


Abstract
A short abnormal polyalanine expansion in the polyadenylate-binding protein nuclear-1 (PABPN1) protein causes oculopharyngeal muscular dystrophy (OPMD). Mutated PABPN1 proteins accumulate as insoluble intranuclear aggregates in muscles of OPMD patients. While the roles of PABPN1 in nuclear polyadenylation and regulation of alternative poly(A) site choice have been established, the molecular mechanisms which trigger pathological defects in OPMD and the role of aggregates remain to be determined. Using exon array, for the first time we have identified several splicing defects in OPMD. In particular, we have demonstrated a defect in the splicing regulation of the muscle-specific Troponin T3 (TNNT3) mutually exclusive exons 16 and 17 in OPMD samples compared to controls. This splicing defect is directly linked to the SC35 (SRSF2) splicing factor and to the presence of nuclear aggregates. As reported here, PABPN1 aggregates are able to trap TNNT3 pre-mRNA, driving it outside nuclear speckles, leading to an altered SC35-mediated splicing. This results in a decreased calcium sensitivity of muscle fibers, which could in turn plays a role in muscle pathology. We thus report a novel mechanism of alternative splicing deregulation that may play a role in various other diseases with nuclear inclusions or foci containing an RNA binding protein.
INTRODUCTION
The poly(A) RNA binding protein, polyadenylate-binding protein nuclear 1 (PABPN1), is a ubiquitous protein, that binds virtually all mRNAs and plays key roles in post-transcriptional processing of RNA (1). PABPN1 activates poly(A) polymerase (PAP) and controls poly(A) tail length on RNA transcripts (2–4). In addition, PABPN1 regulates the use of alternative polyadenylation (APA) sites (5,6), which in turn controls mRNA levels and stability. PABPN1 is also involved in the regulation of the processing of long non-coding RNAs (lncRNAs) (7) and nuclear surveillance, leading to hyperadenylation and decay of RNA (8). It has recently been shown that PABPN1 regulates human telomerase 3′ end RNA maturation (9). A short (1–8) and meiotically stable GCN trinucleotide repeat expansion in the coding region of the PABPN1 gene causes oculopharyngeal muscular dystrophy (OPMD) (10–12), a late and progressive autosomal dominant inherited neuromuscular disorder. OPMD is characterised by progressive eyelid drooping, swallowing difficulties (as pharyngeal and cricopharyngeal muscles are most affected) and proximal limb weakness (13).
OPMD belongs to the group of triplet expansion diseases (14,15); the translation of the PABPN1 allele containing expanded repeats (expPABPN1) leads to a pathogenic polyalanine tract at the N-terminal of the protein and the formation of PABPN1 intranuclear aggregates in muscle fibers (16). These aggregates in addition to PABPN1 contain several proteins such as ubiquitin, proteasome subunits, heat-shock proteins, splicing factors, poly(A) polymerase (PAP) as well as poly(A) RNA (17–19). However these insoluble aggregates also exclude cleavage and polyadenylation specific factor subunits (CPSF), splicing factors such as SC35 and the cytoplasmic poly(A) binding protein (PABPC) (19). The expanded polyalanine tract in PABPN1 is thought to induce protein misfolding and consequently the formation of aggregates, which are targeted to the ubiquitin-proteasome degradation pathway (20,21). Although the presence of aggregates in skeletal muscles is a pathological hallmark of OPMD, their exact role in the disease remains controversial. Several studies have suggested a pathological function for nuclear aggregates e.g. sequestrating essential cellular components including PABPN1 itself (17–18,22). This is supported by the fact that a higher frequency of nuclear aggregates is observed in severe homozygous patients (23) and muscle function is improved when the number of aggregates is reduced (24–26). In contrast, other studies have suggested that the aggregates may just be the result of a cellular defence mechanism (27) and not the direct cause of the disease with the soluble form of the mutated PABPN1 being itself pathogenic (28,29). Apoptosis and deregulation of the ubiquitin-proteasome system have also been proposed as downstream events triggered by the aggregates (20,30). Recently we demonstrated a global deregulation of nuclear-encoded mitochondrial RNA in OPMD patients and animal models, implicating mRNA poly(A) tail regulation via the recruitment of Smaug RNA binding protein as an upstream event (31).
Over the last years, the following observations have suggested that splicing defects may occur in OPMD: (i) the identification of splicing factors in aggregates (17–18,22); (ii) the fact that, using literature-aided analyses, the terms ‘RNA splicing’ and ‘alternative splicing’ were biomedical concepts highly associated with human OPMD-deregulated genes (20); (iii) two recent studies have suggested a role for PABPN1 in splicing regulation (32,33). However, no such mechanism has yet been described. Alternative splicing (AS) produces a highly dynamic proteome, whose diversity is regulated in a developmental and tissue-specific manner (34). More than 90% of human protein-encoding genes create multiple mRNA isoforms by AS. This post-transcriptional mechanism generates diversity and is tightly regulated, with both cis- and trans-acting elements. Defects in RNA splicing processes, such as mutations in cis-acting splicing elements or in trans-acting splicing factors, have emerged as a common disease-causing mechanism (35,36). Skeletal muscle is a highly plastic tissue, adapting its structure and metabolism in response to diverse conditions such as contractile activity, mechanical overload and nutrients. Skeletal muscle is one of the tissues with the highest number of alternative splicing events, highlighting a high degree of complexity (37–39). In particular, alterations of alternative splicing events have been largely described in several muscular dystrophies (40,41).
In this work, we provide the first proof that splicing defects occur in OPMD and describe a novel mechanism involved in at least one of these defects and how this defect is related to muscle function. Interestingly, we demonstrate that in OPMD the mutually exclusive exons of TNNT3 mRNA are misspliced with an imbalanced ratio of the two resulting isoforms. Using a minigene construct, we show that SC35 regulates the splicing of these mutually exclusive exons. We further demonstrated that PABPN1 aggregates are able to sequester a pre-mRNA. Taking these results together, we propose that this splicing defect results from the delocalization of the mRNA, trapped in pathological aggregates, and subsequently misplaced outside SC35-nuclear speckles.
MATERIALS AND METHODS
Patients
For the whole-genome microarray study, we selected skeletal muscle (sternocleidomastoid) biopsies from OPMD patients (n = 4) and control (n = 4) individuals (Table 1). All patients were aged between 54 and 91 years of age at the time of muscle biopsy. For downstream analysis, we used additional muscle biopsies (sternocleidomastoid and quadriceps) from control (aged 44–91) and OPMD (aged 52–82) patients. All samples generated from these biopsies were compared individually and not pooled before the analysis. OPMD patients showed typical clinical phenotype and the nature of the PABPN1 mutation was confirmed by genetic studies. All muscle biopsies were obtained during surgical procedure after informed consent in accordance with the French legislation on ethical rules.
Table 1. Skeletal muscle biopsies from control (CTRL) and aged-matched OPMD patients (n = 4 per group) were used for the study. M: male; F: female.
| Patient | Age | Gender | Genotype |
|---|---|---|---|
| OPMD | 54 | M | (GCN)13/Ala13 |
| OPMD | 61 | F | (GCN)14/Ala14 |
| OPMD | 81 | M | (GCN)12/Ala12 |
| OPMD | 83 | M | (GCN)13/Ala13 |
| CTRL | 44 | F | (GCN)10/Ala10 |
| CTRL | 61 | M | (GCN)10/Ala10 |
| CTRL | 88 | M | (GCN)10/Ala10 |
| CTRL | 91 | M | (GCN)10/Ala10 |
RNA isolation and Affymetrix exon array data processing
Total RNA was extracted from snap frozen skeletal muscle biopsies using Trizol (Invitrogen) according to the manufacturer's instructions. RNA samples were quantified using a ND-1000 NanoDrop spectrophotometer (NanoDrop Technologies) and purity/integrity were assessed using disposable RNA chips (Agilent RNA 6000 Nano LabChip kit) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbrunn, Germany). The average RIN value of total RNAs was 7.16 (6.1–7.8). Each RNA (n = 8; four controls and four OPMD) was analyzed individually. Affymetrix Human Exon 1.0 ST arrays were hybridized by GenoSplice technology (www.genosplice.com) according the Ambion WT protocol (Life technologies, France) and Affymetrix (Santa Clara, CA, USA) labelling and hybridization recommendations. Raw data were controlled with the Expression console (Affymetrix). Affymetrix Human Exon 1.0 ST Array dataset analysis and visualization were made using EASANA® (GenoSplice technology), which is based on the GenoSplice's FAST DB® annotations (42). Exon Array data were normalized using quantile normalization. Background corrections were made with antigenomic probes and probes were selected as described previously (43,44). Only probes targeting exons annotated from FAST DB® transcripts were selected to focus on well-annotated genes whose mRNA sequences are in public databases (42,45). Bad-quality selected probes (e.g. probes labelled by Affymetrix as ‘cross-hybridizing’) and probes whose intensity signal was too low compared to antigenomic background probes with the same GC content were removed from the analysis. Only probes where the detection above background was with a P value ≤0.05 in at least half of the arrays were considered for statistical analysis (43,44). Only genes expressed in at least one compared condition were analyzed. To be considered expressed, the DABG P-value had to be ≤0.05 for at least half of the gene probes. We performed an unpaired Student's t-test to compare gene intensities in the different biological replicates. Genes were considered to be significantly regulated when the fold-change was ≥1.5 and uncorrected P-value ≤0.05. Statistical analyses were also performed using the Student's unpaired t-test on the splicing index to analyze the Exon Array data as described previously (43,44). The splicing index corresponds to a comparison of gene-normalized exon intensity values between the two analyzed experimental conditions. Results were considered statistically significant for uncorrected P-values ≤0.05 and fold-changes ≥2.0.
