules showing a constant high EFRET of ∼0.95 consistent with the absence of U4 were not included in these histogram analyses.
We then analyzed the behavior of the telestem-destabilizing U6 mutants once base-paired to U4. Since it is possible that the U6 mutations could also result in formation of new base-pairing interactions to the 3′ single stranded (ss) RNA region of U4 (the U4 central domain), we first tested whether or not a shorter U4 fragment could be used in the single molecule assay. This U4 fragment (nts 1–64, U41-64) contains only the regions that base-pair with U6 to form U4/U6 stems I and II and the intervening U4 5′ stem loop. We annealed the WT U6 FRET reporter (U625-112) as well as each of the U6 mutants to U41-64 and monitored complex formation by native gel electrophoresis (Supplementary Figure S2B). In agreement with our previous experiments using a similarly truncated U4 (25), the 3′ region of U4 is not essential for stable U4/U6 complex formation with either U625-112, U6MUT1 or U6MUT2.
We examined the smFRET dynamics of WT U625-112 when annealed to U41-64 and compared the results to those obtained with U4WT. We observed similar EFRET states and distributions with U41-64/U625-112 compared to U4WT/U625-112 (cf. Figures 1D and 2A). The lifetime of the high EFRET state (but not the low EFRET state) differed when U625-112 was annealed to U41-64 compared with U4WT. While the distribution of high EFRET lifetimes could be fit to two exponential terms when U4WT was used, only a single lifetime was observed with U41-64 (τ = 1.7 ± 0.1 s, Supplementary Figure S3C and D). These results show that although the 3′ region of U4 (nt 65–160) is not required for promoting transitions in U6 in the di-RNA, it does subtly influence high EFRET state dynamics.
Disruption of either region of the telestem (U41-64/U6MUT1 or U41-64/U6MUT2) dramatically reduced the number of observed smFRET transitions. Both U41-64/U6MUT1 and U41-64/U6MUT2 showed predominantly low EFRET of ∼0.2, the same low EFRET value observed when WT U625-112 was annealed to U4 (Figure 2A–C). The high EFRET state was completely abolished by mutation of nt 36–39 (U6MUT2, Figure 2C), while transient, very short-lived excursions to high EFRET could still be observed upon disruption of nt 30–33 (U6MUT1, Figure 2B). Kinetic analysis of dwell times of the U6MUT1 high EFRET state revealed a single exponential distribution and a decrease in the fitted lifetime compared to U625-112 (τ = 0.6 ± 0.1 s, Supplementary Figure S3E). Therefore, destabilization of the telestem region by mutation results in decreases in both the abundance and lifetime of the high EFRET state.
In addition to telestem-destabilizing mutations, we also designed a series of U6 smFRET reporters of increasing predicted telestem thermodynamic stability (Figure 2D–F). By replacing the G/U wobble pairs at the base of the telestem with G/C pairs (U6BASE), we predicted a modest increase in Tm of ∼7°C using DINAMelt (27). Similarly, we replaced two mismatched base-pairs (A-A and A-G) in the middle of the telestem with two G-C base-pairs (U6ΔBULGE). This modification creates a continuous telestem duplex and results in an increase in predicted Tm of ∼32°C. Our most stabilized U6 combined the mutations in U6BASE and U6ΔBULGE to create a continuous telestem terminating in G-C base-pairs and in an increase in predicted Tm of ∼40°C (U6BASE + ΔBULGE). Finally, a set of U6 mutants were made containing mutations in either the 5′ or 3′ strand of the telestem but without the corresponding base-pair mutation on the opposite strand (U6A34G,A35G, U6G96C,A97C, and U6G96C,A97C,U100C,U101C).
Stabilization of the telestem did not result in any significant changes in distributions of EFRET when the U6 mutants were analyzed in the absence of U4 (Supplementary Figure S5). Further, native PAGE confirmed complex formation with U41-64 for each of the stabilizing mutants (Supplementary Figure S2C). After annealing with U41-64, we observed that mutations that stabilize the telestem also increased the high EFRET state lifetime (Figure 2D–F). The U6BASE mutation most resembled U625-112 and showed dynamic transitions between two EFRET states. In agreement with telestem stabilization, the lifetime of the high EFRET state increased due to the U6BASE mutation (τ = 5.4 ± 0.6 s; Supplementary Figure S3F). The more stabilizing U6ΔBULGE and U6BASE + ΔBULGE mutations impacted the FRET dynamics more significantly than did U6BASE (Figure 2D–F). The most stabilizing U6BASE + ΔBULGE mutation resulted in appearance of predominantly the high EFRET state and very few transitions to low EFRET. This is consistent with U6BASE + ΔBULGE forming a stable telestem while simultaneously being paired to U4. The U6ΔBULGE mutation also showed a much greater abundance of the high EFRET state compared with U625-112, but less so than U6BASE + ΔBULGE. Interestingly, despite observation of U6ΔBULGE molecules possessing either high EFRET or low EFRET, the molecules did not transition between these FRET states during the assay. This finding suggests that the U6ΔBULGE mutation may be stabilizing both the telestem and an alternate structure with low EFRET that does not transition. In agreement with this hypothesis, the U6A34G,A35G RNA, which contains only the 5′-strand mutations found in U6ΔBULGE, also fails to transition to high EFRET (Supplementary Figure S6). Stabilization of the low EFRET state by the A34G and A35G mutations is countered by simultaneous stabilization of the telestem base in the U6BASE + ΔBULGE mutant, in which the low EFRET state is nearly absent (Figure 2F).
In total, our FRET data reveal that the high EFRET state observed in our single-molecule assay is destabilized by mutations that disrupt telestem base-pairing and stabilized by mutations that increase telestem base-pairing. We conclude that transient formation of the telestem occurs in yeast U4/U6 di-RNAs in vitro.
Complementary DNAs accelerate U4/U6 unwinding
The human U4/U6 di-snRNA is destabilized by intramolecular duplex formation in U6, resulting in U4/U6 unwinding and dissociation of U4 from U6 at physiological temperatures (26). Yeast U4/U6, in contrast, remains stably base-paired and appreciable unwinding/melting of the di-snRNA only occurs at temperatures above 50°C (26,28). This finding suggests that if telestem formation occurs in yeast U4/U6 it does not lead to unwinding at temperatures that support yeast growth. We wondered if telestem formation could accelerate yeast U4/U6 unwinding under conditions in which U4/U6 base-pairing had already been partially disrupted. In the spliceosome the ∼250 kDa Brr2 helicase unwinds U4/U6, likely beginning with stem I (15). Stem I disruption could facilitate formation of U6 structures, such as the telestem, that also contribute to U4/U6 unwinding. Since we wished to study the influence of telestem formation in the absence of additional factors such as Brr2, we first developed a simple method to disrupt stem I and unwind U4/U6 that does not rely on proteins or ATP hydrolysis.
Nucleic acid strand exchange can be driven by hybridization of a complementary oligonucleotide that leads to invasion of the duplex and strand displacement (i.e. toehold mediated-strand displacement) (29,30). We tested whether or not this method could be used as a proxy for ATP-dependent Brr2-unwinding of U4/U6 stem I. We designed two DNA oligonucleotides (oligonucleotides A and B) that are each complementary to U4 (Figure 3A). Oligonucleotide A is complementary to both the U4 central domain and U4/U6 stem I (U4 nt 53–91), while oligonucleotide B is complementary to U4/U6 stem II (U4 nt 1–19). These oligonucleotides could be used to disrupt U4/U6 stem I by hybridization of oligonucleotide A to the U4 central domain to form a toehold followed by invasion of stem I to release the corresponding region of U6. Oligonucleotide B may potentially unwind U4/U6 stem II as well as capture free U4.