To filter and classify exons predicted to be differentially included, manual inspection was performed after uploading the Exon Array data into the EASANA® visualization module, which is based on the FAST DB® Client Edition annotations. By computational comparison of publicly available mRNA sequences with genomic sequences, alternative exons were annotated in FAST DB® as alternative first exons, alternative terminal exons, cassette exons, mutually exclusive exons, alternative 5′ donor sites, alternative 3′ acceptor sites, intron retention and exonic internal deletion (Supplementary Figure S1 and Table S1).
RT-PCR and Real-time qRT-PCR
RNA (50–250 ng for muscle biopsies, 1–3 μg for cell pellets) was reverse transcribed using M-MLV reverse transcriptase (Invitrogen) according to the manufacturer's instructions. The splicing of TNNT3 mRNA was observed with 1 μl of cDNA for PCR using Reddy mix polymerase (Thermo Scientific) according to the manufacturer's instructions, and primers located within TNNT3 exon 15, TNNT3 exon 16 and TNNT3 exon 17 (sequences information listed in Supplementary Table S2). The reaction mixture was heated to 94°C for 5 min and followed by 35 PCR cycles: 15 s at 94°C, 15 s at 55°C and 15 s at 72°C followed by a last elongation step at 72°C for 7 min. PCR products from alternatively spliced TNNT3 mRNAs, which are 108 and 131 bp, respectively, were resolved on 5% non-denaturing polyacrylamide ethydium bromide stained gels.
cDNA was used for quantitative PCR reaction using SYBR green mix buffer (LightCycler® 480 Sybr green I Master) in a total reaction volume of 9 μl. The PCR reaction was carried out as follows: 8 min at 95°C followed by 50 cycles of the following: 15 s at 95°C, 15 s at 60°C and 15 s at 72°C. Specificity of the PCR products was checked by melting curve analysis using the following program: 65°C increasing by 0.11°C/s to 97°C. The expression level of each mRNA was normalized to that of human B2M mRNA (β-2 microglobulin) or murine RPLP0 mRNA (large ribosomal protein, subunit P0) expression. Expression levels were calculated according to the ΔΔCt method.
The sequences of primers used for RT-PCR and for real-time qRT-PCR are listed in Supplementary Table S2.
Plasmid construction
The human TNNT3 minigene (3881 bp) was prepared by amplifying the human TNNT3 genomic region between exon 15 and exon 18 with the Platinium® Taq DNA Polymerase High Fidelity (Invitrogen) following the manufacturer's instruction and using the primers FWD: 5′-AAAACTTAAGGGACAAGGCCAAGGAGCTCT-3′ containing the AflII restriction site (in bold) and REV: 5′-ACCGGAATTCTGAAGGGGGTTCTGCAGCTTT-3′ containing the EcoRI site (in bold). The PCR product was digested by AflII and EcoRI and inserted into the pCDNA3.1+ (Invitrogen) at the corresponding restriction sites.
The murine Tnnt3 minigene (3476 bp) was prepared using the same protocol by amplifying the murine Tnnt3 genomic region between exon 15 and 18 using the following primers fwd: 5′-AAAACTTAAGGGACAAGGCCAAGGAACTCTG-3′ and rev: 5′ ACCGGAATTCGGGAGTCTGATAACTTTATTCCT3′.
Cell culture and transfection
Human embryonic kidney HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal calf serum (Life Technologies) and 50 μg/ml gentamicin (Life Technologies) in a 5% CO2 incubator at 37°C. Cells were grown in six-well plates and were transfected at 80% confluence with 5.2 μg of DNA constructs using PEI in 150 mM NaCl. Forty eight hours after transfection, total RNA was immediately extracted using Trizol (Invitrogen).
Human myoblasts LHCNM2 (46) were grown in KMEM 199 Medium (Life Technologies) and DMEM (Life Technologies) in a 1:4 ratio supplemented with 20% foetal calf serum (Life Technologies), 5 ng/ml human epithelial growth factor (Life Technologies), 0.5 ng/ml bFGF, 0.2 μM dexamethasone (Sigma-Aldrich), 50 μg/ml fetuin (Life Technologies) and 5 μg/ml insulin (Life Technologies) in a 5% CO2 incubator at 37°C. Differentiation was induced at confluence by replacing the growth medium with DMEM supplemented with 50 μg/ml of gentamicin. Transfection of human myoblasts were performed with Fugene HD (Promega) following the manufacturer's instructions and 48 h after transfection total RNA was immediately extracted using Trizol (Invitrogen).
Primary H2KB IM2 mouse myoblasts (named ‘control’ hereafter) were conditionally immortalized with a temperature-sensitive SV40 large T-antigen (tsA58) transgene and derived from the ImmortoMouse. Mutated-PABPN1 (H2KB-D7e named ‘Ala17’ hereafter) and wild-type-PABPN1 (H2KB-WTa named ‘Ala10’) stable cells lines were derived from IM2 cells line by transfection with a plasmid expressing expanded-PABPN1 or wild-type-PABPN1 respectively under the control of the human desmin locus control region and promoter to ensure myotube-specific expression of the human PABPN1 transgene (47). Cells were cultivated on matrigel coated support (Corning) in DMEM, supplemented with 20% foetal bovine serum (Invitrogen), 0.5% chicken embryo extract (Seralab), 100 U/ml penicillin-streptomycin (Thermo Scientific), and 20 U/ml interferon-γ (Millipore) at 33°C in a humidified 5% CO2 air atmosphere. For Ala17 and Ala10 cell lines, 500 μg/ml of G418 (Life Technologies) was added to the media. Differentiation was induced when cells were fully confluent (90–100%) by changing medium to DMEM, 10% horse serum and 100 U/ml penicillin-streptomycin at 37°C in a humidified 5% CO2 air atmosphere.
siRNA transfection was performed in HEK293T cells using PEI in 150 mM NaCl with 100 pmol of siRNA with 2 μg of plasmid in a six-well plate. All siRNAs were synthetized by Eurogentec. The control siRNA was against Renilla luciferase (PGL3) from Eurogentec. RNA and proteins were collected 48 h after transfection. For H2KB cells, siRNA transfection was performed using oligofectamine (Life Technologies) according to the manufacturer's instructions in a 12-well plate with a final concentration of 60 pmol of siRNA. siRNA treatment in H2KB cells was performed twice at 48 h interval before switching to differentiation medium. RNA and protein extractions were performed 5 days later.
Mice
A17.1 and A10.1 transgenic mice have been previously described (25,48). Male A17.1, A10.1 mice and wild-type FvB (WT) controls were generated by crossing the heterozygous carrier strain A17.1 and A10.1 obtained from Rubinsztein's group (25) with FvB mice. The mice were genotyped by PCR three to four weeks after birth. Wild-type FvB, A10.1 and A17.1 mice were housed in minimal disease facilities (Royal Holloway, University of London) with food and water ad libitum.
Isometric tension determination
T/pCa relation-ships were established as previously described (49).
Skinning protocol
Muscles were chemically skinned by overnight exposure at 4°C to a skinning solution containing 10 mM MOPS, 170 mM potassium propionate, 2.5 mM magnesium acetate, 5 mM K2 EGTA, 2.5 mM ATP (49). This procedure permeabilizes the sarcolemmal and the transverse tubular membranes and allows the application of activating solutions of various calcium and strontium concentrations (pCa and pSr, with pCa = −log[Ca2+] and pSr = −log[Sr2+]) directly to the contractile proteins. The skinned biopsies were stored at –20°C in a storage solution (glycerol/skinning solution, 50/50, v/v) until analysis.
Solutions
The composition of all solutions was calculated with the Fabiato computer program (50). Different solutions were used in the experiments: a washing solution (10 mM MOPS, 185 mM potassium propionate, 2.5 mM magnesium acetate); a relaxing solution identical to the skinning solution; pCa and pSr activating solutions corresponding to washing solution but with varying concentrations of free Ca2+ or Sr2+ from CaCO3 or SrCl2, respectively, buffered with EGTA and added in proportions to obtain the different pCa values (7.0–4.2) or pSr values (5.0 and 3.4). ATP was added to each solution to a final concentration of 2.5 mM. At the beginning of the experiment, each fiber was bathed for 20 min in 2% Brij58 in relaxing solution, in order to irreversibly eliminate the ability of the sarcoplasmic reticulum of skinned muscles to sequester and release Ca2+.
Force measurements and effect of O-GlcNAcase inhibitors on calcium properties
A 5 mm fiber segment isolated from skinned biopsies was mounted in an experimental chamber. On one end, the fiber was connected to a strain-gauge (force transducer Fort 10, World Precision Instruments). The output of the force transducer was amplified and recorded on a graph recorder (Gould. model 6120) and simultaneously analysed by computer software.