Figure 3.

DNA oligonucleotides complementary to U4 accelerate U4/U6 unwinding. In all experiments U6WT was labeled with [32P] before annealing to excess unlabeled U4WT. U4WT/U6WT and free U6WT were then resolved by 6% native PAGE prior to phosphorimaging. (A) Cartoon showing base-pairing of the unwinding oligonucleotides A and B (red lines) to U4 at nucleotides 55–90 and 1–19, respectively. (B) Oligonucleotides A and B lower the melting temperature of U4/U6. U4WT/U6WT was incubated in the presence or absence of oligonucleotides A and B (30 μM) for 10 min at the indicated temperatures prior to native PAGE. (C) U4WT/U6WT unwinding with oligonucleotides A and B occurs more slowly at 20°C (lanes 1–7) than at 40°C (lanes 9–15), and no unwinding is observed in the absence of the oligonucleotides at either temperature (lanes 8 and 16). (D) Oligonucleotide A by itself does not catalyze unwinding at 40°C (lanes 1–7); however, oligonucleotide B can slowly unwind U4/U6 (lanes 10–16). (E) A truncated oligonucleotide B (B2) that cannot base-pair near the U4/U6 three-helical junction has a reduced rate of U4/U6 unwinding both in the absence (lanes 1–7) and presence of oligonucleotide A (lanes 8–14). (F) Quantification of U4WT/U6WT unwinding by oligonucleotides A, B and B2. Data points represent the results from three separate experiments and lines represent the fits to these data. Fitted rate constants are reported in the main text. Data in (B–F) were acquired in annealing buffer with 400 mM NaCl (see ‘Materials and Methods’ section).
In the absence of oligonucleotides A and B, free U6 did not accumulate at temperatures lower than 60°C in buffer containing 400 mM NaCl, in agreement with previous results (Figure 3B). However, simultaneous addition of both oligonucleotides resulted in a dramatic increase in apparent unwinding at physiological temperatures (Figure 3B). Free U6 now accumulated even at 30°C, similar to results reported for human U4/U6 di-snRNAs in the absence of complementary oligonucleotides (30–35°C) (28). Our data indicate that oligonucleotides complementary to U4 can be used to destabilize yeast U4/U6 and accumulate free U6 and U4 at physiological temperatures.
We next characterized the properties of oligonucleotides A and B that accelerated U4/U6 unwinding. We carried out the assays at 40°C since U4/U6 remains associated at this temperature in the absence of oligonucleotides; however, their addition results in nearly complete unwinding after 60 min with a rate of 0.065 ± 0.003 min−1 compared to 0.014 ± 0.003 min−1 at 20°C (Figure 3C and F). When only oligonucleotide A was added, no U4/U6 unwinding was observed (Figure 3D, lanes 1–7). However, we were able to observe a gel shift when oligonucleotide A was incubated with U4/U6 (Figure 3D, lanes 1–7 versus 8), which indicates that while oligonucleotide A does not by itself unwind U4/U6, it does hybridize to U4. In contrast, oligonucleotide B alone was able to promote U4/U6 unwinding, albeit at a 10-fold slower rate (0.006 ± 0.003 min−1) than oligonucleotides A and B together (Figure 3D, lanes 10–16 and F). We were not able to observe complex formation between U4/U6 and oligonucleotide B by native gel electrophoresis, suggesting that hybridization of B to U4 results in rapid or concomitant U4/U6 unwinding. This result is also consistent with base-pairing between U4 and U6 only in stem I being insufficient to maintain U4/U6 complex stability under these assay conditions. Together, these data suggest that the stem II-complementary oligonucleotide B is responsible for U4/U6 unwinding, but this activity is accelerated by the simultaneous presence of oligonucleotide A and unwinding of U4/U6 stem I.
To confirm that oligonucleotide B was indeed responsible for U4/U6 unwinding and not merely capturing free U4 produced by spontaneous dissociation of U4/U6, we truncated oligonucleotide B by two nucleotides at its 5′ end (oligonucleotide B2, shown in Supplementary Figure S7). We reasoned that if oligonucleotide B was unwinding U4/U6, it may occur by hybridization to create a toehold followed by invasion of stem II and displacement of U6. The truncated oligonucleotide B2 should decrease the likelihood of forming a toehold near the three-helix junction but not change the oligonucleotide's ability to capture free U4. When oligonucleotide B2 was added to U4/U6, no unwinding was observed, in contrast to results obtained with oligonucleotide B (Figure 3E, lanes 1–7 and F). When both oligonucleotide A and oligonucleotide B2 were incubated with U4/U6, unwinding occurred but at a 3-fold slower rate (0.021 ± 0.005 min−1) compared to when oligonucleotides A and B were used (Figure 3E, lanes 8–14 and F). These data support a model in which oligonucleotide B alone disrupts stem II by first hybridizing to U4 near the open end of the 5′ stem loop followed by displacement of U6.
Finally, we examined properties of oligonucleotide A that contribute to unwinding when used in combination with oligonucleotide B. Since we believe that oligonucleotide A is hybridizing to the U4 central domain and this toehold facilitates subsequent invasion of U4/U6 stem I, we predicted that unwinding would be greatly reduced if oligonucleotide A were split into two units (oligonucleotides C and D). Oligonucleotide D is only complementary to U4/U6 stem I and cannot create a toehold, while oligonucleotide C is only complementary to the central domain and cannot invade stem I. When oligonucleotides C or D were used in combination with oligonucleotide B, the unwinding rates were near what was observed in the presence of oligonucleotide B alone (Supplementary Figure S7). No combinations of oligonucleotides B, C and D were as efficient in promoting unwinding as when B was used together with oligonucleotide A. These observations imply that invasion of stem I by oligonucleotide A is facilitated by hybridization to the adjacent region of U4 to create a toehold. In sum, these data demonstrate that U4/U6 unwinding can be driven by U4-complementary oligonucleotides. Unwinding of U4/U6 requires disruption of stem II with oligonucleotide B but not stem I with oligonucleotide A; however, disruption of stem I greatly facilitates unwinding of stem II. It therefore follows that unwinding of U4/U6 stem I by Brr2 in vivo could potentially facilitate unwinding of stem II by some other means.
The telestem accelerates U4/U6 unwinding
Having established the properties of our oligonucleotide-driven U4/U6 unwinding assay, we next tested whether or not the telestem contributes to unwinding. We compared rates of oligonucleotide-mediated unwinding of U4WT/U6WT with a truncated U6 that cannot form the telestem (U4WT/U649-88) and with a U6 mutant containing a telestem stabilized by six contiguous G-C or C-G base pairs (U4WT/U6GC) (Figure 4A, B). The U6GC mutant was prepared by introducing eight mutations into the telestem (U33C/A34G/A35G/G96C/A97C/A99G/U100C/U101C), and this same mutant was previously shown to accelerate Prp24-catalyzed annealing of U4 and U6 in vitro (14).
Figure 4.