At the beginning of each experiment, the fibre was activated with the pSr 5.0 solution, followed by the pSr 3.4 solution, to verify that the tested fibre was slow and not fast. After bathing in the washing solution, the fiber was activated at a level P with various pCa solutions (from 7.0 to 4.8, with a step equal to 0.2 pCa units). Each steady state submaximal P tension was followed immediately by a maximum contraction Po ensured by pCa 4.2 solution, a calcium concentration that will saturate all troponin C sites. The P tensions were expressed as a percentage of the maximal Po tension and reported as Tension/pCa (T/pCa) relationships. Finally, the fibre was relaxed in the relaxing solution. If force declined during a sustained contraction, or decreased by more than 20% during the whole experiment, or if T/pCa was not completely achieved, the fibres were rejected from the analysis. Just after force measurements, each fibre was resuspended in 10 μl of Laemmli buffer, and stored at -20°C until further analysis.
The following parameters were determined from T/pCa curves: the pCa50 value, corresponding to 50% of maximal Ca2+ tension responses, which characterize the affinity of the contractile apparatus for Ca2+; the threshold for activation by Ca2+(pCa threshold) as an indicator of the calcium sensitivity of the contractile system; and the steepness of the T/pCa reflecting the cooperativity between the different regulatory proteins within the thin filament. The steepness of the T/pCa was determined by the Hill coefficients nH, calculated according to the following equation: P/Po = ([Ca2+]/K)nH/[1 + ([Ca2+]/K)nH]), where P/Po is the normalized tension and K is the apparent dissociation constant (pK = −log K = pCa50). All parameters were established independently for each fibre. Data are presented as means ± SEM. Differences between means are considered significant when P < 0.05, according to Student's t-test.
Immunodetection of PABPN1 inclusions on muscle sections
Immunostaining of PABPN1 nuclear aggregates was performed as previously described (51). Briefly, muscle sections (5 or 10 μm) were fixed in 100% cold acetone and pre-incubated in 1 M Potassium Chloride (KCl) solution for 1 h to remove any soluble proteins, before incubation with rabbit polyclonal anti-PABPN1 (1/100, Epitomics) and anti-dystrophin antibody (1/10, NCL-Dys1, Novocastra). Sections were further incubated with respective fluorescent secondary antibodies (Life Technologies) and stained with Hoechst (Sigma-Aldrich) to visualize nuclei. The percentage of muscle fibre nuclei containing PABPN1 aggregates was determined by counting the number of nuclei containing any PABPN1 staining compared to the total number of myonuclei on the section.
PABPN1/SC35 immunostaining
H2KB cells were cultivated on Ibidi 35 mm Dishes (Biovalley). Immunofluorescence was performed at 5 days of differentiation. Cells were washed once with PBS and fixed for 10 min with 4% paraformaldehyde–PBS. Cells were washed with PBS–glycine 0.1 M for 15 min and incubated in 0.2% Triton X-100 PBS, 3% BSA and 5% goat serum for 30 min. Detection of immunocomplexes was performed using anti-PABPN1 (1/100, Epitomics) and anti-SC35 (1/1000, Sigma) for 1 h. Cells were washed 3 times in 0.1% Triton–PBS and incubated with goat anti-mouse and goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 555 respectively (Life Technologies).
Fluorescence in situ hybridization
RNA fluorescence in situ hybridization (FISH) was performed using probes synthesized by PCR with specific primers (one probe for exon 18 and three probes for intron 15, see details in Supplementary Table S2) using the Platinium Taq DNA Polymerase High Fidelity (Invitrogen) with a ratio of 1/6 of DIG-dUTP/dTTP (Roche) and murine Tnnt3 expressing vector as template. PCR products were loaded on agarose gels and purified using the Nucleospin and PCR clean-up kit (Macherey-Nagel).
HEK293T cells were transfected with the murine Tnnt3 minigene expressing vector or with pCDNA3.1 using PEI (2.5 μg of DNA constructs using PEI in 150 mM NaCl). Forty eight hours after transfection, cells were fixed for 10 min in 4% PFA and rinsed with PBS. Cells were then incubated with PBS 1% triton X-100 PBS for 30 min and rinsed three times with PBS. The probe was denatured at 95°C for 10 min and put directly on ice. Cells were then incubated with hybridization buffer (SSC 2×, 40% formamide, 0.2% BSA, 60 ng of yeast RNA) together with 60 ng of Tnnt3 probe at 60°C for 2 h. Then cells were washed with 0.1% Tween 20/PBS buffer for 30 min at 65°C and three times with PBS. This was followed by an incubation in 2% SVF PBS for 30 min and with anti-digoxigenin-Rhodamine Fab Fragments (Roche diagnostics) for 1 h in the dark. Cells were washed three times in PBS and stained with Hoescht, (Sigma Aldrich) to visualize nuclei. All experiments were performed in RNAse free conditions.
H2KB cells (Ala17 and control) were differentiated during 5 days on Ibidi 35mm Dish (Biovalley) and fixed for 10 min in 4% PFA rinsed with PBS. Cells were incubated with PBS 1% triton X-100 for 30 min and rinsed three times with PBS. Cells were incubated with pre-hybridization buffer (SSC 2×, 40% formamide, 0.2% BSA) for 30 min. The probe was denatured at 95°C for 10 min and put immediately on ice. Cells were then incubated with hybridization buffer (SSC 2×, 40% formamide, 0.2% BSA, 60 ng of yeast RNA) together with 60 ng of TNNT3 probe at 60°C for 2 h. The cells were then washed with prehybridization buffer for 30 min at 65°C and three times with PBS 0.1% Tween 20 and three times with PBS.
This was followed by immunofluorescence staining for PABPN1. Briefly, cells were incubated in PBS 2% SVF during 30 min and with PABPN1 primary antibody (Epitomics) and SC35 antibody (Sigma) in the same buffer for 2 h at room temperature. Cell were washed three times with PBS and incubated with the secondary antibodies Alexa Fluor 488 and Alexa fluor 647 goat anti-mouse (Life Technology) and anti-digoxigenin-Rhodamine Fab Fragments (Roche diagnostics) for 1 h in the dark. Cells were stained with Hoescht, (Sigma-Aldrich) to visualize nuclei. All experiments were performed in RNAse free conditions.
Western blotting
Proteins were extracted by sonicating cells in RIPA buffer 0.15 M NaCl, 0.1% SDS, 50 mM Tris (pH 8), 2 mM EDTA and 10% Triton-X-100 with protease inhibitor cocktail (Complete, Roche Diagnostics). Similar quantities of proteins were separated on 4–12% Bis–Tris gels (Invitrogen) and transferred onto a nitrocellulose membrane for 1 h at constant 30 V at 4°C. Membranes were blocked by incubation in 5% milk or BSA in 0.1 M TBS, 0.1% Tween-20 for 1 h at room temperature under agitation. Membranes were stained with primary antibodies directed against PABPN1 (1/1000, Epitomics), SC35 (1/1000, Millipore), SRP30a (1/1000, Santa-Cruz), MBNL1 (1/1000, MB1a antibody, gift from Glenn Morris), Flag-HRP (1/1000, Sigma) and GAPDH–HRP (1/1000, Abcam). Membranes were then incubated with appropriate secondary antibodies conjugated to HRP (except for Flag-HRP and GAPDH-HRP) and the G:Box system (Syngene) was used to detect the signals from the membranes.
Image acquisition and analysis
Images were visualized using an Olympus BX60 microscope (Olympus Optical, Hamburg, Germany), digitalised using a CCD camera (Photometrics CoolSNAP fx; Roper Scientific) and analyzed using MetaView image analysis system (Universal Imaging, Downington, PA, USA), MetaMorph imaging system (Roper Scientific, Tucson, AZ, USA) software, and ImageJ 1.44o (http://imagej.nih.gov/ij) for quantification analysis.
Statistical analysis
All data are presented as mean values ± standard error of the mean (SEM) (cohort size stated per experiment). All statistical analyses were performed using either the Student's t-test, or the ANOVA one-way analysis of variance followed by the Bonferroni post-test, using GraphPad Prism (version 4.0b; GraphPad Software, San Diego California, USA). A difference was considered to be significant at P< 0.05 (*), P< 0.01 (**) or P< 0.001 (***).
RESULTS
TNNT3 exon 16 is downregulated in OPMD
To identify potential splicing defects in OPMD, we performed a genome-wide transcriptomic analysis at the exon level on RNA extracted from skeletal muscle biopsies of OPMD patients (Table 1). For this study, we used sternocleidomastoid muscles as a pre-symptomatic muscle, that show both a typical clinical phenotype and a significant amount of nuclear aggregates compared to age-matched control subjects (Figure 1A). Forty six missplicing events were found in 39 distinct genes, including 12 cassette exon defects (Supplementary Table S1 and Figure S1A). Among them the most significantly deregulated mRNA (highest splicing fold change index) encodes a muscle-specific protein, the human Troponin T type 3 (TNNT3) (Table 2). The TNNT3 transcript contains 18 exons; its pre-mRNA is extensively spliced with two mutually exclusive exons, 16 (α) and 17 (β), included at the 3′ end (52–54) (Figure 1B). Exon 16 was found to be downregulated in OPMD samples (Supplementary Figure S1A), leading to an imbalanced ratio of these mutually exclusive exons. This was confirmed by RT-PCR on sternocleidomastoid and quadriceps muscle biopsies from OPMD patients, with a strong decrease in the level of the exon 16 isoform expression (Figure 1C and D). Splicing defects of RECK exon 8 (Reversion-Inducing-Cysteine-Rich Protein With Kazal Motifs) and ZCCHC11 exon 8 (Zinc Finger CCHC Domain-Containing Protein 11 or TUT4)—which were respectively hits numbers 2 and 3 on the exon array analysis (Table 2 and Supplementary Figure S1A)–were also confirmed by qRT-PCR on sternocleidomastoid muscle biopsies (Supplementary Figure S1B). This is the first time that misregulated alternative splicing events have been described in OPMD.