Telestem formation promotes U4/U6 unwinding. Unwinding assays were carried out at 40°C using oligonucleotides A and B, and U6 RNAs were labeled with [32P] before annealing to excess unlabeled U4WT. U4WT/U6 and free U6 RNAs were then resolved by 6% native PAGE prior to phosphorimaging. (A, top) Cartoon showing U649-88, which cannot form a telestem, annealed to U4WT. (A, bottom) In the absence of the telestem, unwinding of U4WT/U649-88 by oligonucleotides A and B occurs slowly. (B, top) Cartoon showing U6GC, which contains a GC-rich telestem (red), annealed to U4WT. (B, bottom) In the presence of a stabilized telestem, unwinding of U4WT/U6GC by oligonucleotides A and B occurs rapidly. (C) Quantification of unwinding data obtained from U4WT/U6WT, U4WT/U649-88 and U4WT/U6GC by addition of oligonucleotides A and B. Data points represent the results from three separate experiments and lines represent the fits to these data. (D) Unwinding rates for the experiments shown in (C). Error bars represent the error in the fit. (E) U4WT/U6GC unwinding by oligonucleotides A or B alone. (F) U4WT/U6GC unwinding by oligonucleotides B2 alone or A and B2. (G) Quantification of U4WT/U6GC unwinding by oligonucleotides A, B and B2. Fitted unwinding rates are as follows U4WT/U6GC + oligonucleotide A = no unwinding, U4WT/U6GC + oligonucleotide B = 0.013 ± 0.002 min−1, U4WT/U6GC + oligonucleotide B2 = 0.001 ± 0.007 min−1, U4WT/U6GC + oligonucleotides A and B2 = 0.037 ± 0.004 min−1 and U4WT/U6GC + oligonucleotides A and B = 0.216 ± 0.027 min−1. Data points represent the results from three separate experiments and lines represent the fits to these data. Data in (A–G) were acquired in annealing buffer with 400 mM NaCl (see ‘Materials and Methods’ section).
Deletion of the telestem resulted in ∼5-fold decrease in the rate of oligonucleotide-mediated unwinding of U4WT/U649-88 compared to U4WT/U6WT (Figure 4A, C and D). In contrast, stabilization of the telestem with additional G-C base-pairs resulted in ∼3-fold increased rate of unwinding compared to U4WT/U6WT (Figure 4B–D). Consistent with U6GC destabilizing U4/U6, free U6GC also accumulated to a greater extent than U6WT and at lower temperatures when the di-RNAs were denatured by heating (Supplementary Figure S8), reflecting ∼5°C decrease in apparent Tm. Thus, U6GC is able to promote both U4/U6 assembly by Prp24 (14) and U4/U6 oligonucleotide-mediated unwinding.
When U4WT/U6GC was incubated with individual unwinding oligonucleotides A, B or B2, we observed that the rates of unwinding by oligonucleotides B and B2 were faster than in the corresponding experiments with U4WT/U6WT (Figure 4E–G). As with U4WT/U6WT, oligonucleotide A did not induce unwinding of U4WT/U6GC and the fastest rates of unwinding were observed with the combination of oligonucleotides A and B rather than A and B2 (Figure 4E–G). These data indicate that the rate acceleration arising from U6GC is likely due to promotion of stem II unwinding by oligonucleotide B and that stem I disruption is still needed for efficient unwinding of U4WT/U6GC. Thus, the telestem-forming regions of U6 can accelerate the rate of U4/U6 unwinding, and strengthening telestem base-pairing leads to an increase in unwinding rate. These results suggest that telestem formation is an intermediate step during oligonucleotide-mediated unwinding of U4 from U6.
Disruption of U4/U6 stem I results in new U6 conformations
Our bulk unwinding assays imply that both telestem formation and U4/U6 stem I disruption by oligonucleotide A facilitate U4/U6 unwinding by oligonucleotide B. We wondered if oligonucleotide A's influence on the unwinding rate could be explained by stem I disruption leading to new U6 conformations that facilitate stem II unwinding by oligonucleotide B. Since oligonucleotide A forms a stable complex with U4WT/U6WT (Figure 3D), we prepared ternary complexes containing oligonucleotide A, U4WT and the U625-112 smFRET reporter (Figure 5A) and used smFRET to study U625-112 conformation.
Figure 5.

Destabilization of U4/U6 stem I changes U6 smFRET dynamics. (A) Cartoon of the oligonucleotide A/U4WT/U625-112 ternary complex used for smFRET experiments showing disruption of stem I by oligonucleotide A. (B) Histogram of EFRET values obtained from single molecules of oligonucleotide A/U4WT/U625-112 (N = 172, blue) overlaid with the distribution obtained from single molecules of U4WT/U625-112 (Figure 1D, gray). The distribution could be fit to a sum of two Gaussian functions centered at, 0.39 ± 0.01 and 0.91 ± 0.01 (red line). (C) Single molecule EFRET time trajectories predominantly showed reversible excursions between the 0.39 and 0.90 EFRET states. (D) Cartoon of the U41-55/U625-112 di-RNA complex that lacks stem I used for smFRET experiments. (E) Histogram of EFRET values obtained from single molecules of U41-55/U625-112 (N = 105, blue) overlaid with the distribution obtained from single molecules of U4WT/U625-112 (Figure 1D, gray). The distribution could be fit to a sum of two Gaussian functions centered at 0.35 ± 0.01 and 0.90 ± 0.01 (red line). (F) Single molecule EFRET time trajectories predominantly showed reversible excursions between the 0.35 and 0.90 EFRET states. Data in (B, C, E and F) were acquired in imaging buffer with 400 mM NaCl (see ‘Materials and Methods’ section), and molecules showing a constant high EFRET of ∼0.95 consistent with the absence of U4 were not included in these histogram analyses.
The oligonucleotide A/U4WT/U625-112 ternary complex predominantly underwent transitions between two EFRET states: a longer-lived EFRET state of 0.39 and a shorter-lived EFRET of 0.90 (Figure 5B and C). The 0.39 EFRET state had not been previously observed in our experiments with U625-112 either in the presence or absence of U4. This may correspond to a conformation in which U6 has base-paired to form the telestem and additional base-pairs present in free U6 (e.g. the region between the ISL and the telestem, Figure 1A). Addition of oligonucleotide C, which binds to U4 but cannot invade stem I, in lieu of oligonucleotide A does not cause the peaks to shift in the EFRET distribution (Supplementary Figure S9). This finding suggests that oligonucleotide A induces an increased proximity of the FRET pair located at U54 and U90 via disruption of U4/U6 stem I. The higher EFRET value of 0.90 is similar to values obtained for U625-112 in the absence of U4 (EFRET ∼0.95, Figure 1C). However, the 0.90 EFRET state in this experiment rapidly switches back to the 0.39 EFRET state and thus is unlikely to correspond to free U6 unwound from U4. We conclude that U6 molecules showing transitions to and from EFRET of 0.90 likely contain both U4 and oligonucleotide A.
To confirm that the changes in EFRET were due to disruption of stem I by oligonucleotide A, we annealed U625-112 to a shorter U4 fragment (U41-55) lacking the nucleotides responsible for base-pairing with U6 in stem I (Figure 5D). As with the oligonucleotide A/U4WT/U625-112 ternary complex, molecules of U41-55/U625-112 showed fluctuations between a longer lived state with EFRET of 0.35 and a shorter lived state with EFRET of 0.90 (Figure 5E and F). This shows that U625-112 is undergoing a similar set of conformational dynamics when U4/U6 stem I is disrupted by oligonucleotide A or is prevented from forming by truncation of U4.
Together our single molecule data suggest multiple conformations of U6 arise due to U4/U6 stem I disruption and these conformations are not present when stem I is intact. It is possible that the 0.90 EFRET state is an on-pathway intermediate that leads to unwinding, since it exhibits a similar EFRET value as U625-112 in the absence of U4. Oligonucleotide B could promote unwinding by binding to U4/U6 while it transiently forms the telestem and this high EFRET conformation before dislodging U6. Since U4/U6 remains stable with only stem II base-paired, disruption of stem II and likely concomitant U6 ISL formation represent key thermodynamic events to complete folding of U6 into a structure with the static features observed in our smFRET assay of U6 (Figure 1C).