Figure 1.

Splicing of TNNT3 mutually exclusive exons 16 and 17 is deregulated in oculopharyngeal muscular dystrophy. (A) Left: PABPN1 staining following KCl treatment revealed intranuclear aggregates in OPMD muscle biopsies (red: dystrophin, green: PABPN1, blue: dapi). Aggregates are indicated with an arrow. On the bottom right panel, is presented a control biopsy - no aggregates are present. Scale bar = 50 μm. Right: Quantification of nuclear aggregates showed 6.7–16.7% PABPN1 aggregates (mean 12%) in myonuclei from OPMD patients. ** indicates P< 0.01. (B) Schematic representation of the exon structure of TNNT3 pre-mRNA. pA: polyadenylation site. Exons 16 and 17 are highlighted in grey. The two mutually exclusive exons are both 41 bp long encoding each a short 14 amino acid peptide in the C-terminal of the protein. Arrows on exons 15, 16 and 17 indicate the forward (TNNT3-ex15-FWD) and two reverse (TNNT3-ex16-REV-108 and TNNT3-ex17-REV-131) primers used to amplify and discriminate by PCR the two TNNT3 transcript isoforms. (C and D) TNNT3 exon 16 is downregulated in OPMD skeletal muscle biopsies with (C). Left: RT-PCR analysis of endogenous TNNT3 exon 16 and 17 isoforms from human sternocleidomastoid skeletal muscle biopsies of normal adult individuals (CTRL; n = 4) and OPMD patients (OPMD; n = 5). Right: quantification of respective exon 16 and 17 inclusions in TNNT3 mRNA. ***indicates P< 0.001 and (D) RT-PCR analysis of endogenous TNNT3 exon 16 and 17 isoforms from human quadriceps skeletal muscle biopsies of normal adult individuals (CTRL; n = 4) and OPMD patients (OPMD; n = 2).
Table 2. Splicing defects are present in oculopharyngeal muscular dystrophy including alteration of TNNT3 alternative splicing of exon 16 and 17. Exon array revealed several splicing defects, including 12 cassette exon events. TNNT3 was the first hit on the array with the highest splicing fold change index. A manual inspection allowed the filtering and classification of exons predicted to be differentially included.
| GENE STABLE ID | Gene symbol | Fold-change splicing index | P-value splicing index | Confidence |
|---|---|---|---|---|
| GSHG0004534 | TNNT3 | 3,55 | 1,03E-02 | +++ |
| GSHG0030112 | RECK | 2,12 | 2,03E-02 | +++ |
| GSHG0002105 | ZCCHC11 | 2,17 | 1,42E-02 | ++ |
| GSHG0008422 | SPG20 | 2,09 | 1,80E-02 | ++ |
| GSHG0030642 | RFX3 | 2,07 | 3,70E-02 | ++ |
| GSHG0008586 | UGGT2 | 2,04 | 4,80E-02 | ++ |
| GSHG0008374 | USP12 | 2,53 | 1,41E-02 | + |
| GSHG0017444 | SMC6 | 2,49 | 7,97E-04 | + |
| GSHG0022278 | ABCC5 | 2,33 | 3,04E-02 | + |
| GSHG0031771 | WDR44 | 2,24 | 1,24E-02 | + |
| GSHG0007569 | STAC3 | 2,15 | 4,80E-02 | + |
| GSHG0023986 | SLC30A5 | 2,26 | 1,66E-02 | + |
TNNT3 splicing defect is linked to the presence of aggregates
We checked the splicing pattern of Tnnt3 in a murine cell model of OPMD, namely the H2KB-D7e cells (47). Primary H2KB-IM2 cells are conditionally immortalized mouse myoblasts derived from the ImmortoMouse (55) and H2KB-D7e are cells stably derived from the H2KB-IM2 cell line by transduction with an expanded Flag-tagged PABPN1 construct under the control of the human desmin locus control region and promoter. This ensures that the human expPABPN1 transgene will only be expressed in differentiation conditions (Supplementary Figure S2A). For clarity, the H2KB-IM2 and -D7e will be called hereafter ‘control’ and ‘Ala17’ muscle cells respectively. In differentiation conditions, these Ala17 cells contain a large number of nuclear aggregates present in more than 50% of the nuclei ((47) and Figure 2E). In control cells, the Tnnt3 exon 16 isoform is absent at day 1, but progressively increases during differentiation. Interestingly, in Ala17 cells at both 3 and 5 days of differentiation, we observed the same splicing defect as in human OPMD samples with a strong decrease in the level of the Tnnt3 exon 16 isoform compared to control cells (Figure 2A and B). Since the expression of the Tnnt3 exon 16 isoform increases during differentiation we verified that the splicing defect in Ala17 cells was not due to a delay in differentiation. Light microscopy visualization showed comparable fused myoblasts in control and OPMD cell lines at 5 days of differentiation (Supplementary Figure S2B). In addition, myogenin and myosin expression levels were equivalent in both Ala17 and control cells during the time course of differentiation (days 1, 3 and 5). We also checked the alternative splicing of Bin1 exon 10, which is known to be regulated during muscle differentiation (56) and the splicing pattern was similar in both conditions, further confirming that the differentiation kinetics were similar in control and Ala17 cells (Supplementary Figure S2C). To understand the link between PABPN1 nuclear aggregates and the splicing defect, we reduced PABPN1 expression in differentiated Ala17 cells using RNA interference. The reduction of PABPN1 expression by 50% at mRNA and protein level (Figure 2C–D) drastically reduced the percentage of nuclei containing nuclear aggregates from 60% to 10% (Figure 2E). This led to the rescue of the splicing defect in differentiated siRNA-treated Ala17 cells (Figure 2F). Several hypotheses have been made regarding the role of PABPN1 aggregates in the pathogenesis of the disease, one of them being the sequestration of PABPN1 itself, leading to a loss of function. Transfection of siRNA against PABPN1 in control cells—to mimic a loss of function—did not modify the level of exon 16 isoform (Figure 2F). Neither did the addition of a wild-type functional PABPN1 expressing vector in Ala17 cells (data not shown), suggesting that TNNT3 splicing defect is not linked to the level of available PABPN1 itself. To further reinforce the crucial role of the aggregate we checked the splicing defect in H2KB-WTa (Ala10 cells), overexpressing wild-type Flag-tagged PABPN1 at the same level as in Ala17 cells and containing aggregates in differentiation ((47) and Supplementary Figure S3A-B): the splicing defect was confirmed in this differentiated cell line as well (Supplementary Figure S3C and D). Together these results strongly suggest a direct link between the presence of nuclear aggregates and the splicing misregulation of TNNT3.
Figure 2.

Tnnt3 splicing defect is rescued by reducing the amount of aggregates in murine myoblasts. (A) Tnnt3 exon 16 level is downregulated in OPMD murine myoblasts. RT-PCR analysis of endogenous Tnnt3 exon 16 and 17 isoforms from CTRL and OPMD (Ala17) murine myoblasts after 1, 3 and 5 days of differentiation - primary CTRL mouse myoblasts are conditionally immortalized with a temperature-sensitive SV40 large T-antigen (tsA58) transgene and derived from the ImmortoMouse. Mutated-PABPN1 (Ala17) stable cells lines derive from CTRL cells line and express expanded-PABPN1. Below: quantification of respective exon 16 and 17 inclusions in Tnnt3 mRNA. (B and C) siRNA treatment against PABPN1 (siPAB#1 and siPAB#2) in differentiated Ala17 cells reduce the level of PABPN1 at mRNA level, as shown by quantitative RT-PCR (** indicates P< 0.01 compared to siCTRL, transfections are performed in triplicate), and (D) at protein level as shown by representative Western blot. (E) Left: The amount of nuclear aggregates was drastically reduced by siPAB treatment. The histogram indicates the percentage of nuclei containing PABPN1 aggregates classified based on the number of aggregates per nuclei (1, 2, 3 or more than 3). An average of 100 nuclei in myotubes were counted on random fields. Right: Representative immunofluorescence of nuclei with 0, 1, 2, and >3 PABPN1 nuclear aggregates. Scale bar = 5 μm. (F) Rescue of Tnnt3 mRNA splicing defect with siRNA against PABPN1 (siPAB#1 and #2) compared to a siRNA control (siCTRL) in differentiated Ala17 cells. Quantification of RT-PCR analysis of Tnnt3 exon 16 (% exon 16) inclusion. NS indicates non statistical, *P< 0.05, **P< 0.01 compared to the CTRL condition. Transfections were performed in triplicate.