Stabilizing the telestem in vivo confers growth defects
Based on our smFRET and unwinding assays, we hypothesized that telestem stabilization could result in a strong phenotype in yeast due to either an increase in the rate of U4/U6 unwinding or disregulation of this process. This disregulation could potentially impact both spliceosome activation by causing premature U4/U6 unwinding and tri-snRNP stability by disrupting the equilibrium between Brr2-mediated disassembly and Sad1-mediated assembly (31).
Using a previously described yeast strain harboring deletions of both the U4 and U6 snRNA genes along with a complementing URA3-marked plasmid (32), we tested whether or not telestem stabilization confers a growth defect in vivo. We designed a series of U6 genes containing the same stabilized telestems used in smFRET assays (U6BASE, U6ΔBULGE and U6BASE + ΔBULGE; Figure 2D–F) as well as set of alleles containing mutations in either the 5′ or 3′ strand of the telestem but without the corresponding base-pair mutation on the opposite strand (U6A34G,A35G, U6G96C,A97C and U6G96C,A97C,U100C,U101C; Supplementary Figure S6). Each of these mutant U6 alleles was transformed individually into the U4/U6 deletion strain along with the WT U4 gene (32). Transformants were selected on dropout media and single colonies streaked onto medium containing 5-FOA at 30°C to select for loss of the WT U4/U6 URA3-marked plasmid.
U6BASE, U6ΔBULGE, and all of the mutants containing mutations in either the 5′ or 3′ strand of the telestem were viable when plated on 5-FOA. In contrast, our most stabilized telestem construct, U6BASE + ΔBULGE, was lethal after selection for growth on 5-FOA at either 30 (Supplementary Figure S10A) or 37°C (not shown). These results show that yeast viability can be maintained with U6 mutations designed to stabilize either the middle or one end of the telestem, but not when these mutations are present in combination. The U6G96C,A97C,U100C,U101C mutant, which contains all of the substitutions in the 3′ strand of the telestem found in U6BASE + ΔBULGE but lacks the 5′ strand substitutions, was viable on 5-FOA and displayed no detectable growth defect on rich medium (YPD) at temperatures ranging from 16 to 37°C (Figure 6A). This result suggests that lethality of the U6BASE + ΔBULGE mutation arises from stabilization of the telestem rather than disruption of base-pairing interactions between U2 snRNA and the 3′ strand of the telestem that occur in post-B complex spliceosomes (i.e. U2/U6 helix II; Supplementary Figure S10B). Thus, U6BASE + ΔBULGE results in stable telestem formation by smFRET (Figure 2F) and does not support yeast growth. It is possible that a lack of telestem dynamics contributes to this lethality.
Figure 6.

Stabilizing the telestem can interfere with yeast growth but does not prevent U4/U6 formation in vivo. (A) The effects of telestem mutations were assessed by 10-fold serial dilution and plating of yeast containing the indicated U6 alleles onto YPD/agar media followed by incubation of the plates at the indicated temperatures. The U6BASE + ΔBULGE mutation was unable to survive the 5-FOA selection and thus is recessive lethal. (B) Solution hybridization assay of whole-cell RNA. RNA was extracted from the indicated strains at 4°C to preserve U4/U6 base-pairing, hybridized to [32P]-labeled DNA oligonucleotides complementary to U1, U4 or U6 at 37°C, and analyzed by non-denaturing 9% PAGE followed by phosphorimaging. U6-SH probe was used in lanes 2, 6, 8 and 10 and U6-SH-MUT probe was used in lanes 4 and 12 to accommodate the A34G/A35G mutations. U4/U6 exhibits different mobilities when hybridized to the U4 or U6 probes. Multiple U6 bands indicative of structural heterogeneity were observed in some of the mutant strains. (C) Analysis of the fraction of U4 present in each strain as a U4/U6 complex. Also shown are the relative total amounts of U4 and U6, expressed as ratios to U1, normalized to the U6WT strain. Error bars represent SD of data obtained from three replicate experiments.
We next determined if U6BASE or U6ΔBULGE conferred any growth defects to the yeast despite maintaining viability. U6BASE, which is predicted to have the smallest impact on telestem stability, showed a mild growth defect at low temperatures (16°C, Figure 6A). However, it grew comparably to U6WT at higher temperatures. U6ΔBULGE showed a much stronger phenotype. U6ΔBULGE was not viable at 16°C and grew much more poorly than U6WT and U6BASE at other temperatures (Figure 6A). U6 genes containing a subset of bulge mutations that do not result in base-pairing (U6A34G,A35G and U6G96C,A97C) do not show any growth defect at any of the tested temperatures. This finding suggests that the severe growth defect of U6ΔBULGE arises from stabilization of the telestem by base-pairing the bulged nucleotides. Interestingly, while U6BASE showed a mild cold sensitivity, no cold sensitivity was observed with the U6G96C,A97C,U100C,U101C mutant (Figure 6A). This implies that the phenotype conferred by stabilization of the telestem base by U100C, U101C mutations can be suppressed by simultaneous mutation of nucleotides in the 3′ telestem bulge strand (G96C, A97C; see Supplementary Figure S10B for an additional figure illustrating these mutations). The in vivo data demonstrate that telestem stabilization by the U6BASE and U6ΔBULGE mutations maintains yeast viability but results in growth defects that are roughly proportional to the predicted increase in stability as well as the extent to which the mutations perturb smFRET dynamics.
U4/U6 di-snRNA is maintained after telestem stabilization in vivo
We hypothesized that the phenotypes observed for U6BASE and U6ΔBULGE could be related to destabilization of U4/U6 in cells due to telestem stabilization. However, telestem stabilization was previously shown to increase the rate of Prp24-dependent U4/U6 annealing in vitro (14), so these two effects could offset one another. To determine the effect of U6BASE and U6ΔBULGE on steady-state U4/U6 levels, we extracted RNA from cultures of the mutant U6 strains at low temperature to maintain U4/U6 base-pairing and quantified the amount of U4, U6 and U4/U6 in each strain by solution hybridization and non-denaturing PAGE (33). RNAs were isolated after growth of yeast cells at 30°C since the U6ΔBULGE-containing strain grows very poorly at other temperatures. Defects in U4/U6 di-snRNA formation due to conditional mutations in U6 or U4 have been detected previously at permissive temperature (32,34).
Bands corresponding to U4/U6 were observed in RNA extracted from each telestem mutant (Figure 6B). The U4/U6 hybrids containing the U6 probe all migrated at similar rates, but U4/U6ΔBULGE hybridized to the U4 probe migrated slightly faster than the other U4/U6 complexes. This altered mobility suggests an altered, stable structure, consistent with the smFRET data (Figure 2E). U6BASE, U6ΔBULGE and U6G96C,A97C,U100C,U101C all migrated as doublets when hybridized to the U6 probe (Figure 6B), indicative of structural heterogeneity. When the amounts of U4 and U6 relative to U1 snRNA were compared between the strains, we discovered that mutation of U6 generally resulted in an increase in total U6 levels, with U6BASE and U6ΔBULGE showing the largest increases of ∼2- and 3-fold, respectively (Figure 6C). Interestingly, while U4 levels remained constant in most of the U6 telestem mutant strains, U6ΔBULGE also showed ∼3-fold increase in U4 RNA.