SC35 splicing factor regulates TNNT3 mutually exclusive exons
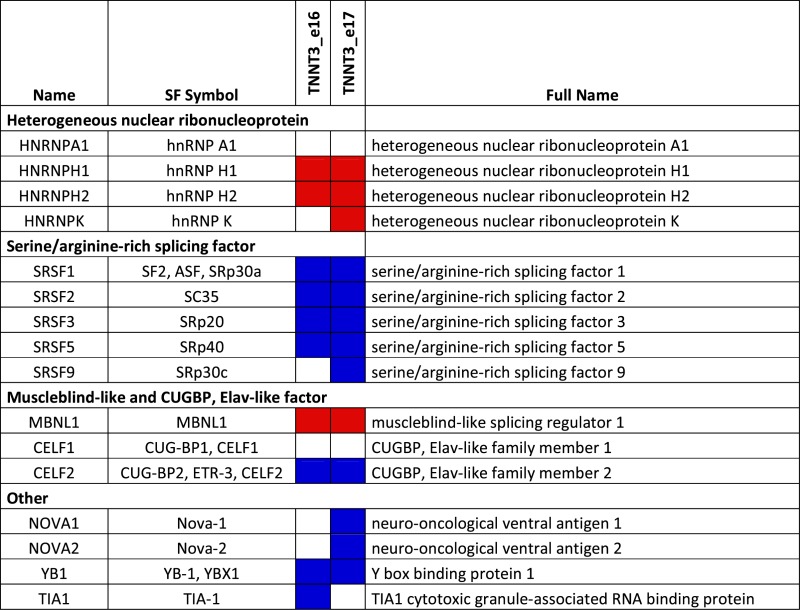
To determine which splicing factor is involved in the regulation of TNNT3 mutually exclusive exons, a minigene containing exons 16 and 17 of human TNNT3 bordered by exon 15, 18 and their intronic regions (Figure 3A) was cloned from human genomic DNA in a plasmid expression vector. This minigene construct was co-transfected into HEK293T cells—which do not express TNNT3—together with various vectors expressing well-characterized splicing regulatory factors. These included members of the hnRNP, SR, MBNL and CELF families and were selected using prediction software (SpliceAid2 and ESEFinder publically available at http://193.206.120.249/splicing_tissue.html and http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home respectively) (57,58) (Table 3). Fifteen selected splicing factors were tested by co-transfection with the TNNT3 minigene expression vector. GFP was used to monitor expression and transfection efficiency (Supplementary Figure S4A), and we further confirmed by western-blotting the level of expression in HEK293T cells for several of them (Figure 3D and Supplementary Figure S4B). Of the 15 splicing factors, only the co-expression of SC35 (also called SRSF2) or hnRNPK together with the TNNT3 minigene expression vector induced a significant increase in exon 16 inclusion (Figure 3B). Using the same approach, we also confirmed that PABPN1 overexpression itself did not modify the level of exon 16 (Supplementary Figure S4C and D). As the splicing defect is present in both human and murine OPMD samples (Figures 1C and D and 2A), the same approach was applied using a murine Tnnt3 exons 15–18 minigene expression vector. In this situation, only the co-transfection of the SC35 expression vector significantly modified the inclusion of exon 16 (Figure 3C). Finally in human myoblasts, only the transfection of the SC35 expression vector led to the upregulation of the exon 16 isoform when co-transfected with the human TNNT3 minigene expression vector (Figure 3E), confirming that SC35 is the most probable splicing factor candidate for the regulation of exon 16 inclusion. SC35 is also a well-known constituent of nuclear speckles (59) involved in pre-mRNA splicing (60). The transfection of cells with SC35 results in an upregulation of the exon 16 isoform, which is downregulated in OPMD patients or cells. Although the level of SC35 itself is not downregulated in OPMD samples when compared to control samples (Figure 3F), downregulation of the exon 16 isoform can be mimicked by depletion of SC35 expression level using RNA interference in HEK293T cells transfected with the TNNT3 minigene (Figure 3G–I). In contrast, the downregulation of SRP30a (ASF/SF2, a splicing factor related to SC35 (61)) or MBNL1 (which regulates the splicing of TNNT3 fetal exon (62,63)) by RNA interference did not recapitulate this splicing defect found in OPMD or in SC35-knockdown cells (Supplementary Figure S4E and F). These results demonstrate that the splicing regulation of TNNT3 mutually exclusive last exons is strongly dependent on SC35 and suggest that in OPMD TNNT3 pre-mRNA and SC35 splicing factor are somehow dissociated.
Figure 3.

SC35 regulates alternative splicing of TNNT3 exon 16 and 17. (A) Schematic representation of the human TNNT3 minigene containing both exons 16 and 17 bordered by exons 15 and 18 including full downstream and upstream intronic sequences (sizes are indicated in italic above). (B) and (C) SC35 overexpression increases inclusion of TNNT3 exon 16. Quantification of RT-PCR analysis of TNNT3 exon 16 (% exon 16) inclusion following respectively human (B) and murine (C) exons 15–18 minigene expressing vector cotransfection in HEK293T cells together with 15 different vectors expressing splicing factors. Histograms represent the mean of at least three independent transfections and are depicted as the percentage of mRNA containing TNNT3 exon 16. Error bars indicate S.D. and # indicates statistical ANOVA difference compared to the minigene only condition. (D) A western-blot showing SC35 overexpression in HEK293T cells transfected with SC35 expressing vector fused with GFP compared to untransfected cells. (E) SC35 overexpression increases inclusion of TNNT3 exon 16 in human myoblasts. RT-PCR analysis after transfection of the human TNNT3 minigene into the human myoblast cell line LHCNM2 together with several splicing factors. (F) RT-qPCR on human muscle biopsies demonstrating that the expression level of SC35 mRNA is similar in control and OPMD samples. (G) Representative Western blot showing SC35 extinction in HEK293T cells transfected with siRNA against SC35 compared to HEK293T cells transfected with siRNA CTRL. (H–I) SC35 extinction mimics theTNNT3 splicing defect observed in OPMD. RT-PCR analysis of the TNNT3 minigene cotransfected with a siRNA against SC35 (siSC35#1 and #2), a siRNA against luciferase as a control (siCTRL), or with a plasmid expressing SC35 (+SC35). Quantification of TNNT3 exon 16 inclusion is presented in (I). *** indicates P< 0.001 compared to the siCTRL condition.
Table 3. List of splicing factors (SF) able to regulate exon 16 and 17 of TNNT3 pre-mRNA using prediction sites. Splicing enhancers are in blue and splicing silencers are in red.
|
TNNT3 pre-mRNA is trapped in nuclear aggregates
Aggregates are formed by the accumulation of misfolded PABPN1 in muscle nuclei. Interestingly, it is already known that these PABPN1 aggregates are delocalized from SC35-nuclear speckles (19,47,64). By immunostaining we confirmed the delocalization of PABPN1 aggregates and SC35 nuclear splicing domains in differentiated Ala17 OPMD cells as opposed to the co-localization of PABPN1 and SC35 observed in control cells (Figure 4A and Supplementary Figure S5A). A recent study proposed that growing PABPN1 aggregates gradually deplete SC35-nuclear speckles of poly(A) RNA/PABPN1 complexes, thus interfering with the normal trafficking and post-transcriptional processing of poly(A) mRNA in speckles (64). We took advantage of the Ala17 cells where Tnnt3 splicing defect and nuclear aggregates are both present and we analysed the localization of Tnnt3 pre-mRNA in these cells, by performing fluorescent in situ hybridization (FISH). Using probes targeting specifically exon 18 (ex18) or intron 15 (int15) of Tnnt3 pre-mRNA (Supplementary Figure S6), we observed a co-localization of Tnnt3 pre-mRNA with PABPN1 in nuclear aggregates (Figure 4B and C and Supplementary Figure S5B). Hence, the pre-mRNA of Tnnt3 is physically delocalized from SC35-nuclear speckles. Therefore, we speculated that the splicing defect is a consequence of the spatial dissociation of SC35 and Tnnt3 pre-mRNA, the latter being trapped in PABPN1 aggregates.
Figure 4.

Tnnt3 pre-mRNA is trapped in PABPN1 aggregates. (A) PABPN1 is localized in SC35-nuclear speckles in control conditions and delocalized from SC35-nuclear speckles with nuclear aggregate formation in OPMD (Ala17). Immunofluorescence of PABPN1 and SC35 in control and OPMD (Ala17) H2KB cell lines. Scale bar = 5 μm. Single cell magnified of Ctrl and Ala17 cells are presented in Supplementary Figure S5. (B and C) Tnnt3 pre-mRNA is present in PABPN1 nuclear aggregates. (B) Representative image of RNA fluorescence in situ hybridization (FISH) using a Digoxigenin probe targeting exon 18 of TNNT3 mRNA (red) coupled to immunofluorescence detection of PABPN1 (green) and counterstained with Hoechst (blue) on fixed 5-day differentiated Ala17 cells. Scale bar = 10 μm. (C) Different representative images of FISH using a probe targeting exon 18 (ex18, left) or intron 15 (int15, right) of TNNT3 pre-mRNA coupled to immunofluorescence detection of PABPN1 on fixed 5-day differentiated Ala17 cells. Pre-mRNA detection is indicated with white arrows. Scale bar = 20 μm.