We confirmed these results by extracting RNAs from each strain under fully denaturing conditions and quantifying RNA levels by primer extension and RNA lengths by Northern blot (Supplementary Figure S11). Primer extension revealed an increase in U6 snRNA level of ∼ 3-fold in the U6ΔBULGE strain in comparison to U6WT after normalization to the U1 snRNA (Supplementary Figure S11A and B). Both U6ΔBULGE and U6BASE mutations also showed two U6 bands by northern blotting, suggesting formation of a truncated U6 product (Supplementary Figure S11C). Shorter U6 products have been observed previously in the presence of mutations that are proposed to disrupt binding of U6 snRNP proteins (22). Primer extension further confirmed that the amount of U4 snRNA was also increased in the U6ΔBULGE strain (∼2-fold). U5 snRNA expression was not increased, suggesting that U5 may become limiting for tri-snRNP assembly in these strains. Thus, we conclude that the U6ΔBULGE strain contains 2- to 3-fold more U4/U6 complex than a WT strain, possibly due to selection for amplification of the CEN plasmids containing the U4 and U6 alleles.
All telestem stabilization mutants showed a similar fraction of U4/U6 being formed relative to total U4. The fraction of U4 incorporated into U4/U6 ranged from 63–79% (Figure 6C). No strong correlation is apparent between these values and observable growth phenotypes. In particular, U6ΔBULGE, which has a severe growth defect, exhibits a WT fraction of U4 in U4/U6. These results indicate that cells containing telestem-stabilizing mutations maintain a normal level of U4/U6 base-pairing, suggesting that Prp24 and/or U4/U6 di-snRNP proteins can overcome the U4/U6-destabilizing effect of these mutations on the protein-free RNAs. However, the elevated overall level of U4/U6ΔBULGE complex suggests that a later step in spliceosome assembly, such as U5 snRNP binding or tri-snRNP stability, may be limiting for growth in this strain and lead to the observed growth defect.
Telestem stabilization results in U4/U6 di-snRNP accumulation
To investigate the distribution of U4/U6 among different snRNPs, we used native gel electrophoresis followed by Northern blotting to detect free mono-snRNPs as well as snRNP complexes, including the U4/U6 di-snRNP and U4/U6.U5 tri-snRNP. Since the viable U6ΔBULGE mutation showed the strongest growth and single molecule phenotypes, we focused on analysis of this mutant in comparison with a WT strain.
When cell extracts prepared from yeast containing U6ΔBULGE and grown at 30°C were subjected to native PAGE, two major species with distinct mobilities were detected with a U4 probe (Figure 7A, lanes 3 and 4). Both species are snRNPs since Proteinase K treatment of the extract resulted in faster migrating bands, identified as U4/U6 di-snRNA and U4 snRNA (Figure 7A, lanes 5 and 6). The slower migrating snRNP species corresponds to tri-snRNP since it is also detected with oligonucleotides complementary to U5 and U6 (Figure 7B and C). The faster migrating snRNP species was much more abundant in extracts prepared from U6ΔBULGE than U6WT strains and comigrated with a band also detected by the U6 probe. The abundance of this faster migrating species was increased in U6WT extracts by incubation of the extract with ATP on ice (Supplementary Figure S12). The data are consistent with the faster migrating species corresponding to the U4/U6 di-snRNP, since U6ΔBULGE strains have a higher steady state level of U4/U6 than U6WT strains (Figure 6). Neither extract contains detectable free U4 snRNP, indicating either that the U4/U6ΔBULGE snRNP is stable or that Prp24 rapidly reassembles any dissociated U4 and U6ΔBULGE.
Figure 7.

Telestem stabilization results in accumulation of U4/U6 di-snRNPs. Whole cell extracts from the U6WT and U6ΔBULGE strains were incubated at 30°C with or without ATP prior to separation of the snRNPs by 4% native PAGE. The RNA was transferred to a membrane and then probed with [32P]-labeled oligonucleotides complementary to U4, U6 or U5 snRNAs. To identify the location of snRNA-only species, some samples were also treated with Proteinase K prior to electrophoresis. (A–C) Results from the same membrane probed for U4 (A), U6 (B) or U5 (C) snRNAs. Bands corresponding to various snRNP complexes and the free snRNAs are noted.
Further analysis of blots probed with oligonucleotides complementary to U2, U5 and U6 snRNAs revealed other features resulting from the U6ΔBULGE mutation. No changes in U2-containing snRNP complexes were observed between the U6WT and U6ΔBULGE strains (data not shown). Free U5 snRNP was likely present at similar levels; however, U5 bands appear more diffuse in the mutant strain (Figure 7C, lanes 1–4). The U6 probe revealed an abundance of free U6 snRNA that was not incorporated into snRNPs in the U6ΔBULGE extract (Figure 7B, lanes 3, 4 and 6), similar to what was seen previously with U6 RNA containing the A91G mutation in the telestem and A62G mutation in the ISL (22). In contrast to the sole U6 snRNP species apparent in U6WT extract, multiple U6 snRNP species were observed in U6ΔBULGE extract, including bands with slightly slower mobility than those observed with U6WT. Some of these snRNP complexes may contain the truncated form of U6ΔBULGE we observed under denaturing conditions (Supplementary Figure S11), while others might lack U6 snRNP proteins (Prp24 or the Lsm2-8 ring).
The amount of U4 and U5 snRNA in tri-snRNP containing U6ΔBULGE seems comparable to that in tri-snRNP containing U6WT; however, the amount of U6ΔBULGE in these complexes appears less than for U6WT (∼44% of the level found in U6WT, Figure 7B). To determine if this reflects a change in U6 abundance rather than a difference in probe hybridization to U6ΔBULGE versus U6WT, we also analyzed snRNP distribution by glycerol gradient centrifugation followed by fractionation and detection of the snRNAs by primer extension (data not shown). These experiments confirmed the results shown in Figure 7B: U6ΔBULGE was present in the tri-snRNP however the amount of U6 relative to U4 was decreased by ∼50% in comparison to U6WT. Thus, telestem stabilization via the U6ΔBULGE mutation results in heterogeneous U6 snRNP complexes, but these complexes do not prevent accumulation of U4/U6. Tri-snRNPs still form with U6ΔBULGE, but the mutations may also impact tri-snRNP stability. The most striking differences between the U6WT and U6ΔBULGE strains are that the U6ΔBULGE extract contains large amounts of U4/U6 di-snRNP and free U6 snRNA. Accumulation of U4/U6 indicates that while the telestem favors U4/U6 formation (14), increasing telestem base-pairing does not result in greater accumulation of the tri-snRNP.
DISCUSSION
It has long been appreciated that U6 snRNA undergoes astonishingly extensive structural transitions during tri-snRNP assembly and spliceosomal activation. However, little is known about the molecular mechanism of these events. Here we use a combination of smFRET, ensemble duplex unwinding and in vivo assays to study the influence of the U6 telestem on RNA dynamics, U4/U6 stability and endogenous yeast snRNP complexes.
Our data reveal that U6 transiently forms the telestem while base-paired with U4 RNA, the telestem promotes U4/U6 oligonucleotide-driven unwinding and disruption of U4/U6 stem I induces structural changes in U6 and facilitates unwinding of stem II. Mutations that are predicted to stabilize or destabilize the telestem alter smFRET dynamics and U4/U6 unwinding, although we cannot exclude the possibility that some mutations may create aberrant structures or alter other existing structures, as we suspect for the U6-A34G,A35G mutation (see above). The impacts of telestem stabilization on yeast growth agree with observations from smFRET, in that mutations that result in the largest accumulation of FRET signal from the telestem are the most deleterious to yeast growth. Mutations that stabilize the telestem result in accumulation of U4/U6 di-snRNPs in vivo rather than unwound mono-snRNPs, suggesting defects in later stages of spliceosome assembly. Together our data illustrate the destabilizing influence of the telestem on U4/U6 complexes in vitro as well as provide new mechanistic insights into U4/U6 unwinding and features of cellular regulation of tri-snRNP components. These results allow us to propose a model in which U4/U6 stem I disruption and telestem formation contribute to U4/U6 di-snRNA and di-snRNP unwinding (Figure 8).