Calcium sensitivity is decreased in slow fibers of OPMD muscles
TNNT3 pre-mRNA is intensively spliced and finely regulated; it is suggested that the abundance in the ratio of the different isoforms participates to muscle plasticity (54). In order to investigate the physiopathological consequences of this splicing defect, we used a mouse model of OPMD, the A17.1 mouse. This model was generated by the expression of alanine expanded mammalian PABPN1 (PABPN1-17ala) specifically in muscles; it reproduces the disease characteristics, namely progressive muscle weakness and formation of nuclear aggregates containing mutant PABPN1 in muscles (25,48). Interestingly, we found the same splicing defect in the slow soleus muscle of this A17.1 OPMD mouse model (Figure 5A), where nuclear aggregates are also present (Figure 5B), whereas this splicing defect was absent in the soleus of the control A10.1 mouse (overexpressing wild-type PABPN1 (25)), where nuclear aggregates are absent (Supplementary Figure S7). These data strengthen the claim of a direct link between the splicing defect and the presence of nuclear aggregates. TNNT3 is a subunit of the trimeric troponin complex, which regulates skeletal muscle contraction in response to calcium (65). In vitro assays with recombinant proteins already demonstrated that a difference in the ratio of isoforms including exon 16 or 17 led to alterations in muscle fibre calcium affinity, a key parameter involved in the modulation of force contraction (54,66). Indeed, while numerous reports have demonstrated the predominant role of the calcium sensor Troponin C of the troponin complex in the modulation of calcium sensitivity and affinity, it has been clearly established that TNNT3 is also involved in the modulation of these mechanisms (67). We took advantage of the soleus of the A17.1 mouse, which presents the unbalanced ratio of Tnnt3 mutually exclusive exons to look at the functional consequences of the TNNT3 splicing defect. We first calculated the pCa/tension relationship in order to analyse whether the activation parameters of muscle fibre tension by calcium could be altered in OPMD muscles. The Tension/pCa relationship performed on isolated skinned fibres from murine soleus muscles revealed a decrease in the calcium affinity of slow OPMD muscle fibres compared to slow control muscle fibres (pCa of 5.85 ± 0.04 and 6.02 ± 0.04 respectively, P< 0.05) (Figure 5C). This decrease in calcium affinity can be clearly related to the expression of the exon 17 containing isoform instead of the exon 16 isoform, since previous data has shown that the Tnnt3 exon 17 containing isoform had a lower calcium affinity than the exon 16 isoform (66). Our data demonstrate that a decrease in calcium handling is a functional consequence of the decrease in exon 16 Tnnt3 isoform in slow fibres, which are the predominant fibre type in many human skeletal muscles.
Figure 5.

Splicing defect in OPMD mouse induces decreased calcium sensitivity. (A) Tnnt3 splicing defect is present in OPMD mouse model. RT-PCR analysis of endogenous Tnnt3 exons 16 and 17 isoforms in soleus (Sol) muscles of wild-type (WT) and A17.1 (OPMD) mice. (B) PABPN1 staining following KCl treatment revealed intranuclear aggregates in the soleus muscle of A17.1 mouse (red: dystrophin, green: PABPN1, blue: dapi), whereas no aggregate are observed in control mice. Aggregates are indicated with an arrow (see also inset boxes). Scale bar = 30 μm. (C) Deregulation of Tnnt3 exon 16 and 17 induces decreased calcium sensitivity in mice slow fibers, as measured by pCa/Tension relationship. * indicates P< 0.05, ** indicates P< 0.01.
DISCUSSION
Like many triplet expansion disorders, OPMD is characterized by the presence of insoluble nuclear aggregates in muscle fibres. However the exact role and contribution of these PABPN1 aggregates to the clinical phenotype in OPMD is still debated (1). In this study, we clearly illustrate that PABPN1 nuclear aggregates have at least one toxic function as they lead to a specific splicing defect: PABPN1 aggregates are able to trap the muscle-specific TNNT3 pre-mRNA, driving it outside of the nuclear speckles, leading to altered SC35-mediated splicing. This trapping leads to a defect in the splicing regulation of the TNNT3 mutually exclusive exons 16 and 17 in OPMD samples compared to controls. This is the first demonstration of splicing defects in OPMD. Poly(A) RNA were previously observed in PABPN1 nuclear aggregates using oligo-dT probes (18–19,64); here we additionally demonstrate that PABPN1 nuclear aggregates are able to sequester a pre-mRNA leading to its missplicing. Whether this muscle-specific RNA plays a role as a potential scaffold for aggregate formation in a similar way to architectural RNAs (arcRNA) for nuclear domains such as paraspeckles or stress bodies (68) remains to be further investigated. Nuclear speckles are essential nuclear compartments involved in post-transcriptional processing of mRNA. These nuclear structures are enriched in poly(A) RNA, in many pre-mRNA splicing factors including snRNP (small nuclear ribonucleoprotein particles) and in SR splicing factors such as SC35 (69). PABPN1 is found in the nucleoplasm excluding the nucleolus and is concentrated mainly in nuclear speckles where it participates in mRNA metabolism. This localization of PABPN1 in nuclear speckles requires the binding to poly(A) RNA (19,29,70–71). Previous studies have shown that PABPN1 nuclear aggregates originate at the edge of the speckles and gradually delocalize outside of these compartments, together with a gradual depletion of poly(A) RNA from speckles (19,64). It has been suggested that Pab2 (fission yeast ortholog of mammalian PABPN1) associates with pre-mRNA co-transcriptionally prior to 3′ end processing/polyadenylation (72). In addition, pre-mRNA introns located at the 3′ end of multiple intron transcripts are more frequently spliced post-transcriptionally, presumably because transcription termination and cleavage at the polyadenylation site occurs before the splicing machinery manages to catalyse their removal (73). It is well established that the different steps of pre-mRNA processing (5′ cap, splicing, 3′ end processing) are connected and influence each other (74,75). The splicing defect that we identified is at the 3′ end of the TNNT3 pre-mRNA and we therefore propose that during the processing of TNNT3 pre-mRNA, the binding of PABPN1 to its 3′ end for further cleavage and polyadenylation induces, in the case of OPMD, a gradual delocalization of the complex pre-mRNA/PABPN1 from SC35-nuclear speckles. This delocalization induces a missplicing of the last exons of TNNT3 with the production of a modified protein that has a negative effect on muscle contraction. Indeed, the imbalanced ratio of TNNT3 exons 16 and 17 induced a decrease in calcium sensitivity with physiopathological consequences at the level of the muscle fibre (see Figure 6 for a molecular model).
Figure 6.

Molecular mechanism of splicing deregulation in OPMD. Molecular model of TNNT3 mRNA splicing defect in OPMD: in healthy condition, mutually exclusives exons of TNNT3 pre-mRNA are regulated by SC35 splicing factor favouring exon 16 inclusion. In OPMD condition, TNNT3 pre-mRNA is delocalized from SC35 nuclear speckles by PABPN1 nuclear aggregates trapping resulting in a missplicing of exon 16 and a decreased calcium affinity of muscle fibres.
In this study, we have exploited different OPMD cell and animal models to decipher the exact mechanism, which controls the splicing defect observed in TNNT3. These models, based on expanded PABPN1 overexpression, reproduce pathological characteristics found in OPMD patients, including the presence of nuclear aggregates. The overexpression is not ideal, since this is absent in OPMD patients (76). In the present study, we therefore performed several experiments to clearly dissociate aggregation from overexpression in these models. Using both RNAi and transfection systems to modify the level of PABPN1, we confirmed that the splicing defect reproduced in OPMD models is solely due to the presence of aggregates and not to the level of PABPN1 itself. Indeed, neither the reduction of PABPN1 levels in control IM2 cells, nor the transfection in Ala17 cells with an exogenous PABPN1 expression vector to potentially compensate for a loss of function, nor the silencing or overexpression of PABPN1 in HEK293T cells transfected with the TNNT3 minigene, were able to modify the splicing pattern of TNNT3. In addition, since PABPN1 itself is an aggregate-prone protein, it is well known that the artificial overexpression of wild-type PABPN1 protein is able to induce in cells the formation of nuclear aggregates (77,78). For example, it has previously been demonstrated that the H2KB-WTa cells (named Ala10 cells here), overexpress wild-type PABPN1 and do contain around 20% nuclear aggregates (47). That the same phenotypes can be induced following expression of normal or mutant protein in Drosophila models of other disorders has also been reported previously (e.g. Parkinson's disease (79)). We used this cell model to further verify our mechanism and as expected, since these cells do contain aggregates, we confirmed the splicing defect in these cells. Interestingly in the soleus of A10.1 mice we were unable to detect aggregates and logically the splicing defect was absent. Altogether these data again emphasize the pivotal role of nuclear aggregates in the mechanism.
Similar findings regarding alanine repeats and subnuclear protein localization have been recently observed with the RNA-binding motif protein 4 (RBM4) (80). Alternative splicing defects have previously been observed in other trinucleotide repeat disorders including myotonic dystrophy (41). However, this is the first time that a splicing defect, with a misregulation of mutually exclusive last exons of the muscle specific TNNT3 mRNA, has been identified in OPMD. In the spliceopathies described so far, alternative splicing misregulation is in most cases due to an abnormal activity of an RNA splicing factor, for example, functional loss of MBNL proteins due to their sequestration by nuclear mutant RNA in myotonic dystrophy (81). This abnormal activity often results from splicing factors being misplaced from their natural location within the cell, highlighting the importance of the spatial distribution of the intracellular mRNA and splicing factors. We describe here a different mechanism directly linked to the presence of nuclear aggregates: the splicing defect is due to the spatial separation of the deregulated RNA from its splicing factor. Although the generic nature of this alternative mechanism remains to be demonstrated, it may play a role in various other diseases with nuclear inclusions or foci containing an RNA binding protein, as shown here for OPMD.
Supplementary Material
Acknowledgments
We are grateful to D. Auboeuf, D. Weil and N. Charlet-Berguerand for their gifts of splicing factor expressing vectors. We thank I. Behm for the ASF/SF2 antibody and N. Vignier for qPCR primers. We thank Glenn Morris at the Centre for Inherited Neuromuscular Disease for supplying antibody Mb1a to detect MBNL1. We thank J. Marie for fruitful discussion and careful reading of the manuscript. We thank N. Beltchenko for the construction of the murine Tnnt3 minigene and Elisa Negroni for image processing. The authors acknowledge the OPMD patients for their collaboration.