Figure 8.

Model for competing U6 intra- and intermolecular interactions involved in U4/U6 unwinding and spliceosome activation. Telestem base-pairing and additional structures formed upon disruption of U4/U6 stem I promote destabilization of stem II by oligonucleotides, resulting in unwinding and formation of the extensively base-paired U6 snRNA. During spliceosome activation, disruption of U4/U6 stem I by the Brr2 helicase may facilitate formation of competing intermolecular base-pairing interactions between U6, U2 and the pre-mRNA. These interactions may similarly promote destabilization of U4/U6 stem II, resulting in concomitant formation of the U2/U6 catalytic core of the spliceosome.
The telestem promotes di-snRNP assembly and disassembly
The telestem promotes Prp24-catalyzed U4/U6 annealing in vitro, likely by facilitating formation of an ‘electropositive groove’ on Prp24 that positions U4 for formation of U4/U6 stem I (14). Our in vitro data (Figures 1–4) suggest that telestem formation may be a double-edged sword: promoting Prp24 annealing activity while also destabilizing U4/U6 di-snRNAs. Thus, disruption of the telestem subsequent to U4/U6 annealing may be critical for preventing unwinding from competing with U4/U6 di-snRNP formation. Indeed, this step has been previously proposed to occur during U4/U6 di-snRNP assembly by Didychuk et al. (14). When the telestem is strongly stabilized in vivo, it confers growth defects and results in accumulation of U4/U6 di-snRNP that appears to be less efficiently incorporated into tri-snRNP (Figures 6 and 7). Together, these results indicate that normally the telestem forms only transiently to promote the structural transitions of the U6 snRNA.
Our observations also suggest previously unrecognized functions for splicing factors in stabilizing U6-containing di-snRNAs by suppressing telestem formation. Recently, cryo-EM structures of the tri-snRNP (35,36), as well as the crystal structure of a fragment of the U4/U6 di-snRNP protein Prp3 (37), have revealed that Prp3 associates with U4/U6 in both the stem II region and by binding the unpaired region of U6 located between U4/U6 stem II and the 3′ strand of the telestem. This positioning suggests that Prp3 may promote U4/U6 stability by preventing telestem formation. The 5′ strand of the telestem may also be sequestered in the tri-snRNP in the ACAGAGA stem loop, which is stabilized by the U5 protein Dib1 (36). Consequently, tri-snRNP assembly may require telestem disruption and interaction of those regions of U6 with tri-snRNP-specific proteins. Hyperstabilization of the telestem may inhibit tri-snRNP assembly by preventing such interactions.
Within activated spliceosomes, the U2 snRNA base-pairs with U6 and U2/U6 helix II contains the 3′ telestem strand and is mutually exclusive with telestem base-pairing (38). It is possible that U2/U6 helix II may be important for preventing U6 telestem formation and premature U2/U6 disassembly during the catalytic steps of splicing. Evidence for such a function has been obtained by deletion of the nonessential yeast splicing factor Slt11 and observing that U2 mutations that disrupt U2/U6 helix II are lethal unless the complementary U6 strand had also been mutated to restore pairing (39,40). The U6 mutations that maintain viability in the U2-mutant/slt11Δ background also disrupt the telestem and implicate U2/U6 helix II in antagonizing telestem formation in the absence of Stl11. A role for U2/U6 helix II in antagonizing U6 intramolecular pairing has been previously proposed in studies of the human snRNAs (26). Our data in combination with those obtained by others using slt11Δ yeast strains suggest that this stabilizing function of U2/U6 helix II may also be present in yeast.
In addition to U2/U6 helix II, protein components of catalytic spliceosomes may also antagonize telestem formation. In a cryo-EM structure of a Schizosaccharomyces pombe spliceosome complex, the Slt11 homolog, Cwf5, abuts the base of a small loop in the U6 snRNA formed from the 5′ strand of the telestem (8). These telestem nucleotides also appear to be bound by the Prp19 complex protein Cwf2 (homolog of S. cerevisiae Cwc2). Thus, sequestration of both the 5′ and 3′ strands of the telestem may occur in both the tri-snRNP and the spliceosome but by different factors. How these factors are exchanged during activation is unclear.
Finally, we note that while U2/U6 helix II may promote U4/U6 stability by antagonizing telestem formation, it is also possible that the telestem could promote U2/U6 unwinding by formation of a structure that competes with U2/U6 helix II during spliceosome disassembly. This reversal of roles may reflect an economic solution by the spliceosome for targeting U6 between different complexes through cycles of intra- and intermolecular duplex formation within the telestem region.
Implications for U4/U6 unwinding in the spliceosome
It is unclear what happens when Brr2 encounters the U4/U6 three-helix junction during spliceosomal activation and how unwinding of U4/U6 stem II takes place. Multiple models have been proposed including both direct and indirect unwinding of stem II by Brr2 (15). Indirect unwinding of stem II—that is, Brr2 unwinding of stem I leading to events that destabilize stem II without further Brr2 translocation—is supported primarily by failure to detect crosslinks between Brr2 and any region of stem II (41). Our ability to dissociate U4/U6 di-snRNAs with stem II complementary oligonucleotides demonstrates that ATP hydrolysis and Brr2 helicase activity is not an obligate requirement for U4/U6 stem II disruption. How can stem II be unwound without RNA displacement by Brr2?
One possibility is that the U6 telestem forms transiently during spliceosome activation, likely requiring prior release of proteins such as Prp3 or Dib1 (35,36). Telestem formation then leads to stem II disruption. A similar model has previously been proposed for human U4/U6 in which the intrinsic instability of the di-snRNA may alter the requirement for helicase-dependent unwinding (26). Alternatively, the destabilizing influence of the telestem could be recapitulated through intermolecular base-pairing interactions between U6, U2 and the pre-mRNA substrate. Our smFRET experiments support formation of additional U6 structures while U4/U6 stem II remains paired (Figure 5), and it is possible such structures form concertedly with U4/U6 stem I disruption. These observations suggest a model in which U4/U6 unwinding is coupled to formation of a competing structure—the U2/U6 di-snRNA—that in turn promotes U4/U6 stem II destabilization (Figure 8). The net result is concerted U4/U6 unwinding and U2/U6 active site formation.
One feature of this model is that it predicts formation of a U2/U6/U4 ternary complex during spliceosomal activation. Even though this species has not been detected in yeast spliceosomes, evidence exists from psoralen crosslinking for such a complex in humans (42,43). Furthermore, an intermediate has been detected during unwinding of the minor spliceosome in which U4atac/U6atac stem II remains intact, stem I has been unwound and U12/U6atac helix Ia has formed (44). In yeast, it has previously been observed that an allele of BRR2 (slt22-1, corresponding to an E909K mutation in the N-terminal unwinding cassette) is synthetically lethal in combination with U2 snRNA mutations that destabilize U2/U6 helix Ib and helix II as well as mutations in U6 that form the 3′ strand of the telestem or, alternatively, U2/U6 helix II (45). This finding implies that Brr2's unwinding activity is coupled with U2/U6 base-pairing. Together these results suggest U2 may begin forming base-pairing interactions with multiple regions of U6 while the ISL region of U6 remains base-paired to U4 in U4/U6 stem II and that unwinding of U4/U6 stem I by Brr2 is required for a subset of these interactions with U2 to form (Figure 8).
Our results also have implications for interpreting in vitro assays of Brr2-catalyzed unwinding of purified U4/U6 di-snRNAs that contain the telestem region of U6. We have clearly demonstrated that the telestem promotes oligonucleotide-catalyzed unwinding of U4/U6 stem II in vitro (Figures 1–5), so it is expected to also contribute to Brr2-catalyzed U4/U6 unwinding in vitro, as long as Brr2 itself does not block telestem formation. Indeed, it has already been shown that deletion of U6 nt 1–54, which would prevent telestem formation, results in a decrease in Brr2 unwinding rate (46). More recently, it has been postulated that a competing structure in U6 actively participates in releasing of U4/U6 di-snRNP proteins and U4 unwinding in a model system (47). This competing structure may initially be the telestem, followed by the ISL. Finally, our model predicts that Brr2 helicase assays in which disruption of U4/U6 is coupled to formation of U2/U6 would be most relevant to spliceosome activation.
Telestem stabilization results in changes to cellular snRNP distribution
Expression of U6ΔBULGE results in U4/U6 di-snRNP accumulation and free U6 ΔBULGE RNA, but not U4 mono-snRNP (Figure 7). We also observe that the U6ΔBULGE extract contains free U5 snRNPs but similar levels of tri-snRNP to other strains. It is not clear what is preventing additional tri-snRNP assembly with U6ΔBULGE and it is possible that the poor growth of the strain leads to secondary effects that alter snRNP distribution in extract. Alternatively, not all free U5 snRNPs are competent for tri-snRNP formation with U4/U6ΔBULGE. This may be due to composition of the U5 particles themselves (i.e. Aar2-containing U5 snRNPs may not be competent for U4/U6 di-snRNP association (48)) or possibly a factor involved in tri-snRNP assembly has become limiting in U6ΔBULGE extract [e.g. Sad1 (31) or limiting U5 snRNP itself]. Additionally, U4/U6ΔBULGE itself could be disruptive to the tri-snRNP or introduce a block in assembly by altering interactions with tri-snRNP proteins such as Prp3 and Dib1. The amount of U6 is decreased in U6ΔBULGE tri-snRNP (or tri-snRNP-like) complexes, suggesting disruption of tri-snRNP stability. These effects could have resulted from persistence of the telestem after U4/U6 di-snRNP assembly to the detriment of subsequent steps in tri-snRNP assembly. Nonetheless, the destabilizing influence of the telestem on U4/U6 we observe in vitro appears to be mitigated in cell extracts in which telestem stabilization leads to accumulation of U4/U6.
Since our native gel data (Figure 7) probe the steady state level of U4/U6 in extract, telestem stabilization may be changing the rate of U4/U6 unwinding but not the equilibrium concentration of snRNP complexes. Free snRNPs could be rapidly re-annealed by Prp24 (a process also stimulated by the telestem) and result in apparent U4/U6 accumulation if subsequent steps in tri-snRNP assembly become rate-limiting. Further investigation into how telestem stabilization changes discrete steps in tri-snRNP assembly and spliceosome activation may reveal additional insights into the impact of these mutations.
Supplementary Material
Acknowledgments
We thank members of the Hoskins, Brow and Butcher laboratories as well as Peter Šulc for helpful discussions. We thank the Wildonger lab for equipment access and Dr Ray O'Keefe for primer extension protocols. We thank the laboratories of Taekjip Ha and Nils Walter for smFRET analysis software as well as Fatemehsadat Jamalidinan for computer software development.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R00 GM086471, R01 GM112735 to A.A.H., T32-GM08293 to M.R., R35 GM118131 to S.E.B., R35 GM118075 to D.A.B.]; Shaw Scientist Award (to A.A.H.); Beckman Young Investigator Awards (to A.A.H.); University of Wisconsin-Madison startup funding; Wisconsin Alumni Research Foundation (WARF); Department of Biochemistry (to A.A.H.). Funding for open access charge: University startup funds (to A.A.H.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Wahl M.C., Will C.L., Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Hoskins A.A., Gelles J., Moore M.J. New insights into the spliceosome by single molecule fluorescence microscopy. Curr. Opin. Chem. Biol. 2011;15:864–870. doi: 10.1016/j.cbpa.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brow D.A. Allosteric cascade of spliceosome activation. Annu. Rev. Genetics. 2002;36:333–360. doi: 10.1146/annurev.genet.36.043002.091635. [DOI] [PubMed] [Google Scholar]
- 4.Fabrizio P., Dannenberg J., Dube P., Kastner B., Stark H., Urlaub H., Lührmann R. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol. Cell. 2009;36:593–608. doi: 10.1016/j.molcel.2009.09.040. [DOI] [PubMed] [Google Scholar]
- 5.Karaduman R.R., Fabrizio P.P., Hartmuth K.K., Urlaub H.H., Lührmann R.R. RNA structure and RNA-protein interactions in purified yeast U6 snRNPs. J. Mol. Biol. 2006;356:15–15. doi: 10.1016/j.jmb.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Montemayor E.J., Curran E.C., Liao H.H., Andrews K.L., Treba C.N., Butcher S.E., Brow D.A. Core structure of the U6 small nuclear ribonucleoprotein at 1.7-Å resolution. Nat. Struct. Mol. Biol. 2014;21:544–551. doi: 10.1038/nsmb.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fica S.M., Tuttle N., Novak T., Li N.-S., Lu J., Koodathingal P., Dai Q., Staley J.P., Piccirilli J.A. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503:229–234. doi: 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan C., Hang J., Wan R., Huang M., Wong C.C.L., Shi Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science. 2015;349:1182–1191. doi: 10.1126/science.aac7629. [DOI] [PubMed] [Google Scholar]
- 9.Staley J.P., Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 10.Hausner T.P., Giglio L.M., Weiner A.M. Evidence for base-pairing between mammalian U2 and U6 small nuclear ribonucleoprotein particles. Genes Dev. 1990;4:2146–2156. doi: 10.1101/gad.4.12a.2146. [DOI] [PubMed] [Google Scholar]
- 11.Datta B., Weiner A.M. Genetic evidence for base pairing between U2 and U6 snRNA in mammalian mRNA splicing. Nature. 1991;352:821–824. doi: 10.1038/352821a0. [DOI] [PubMed] [Google Scholar]
- 12.Wu J.A., Manley J.L. Base pairing between U2 and U6 snRNAs is necessary for splicing of a mammalian pre-mRNA. Nature. 1991;352:818–821. doi: 10.1038/352818a0. [DOI] [PubMed] [Google Scholar]
- 13.Madhani H.D., Guthrie C. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell. 1992;71:803–817. doi: 10.1016/0092-8674(92)90556-r. [DOI] [PubMed] [Google Scholar]
- 14.Didychuk A.L., Montemayor E.J., Brow D.A., Butcher S.E. Structural requirements for protein-catalyzed annealing of U4 and U6 RNAs during di-snRNP assembly. Nucleic Acids Res. 2016;44:1398–1410. doi: 10.1093/nar/gkv1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen K.H., Staley J.P. Spliceosome activation: U4 is the path, stem I is the goal, and Prp8 is the keeper. Let's cheer for the ATPase Brr2! Genes Dev. 2012;26:2461–2467. doi: 10.1101/gad.207514.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raghunathan P.L., Guthrie C. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 1998;8:9. doi: 10.1016/s0960-9822(07)00345-4. [DOI] [PubMed] [Google Scholar]