Footnotes
Present address: Capucine Trollet, Myology Research Center, UMRS 974 UPMC – INSERM – FRE 3617 CNRS – AIM, 47, bld de l'hôpital – G.H. Pitié-Salpétrière – Bâtiment Babinski, 75651 Paris cedex 13, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre National pour la Recherche Scientifique, the Association Française contre les Myopathies [Research Programs 15123 and 17110]; University Paris VI Pierre et Marie Curie; Institut National de la Santé et de la Recherche Médicale, the Fondation de l'Avenir [project ET1-622]; P.K. has a salary from the Ministère de l'Education Nationale de la Recherche et de la Technologie. Funding for open access charge: AFM eOPMD grant Research Program 17110.
Conflict of interest statement. None declared.
REFERENCES
- 1.Banerjee A., Apponi L.H., Pavlath G.K., Corbett A.H. PABPN1: molecular function and muscle disease. FEBS J. 2013;280:4230–4250. doi: 10.1111/febs.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eckmann C.R., Rammelt C., Wahle E. Control of poly(A) tail length. Wiley Interdiscip. Rev. RNA. 2011;2:348–361. doi: 10.1002/wrna.56. [DOI] [PubMed] [Google Scholar]
- 3.Apponi L.H., Leung S.W., Williams K.R., Valentini S.R., Corbett A.H., Pavlath G.K. Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum. Mol. Genet. 2009;19:1058–1065. doi: 10.1093/hmg/ddp569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benoit B., Mitou G., Chartier A., Temme C., Zaessinger S., Wahle E., Busseau I., Simonelig M. An essential cytoplasmic function for the nuclear poly(A) binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev. Cell. 2005;9:511–522. doi: 10.1016/j.devcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 5.de Klerk E., Venema A., Anvar S.Y., Goeman J.J., Hu O., Trollet C., Dickson G., den Dunnen J.T., van der Maarel S.M., Raz V., et al. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res. 2012;40:9089–9101. doi: 10.1093/nar/gks655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenal M., Elkon R., Loayza-Puch F., van Haaften G., Kuhn U., Menzies F.M., Vrielink J.A., Bos A.J., Drost J., Rooijers K., et al. The poly(a)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell. 2012;149:538–553. doi: 10.1016/j.cell.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 7.Beaulieu Y.B., Kleinman C.L., Landry-Voyer A.M., Majewski J., Bachand F. Polyadenylation-dependent control of long noncoding RNA expression by the poly(A)-binding protein nuclear 1. PLoS Genet. 2012;8:e1003078. doi: 10.1371/journal.pgen.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bresson S.M., Conrad N.K. The human nuclear poly(a)-binding protein promotes RNA hyperadenylation and decay. PLoS Genet. 2013;9:e1003893. doi: 10.1371/journal.pgen.1003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen D., Grenier St-Sauveur V., Bergeron D., Dupuis-Sandoval F., Scott M.S., Bachand F. A Polyadenylation-Dependent 3′ End Maturation Pathway Is Required for the Synthesis of the Human Telomerase RNA. Cell Rep. 2015;13:2244–2257. doi: 10.1016/j.celrep.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Brais B., Bouchard J.P., Xie Y.G., Rochefort D.L., Chretien N., Tome F.M., Lafreniere R.G., Rommens J.M., Uyama E., Nohira O., et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 11.Richard P., Trollet C., Gidaro T., Demay L., Brochier G., Malfatti E., Tome F.M.S., Fardeau M., Laforet P., Romero N., et al. PABPN1 (GCN)11 as a Dominant Allele in Oculopharyngeal Muscular Dystrophy –Consequences in Clinical Diagnosis and Genetic Counselling. J. Neuromuscul. Dis. 2015;2:175–180. doi: 10.3233/JND-140060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jouan L., Rocheford D., Szuto A., Carney E., David K., Dion P.A., Rouleau G.A. An 18 Alanine Repeat in a Severe Form ofOculopharyngeal Muscular Dystrophy. Can J Neurol Sci. 2014;41:508–511. doi: 10.1017/s0317167100018588. [DOI] [PubMed] [Google Scholar]
- 13.Trollet C., Gidaro T., Klein P., Perie S., Butler-Browne G., Lacau St Guily J. GeneReview. Seattle: University of Washington; 1993. Oculopharyngeal muscular dystrophy. [Google Scholar]
- 14.Lopez Castel A., Cleary J.D., Pearson C.E. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell. Biol. 2010;11:165–170. doi: 10.1038/nrm2854. [DOI] [PubMed] [Google Scholar]
- 15.Galka-Marciniak P., Urbanek M.O., Krzyzosiak W.J. Triplet repeats in transcripts: structural insights into RNA toxicity. Biol. Chem. 2012;393:1299–1315. doi: 10.1515/hsz-2012-0218. [DOI] [PubMed] [Google Scholar]
- 16.Tome F.M., Fardeau M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol. 1980;49:85–87. doi: 10.1007/BF00692226. [DOI] [PubMed] [Google Scholar]
- 17.Corbeil-Girard L.P., Klein A.F., Sasseville A.M., Lavoie H., Dicaire M.J., Saint-Denis A., Page M., Duranceau A., Codere F., Bouchard J.P., et al. PABPN1 overexpression leads to upregulation of genes encoding nuclear proteins that are sequestered in oculopharyngeal muscular dystrophy nuclear inclusions. Neurobiol. Dis. 2005;18:551–567. doi: 10.1016/j.nbd.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Calado A., Tome F.M., Brais B., Rouleau G.A., Kuhn U., Wahle E., Carmo-Fonseca M. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum. Mol. Genet. 2000;9:2321–2328. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- 19.Tavanez J.P., Calado P., Braga J., Lafarga M., Carmo-Fonseca M. In vivo aggregation properties of the nuclear poly(A)-binding protein PABPN1. RNA. 2005;11:752–762. doi: 10.1261/rna.7217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anvar S.Y., 't Hoen P.A., Venema A., van der Sluijs B., van Engelen B., Snoeck M., Vissing J., Trollet C., Dickson G., Chartier A., et al. Deregulation of the ubiquitin-proteasome system is the predominant molecular pathology in OPMD animal models and patients. Skelet Muscle. 2011;1:15. doi: 10.1186/2044-5040-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raz V., Buijze H., Raz Y., Verwey N., Anvar S.Y., Aartsma-Rus A., van der Maarel S.M. A novel feed-forward loop between ARIH2 E3-ligase and PABPN1 regulates aging-associated muscle degeneration. Am. J. Pathol. 2014;184:1119–1131. doi: 10.1016/j.ajpath.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Fan X., Messaed C., Dion P., Laganiere J., Brais B., Karpati G., Rouleau G.A. HnRNP A1 and A/B interaction with PABPN1 in oculopharyngeal muscular dystrophy. Can. J. Neurol. Sci. 2003;30:244–251. doi: 10.1017/s0317167100002675. [DOI] [PubMed] [Google Scholar]
- 23.Blumen S.C., Brais B., Korczyn A.D., Medinsky S., Chapman J., Asherov A., Nisipeanu P., Codere F., Bouchard J.P., Fardeau M., et al. Homozygotes for oculopharyngeal muscular dystrophy have a severe form of the disease. Ann. Neurol. 1999;46:115–118. [PubMed] [Google Scholar]
- 24.Davies J.E., Sarkar S., Rubinsztein D.C. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2006;15:23–31. doi: 10.1093/hmg/ddi422. [DOI] [PubMed] [Google Scholar]
- 25.Davies J.E., Wang L., Garcia-Oroz L., Cook L.J., Vacher C., O'Donovan D.G., Rubinsztein D.C. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med. 2005;11:672–677. doi: 10.1038/nm1242. [DOI] [PubMed] [Google Scholar]
- 26.Chartier A., Raz V., Sterrenburg E., Verrips C.T., van der Maarel S.M., Simonelig M. Prevention of oculopharyngeal muscular dystrophy by muscular expression of Llama single-chain intrabodies in vivo. Hum. Mol. Genet. 2009;18:1849–1859. doi: 10.1093/hmg/ddp101. [DOI] [PubMed] [Google Scholar]
- 27.Casafont I., Berciano M.T., Lafarga M. Bortezomib induces the formation of nuclear poly(A) RNA granules enriched in Sam68 and PABPN1 in sensory ganglia neurons. Neurotox Res. 2010;17:167–178. doi: 10.1007/s12640-009-9086-1. [DOI] [PubMed] [Google Scholar]
- 28.Catoire H., Pasco M.Y., Abu-Baker A., Holbert S., Tourette C., Brais B., Rouleau G.A., Parker J.A., Neri C. Sirtuin inhibition protects from the polyalanine muscular dystrophy protein PABPN1. Hum. Mol. Genet. 2008;17:2108–2117. doi: 10.1093/hmg/ddn109. [DOI] [PubMed] [Google Scholar]