- 17.Noble S.M., Guthrie C. Identification of novel genes required for yeast pre-mRNA splicing by means of cold-sensitive mutations. Genetics. 1996;143:67–80. doi: 10.1093/genetics/143.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodgers M.L., Tretbar U.S., Dehaven A., Alwan A.A., Luo G., Mast H.M., Hoskins A.A. Conformational dynamics of stem II of the U2 snRNA. RNA. 2016;22:225–236. doi: 10.1261/rna.052233.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bronson J.E., Fei J., Hofman J.M., Gonzalez R.L., Wiggins C.H. Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys. J. 2009;97:3196–3205. doi: 10.1016/j.bpj.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinney S.A., Joo C., Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanco M., Walter N.G. Analysis of complex single-molecule FRET time trajectories. Meth. Enzymol. 2010;472:153–178. doi: 10.1016/S0076-6879(10)72011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burke J.E., Butcher S.E., Brow D.A. Spliceosome assembly in the absence of stable U4/U6 RNA pairing. RNA. 2015;21:923–934. doi: 10.1261/rna.048421.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardin J.W., Warnasooriya C., Kondo Y., Nagai K., Rueda D. Assembly and dynamics of the U4/U6 di-snRNP by single-molecule FRET. Nucleic Acids Res. 2015;43:10963–10974. doi: 10.1093/nar/gkv1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornilescu G., Didychuk A.L., Rodgers M.L., Michael L.A., Burke J.E., Montemayor E.J., Hoskins A.A., Butcher S.E. Structural analysis of multi-helical RNAs by NMR-SAXS/WAXS: application to the U4/U6 di-snRNA. J. Mol. Biol. 2016;428:777–789. doi: 10.1016/j.jmb.2015.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brow D.A., Vidaver R.M. An element in human U6 RNA destabilizes the U4/U6 spliceosomal RNA complex. RNA. 1995;1:122–131. [PMC free article] [PubMed] [Google Scholar]
- 27.Markham N.R., Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brow D.A., Guthrie C. Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature. 1988;334:213–218. doi: 10.1038/334213a0. [DOI] [PubMed] [Google Scholar]
- 29.Srinivas N., Ouldridge T.E., Sulc P., Schaeffer J.M., Yurke B., Louis A.A., Doye J.P.K., Winfree E. On the biophysics and kinetics of toehold-mediated DNA strand displacement. Nucleic Acids Res. 2013;41:10641–10658. doi: 10.1093/nar/gkt801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Šulc P., Ouldridge T.E., Romano F., Doye J.P.K., Louis A.A. Modelling toehold-mediated RNA strand displacement. Biophys J. 2015;108:1238–1247. doi: 10.1016/j.bpj.2015.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y.-H., Chung C.-S., Kao D.-I., Kao T.-C., Cheng S.-C. Sad1 counteracts Brr2-mediated dissociation of U4/U6.U5 in Tri-snRNP homeostasis. Mol. Cell. Biol. 2014;34:210–220. doi: 10.1128/MCB.00837-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McManus C.J., Schwartz M.L., Butcher S.E., Brow D.A. A dynamic bulge in the U6 RNA internal stem-loop functions in spliceosome assembly and activation. RNA. 2007;13:2252–2265. doi: 10.1261/rna.699907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z., Brow D.A. A rapid assay for quantitative detection of specific RNAs. Nucleic Acids Res. 1993;21:4645–4646. doi: 10.1093/nar/21.19.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vidaver R.M., Fortner D.M., Loos-Austin L.S., Brow D.A. Multiple functions of Saccharomyces cerevisiae splicing protein Prp24 in U6 RNA structural rearrangements. Genetics. 1999;153:1205–1218. doi: 10.1093/genetics/153.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan R., Yan C., Bai R., Wang L., Huang M., Wong C.C.L., Shi Y. The 3.8 Å structure of the U4/U6.U5 tri-snRNP: insights into spliceosome assembly and catalysis. Science. 2016;351:466–475. doi: 10.1126/science.aad6466. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen T.H.D., Galej W.P., Bai X.-C., Oubridge C., Newman A.J., Scheres S.H.W., Nagai K. Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 Å resolution. Nature. 2016;530:298–302. doi: 10.1038/nature16940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu S., Mozaffari-Jovin S., Wollenhaupt J., Santos K.F., Theuser M., Dunin-Horkawicz S., Fabrizio P., Bujnicki J.M., Lührmann R., Wahl M.C. A composite double-/single-stranded RNA-binding region in protein Prp3 supports tri-snRNP stability and splicing. Elife. 2015;4:e07320. doi: 10.7554/eLife.07320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madhani H.D., Guthrie C. Dynamic RNA-RNA interactions in the spliceosome. Annu. Rev. Genet. 1994;28:1–26. doi: 10.1146/annurev.ge.28.120194.000245. [DOI] [PubMed] [Google Scholar]
- 39.Field D.J., Friesen J.D. Functionally redundant interactions between U2 and U6 spliceosomal snRNAs. Genes Dev. 1996;10:489–501. doi: 10.1101/gad.10.4.489. [DOI] [PubMed] [Google Scholar]
- 40.Xu D., Friesen J.D. Splicing factor Slt11p and its involvement in formation of U2/U6 helix II in activation of the yeast spliceosome. Mol. Cell. Biol. 2001;21:1011–1023. doi: 10.1128/MCB.21.4.1011-1023.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hahn D., Kudla G., Tollervey D., Beggs J.D. Brr2p-mediated conformational rearrangements in the spliceosome during activation and substrate repositioning. Genes Dev. 2012;26:2408–2421. doi: 10.1101/gad.199307.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wassarman D.A., Steitz J.A. A base-pairing interaction between U2 and U6 small nuclear RNAs occurs in >150S complexes in HeLa cell extracts: implications for the spliceosome assembly pathway. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7139–7143. doi: 10.1073/pnas.90.15.7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider M., Will C.L., Anokhina M., Tazi J., Urlaub H., Lührmann R. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol. Cell. 2010;38:223–235. doi: 10.1016/j.molcel.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 44.Frilander M.J., Steitz J.A. Dynamic exchanges of RNA interactions leading to catalytic core formation in the U12-dependent spliceosome. Mol. Cell. 2001;7:217–226. doi: 10.1016/s1097-2765(01)00169-1. [DOI] [PubMed] [Google Scholar]
- 45.Xu D., Nouraini S., Field D., Tang S.J., Friesen J.D. An RNA-dependent ATPase associated with U2/U6 snRNAs in pre-mRNA splicing. Nature. 1996;381:709–713. doi: 10.1038/381709a0. [DOI] [PubMed] [Google Scholar]
- 46.Mozaffari-Jovin S., Santos K.F., Hsiao H.H., Will C.L., Urlaub H., Wahl M.C., Luhrmann R. The Prp8 RNase H-like domain inhibits Brr2-mediated U4/U6 snRNA unwinding by blocking Brr2 loading onto the U4 snRNA. Genes Dev. 2012;26:2422–2434. doi: 10.1101/gad.200949.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Theuser M., Höbartner C., Wahl M.C., Santos K.F. Substrate-assisted mechanism of RNP disruption by the spliceosomal Brr2 RNA helicase. Proc. Natl. Acad. Sci. U.S.A. 2016;113:7798–7803. doi: 10.1073/pnas.1524616113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber G., Cristão V.F., Santos K.F., Jovin S.M., Heroven A.C., Holton N., Lührmann R., Beggs J.D., Wahl M.C. Structural basis for dual roles of Aar2p in U5 snRNP assembly. Genes Dev. 2013;27:525–540. doi: 10.1101/gad.213207.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.