- 29.Messaed C., Dion P.A., Abu-Baker A., Rochefort D., Laganiere J., Brais B., Rouleau G.A. Soluble expanded PABPN1 promotes cell death in oculopharyngeal muscular dystrophy. Neurobiol. Dis. 2007;26:546–557. doi: 10.1016/j.nbd.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Davies J.E., Rubinsztein D.C. Over-expression of BCL2 rescues muscle weakness in a mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2011;20:1154–1163. doi: 10.1093/hmg/ddq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chartier A., Klein P., Pierson S., Barbezier N., Gidaro T., Casas F., Carberry S., Dowling P., Maynadier L., Bellec M., et al. Mitochondrial dysfunction reveals the role of mRNA poly(A) tail regulation in oculopharyngeal muscular dystrophy pathogenesis. PLoS Genet. 2015;11:e1005092. doi: 10.1371/journal.pgen.1005092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergeron D., Pal G., Beaulieu Y.B., Chabot B., Bachand F. Regulated Intron Retention and Nuclear Pre-mRNA Decay Contribute to PABPN1 Autoregulation. Mol. Cell. Biol. 2015;35:2503–2517. doi: 10.1128/MCB.00070-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muniz L., Davidson L., West S. Poly(A) polymerase and the nuclear poly(A) binding protein, PABPN1, coordinate the splicing and degradation of a subset of human Pre-mRNAs. Mol. Cell. Biol. 2015;35:2218–2230. doi: 10.1128/MCB.00123-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J., Zhang J., Li K., Zhao W., Cui Q. SpliceDisease database: linking RNA splicing and disease. Nucleic Acids Res. 2012;40:D1055–1059. doi: 10.1093/nar/gkr1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G.S., Cooper T.A. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 37.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 38.Castle J.C., Zhang C., Shah J.K., Kulkarni A.V., Kalsotra A., Cooper T.A., Johnson J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008;40:1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pistoni M., Ghigna C., Gabellini D. Alternative splicing and muscular dystrophy. RNA Biol. 2010;7:441–452. doi: 10.4161/rna.7.4.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T., et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013;74:862–872. doi: 10.1002/ana.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de la Grange P., Dutertre M., Martin N., Auboeuf D. FAST DB: a website resource for the study of the expression regulation of human gene products. Nucleic Acids Res. 2005;33:4276–4284. doi: 10.1093/nar/gki738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang E., Aslanzadeh V., Papa F., Zhu H., de la Grange P., Cambi F. Global profiling of alternative splicing events and gene expression regulated by hnRNPH/F. PLoS One. 2012;7:e51266. doi: 10.1371/journal.pone.0051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gandoura S., Weiss E., Rautou P.E., Fasseu M., Gustot T., Lemoine F., Hurtado-Nedelec M., Hego C., Vadrot N., Elkrief L., et al. Gene- and exon-expression profiling reveals an extensive LPS-induced response in immune cells in patients with cirrhosis. J. Hepatol. 2013;58:936–948. doi: 10.1016/j.jhep.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 45.de la Grange P., Dutertre M., Correa M., Auboeuf D. A new advance in alternative splicing databases: from catalogue to detailed analysis of regulation of expression and function of human alternative splicing variants. BMC Bioinformatics. 2007;8:180. doi: 10.1186/1471-2105-8-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mamchaoui K., Trollet C., Bigot A., Negroni E., Chaouch S., Wolff A., Kandalla P.K., Marie S., Di Santo J., St Guily J.L., et al. Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet Muscle. 2011;1:34. doi: 10.1186/2044-5040-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raz V., Routledge S., Venema A., Buijze H., van der Wal E., Anvar S., Straasheijm K.R., Klooster R., Antoniou M., van der Maarel S.M. Modeling oculopharyngeal muscular dystrophy in myotube cultures reveals reduced accumulation of soluble mutant PABPN1 protein. Am. J. Pathol. 2011;179:1988–2000. doi: 10.1016/j.ajpath.2011.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trollet C., Anvar S.Y., Venema A., Hargreaves I.P., Foster K., Vignaud A., Ferry A., Negroni E., Hourde C., Baraibar M.A., et al. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum. Mol. Genet. 2010;19:2191–2207. doi: 10.1093/hmg/ddq098. [DOI] [PubMed] [Google Scholar]
- 49.Mounier Y., Holy X., Stevens L. Compared properties of the contractile system of skinned slow and fast rat muscle fibres. Pflugers Arch. 1989;415:136–141. doi: 10.1007/BF00370583. [DOI] [PubMed] [Google Scholar]
- 50.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 51.Gidaro T., Negroni E., Perie S., Mirabella M., Laine J., Lacau St Guily J., Butler-Browne G., Mouly V., Trollet C. Atrophy, fibrosis, and increased PAX7-positive cells in pharyngeal muscles of oculopharyngeal muscular dystrophy patients. J. Neuropathol. Exp. Neurol. 2013;72:234–243. doi: 10.1097/NEN.0b013e3182854c07. [DOI] [PubMed] [Google Scholar]
- 52.Breitbart R.E., Nadal-Ginard B. Developmentally induced, muscle-specific trans factors control the differential splicing of alternative and constitutive troponin T exons. Cell. 1987;49:793–803. doi: 10.1016/0092-8674(87)90617-9. [DOI] [PubMed] [Google Scholar]
- 53.Breitbart R.E., Nguyen H.T., Medford R.M., Destree A.T., Mahdavi V., Nadal-Ginard B. Intricate combinatorial patterns of exon splicing generate multiple regulated troponin T isoforms from a single gene. Cell. 1985;41:67–82. doi: 10.1016/0092-8674(85)90062-5. [DOI] [PubMed] [Google Scholar]
- 54.Schilder R.J., Kimball S.R., Jefferson L.S. Cell-autonomous regulation of fast troponin T pre-mRNA alternative splicing in response to mechanical stretch. Am. J. Physiol. Cell Physiol. 2013;303:C298–C307. doi: 10.1152/ajpcell.00400.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jat P.S., Noble M.D., Ataliotis P., Tanaka Y., Yannoutsos N., Larsen L., Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc. Natl. Acad. Sci. U.S.A. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wechsler-Reya R.J., Elliott K.J., Prendergast G.C. A role for the putative tumor suppressor Bin1 in muscle cell differentiation. Mol. Cell. Biol. 1998;18:566–575. doi: 10.1128/mcb.18.1.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piva F., Giulietti M., Burini A.B., Principato G. SpliceAid 2: a database of human splicing factors expression data and RNA target motifs. Hum. Mutat. 2012;33:81–85. doi: 10.1002/humu.21609. [DOI] [PubMed] [Google Scholar]
- 58.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spector D.L., Fu X.D., Maniatis T. Associations between distinct pre-mRNA splicing components and the cell nucleus. EMBO J. 1991;10:3467–3481. doi: 10.1002/j.1460-2075.1991.tb04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu X.D. Specific commitment of different pre-mRNAs to splicing by single SR proteins. Nature. 1993;365:82–85. doi: 10.1038/365082a0. [DOI] [PubMed] [Google Scholar]
- 61.Wang J., Manley J.L. Overexpression of the SR proteins ASF/SF2 and SC35 influences alternative splicing in vivo in diverse ways. RNA. 1995;1:335–346. [PMC free article] [PubMed] [Google Scholar]
- 62.Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 63.Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A., Swanson M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bengoechea R., Tapia O., Casafont I., Berciano J., Lafarga M., Berciano M.T. Nuclear speckles are involved in nuclear aggregation of PABPN1 and in the pathophysiology of oculopharyngeal muscular dystrophy. Neurobiol. Dis. 2012;46:118–129. doi: 10.1016/j.nbd.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 65.Perry S.V. Troponin T: genetics, properties and function. J. Muscle Res. Cell Motil. 1998;19:575–602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- 66.Chaudhuri T., Mukherjea M., Sachdev S., Randall J.D., Sarkar S. Role of the fetal and alpha/beta exons in the function of fast skeletal troponin T isoforms: correlation with altered Ca2+ regulation associated with development. J. Mol. Biol. 2005;352:58–71. doi: 10.1016/j.jmb.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 67.Gordon A.M., Homsher E., Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 68.Chujo T., Yamazaki T., Hirose T. Architectural RNAs (arcRNAs): a class of long noncoding RNAs that function as the scaffold of nuclear bodies. Biochim. Biophys. Acta. 2016;1859:139–146. doi: 10.1016/j.bbagrm.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 69.Lamond A.I., Spector D.L. Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell. Biol. 2003;4:605–612. doi: 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 70.Calado A., Carmo-Fonseca M. Localization of poly(A)-binding protein 2 (PABP2) in nuclear speckles is independent of import into the nucleus and requires binding to poly(A) RNA. J. Cell Sci. 2000;113:2309–2318. doi: 10.1242/jcs.113.12.2309. [DOI] [PubMed] [Google Scholar]
- 71.Calado A., Kutay U., Kuhn U., Wahle E., Carmo-Fonseca M. Deciphering the cellular pathway for transport of poly(A)-binding protein II. RNA. 2000;6:245–256. doi: 10.1017/s1355838200991908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lemieux C., Bachand F. Cotranscriptional recruitment of the nuclear poly(A)-binding protein Pab2 to nascent transcripts and association with translating mRNPs. Nucleic Acids Res. 2009;37:3418–3430. doi: 10.1093/nar/gkp207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Girard C., Will C.L., Peng J., Makarov E.M., Kastner B., Lemm I., Urlaub H., Hartmuth K., Luhrmann R. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun. 2012;3:994. doi: 10.1038/ncomms1998. [DOI] [PubMed] [Google Scholar]
- 74.Proudfoot N.J. Ending the message: poly(A) signals then and now. Genes Dev. 2011;25:1770–1782. doi: 10.1101/gad.17268411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Proudfoot N.J., Furger A., Dye M.J. Integrating mRNA processing with transcription. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 76.Anvar S.Y., Raz Y., Verway N., van der Sluijs B., Venema A., Goeman J.J., Vissing J., van der Maarel S.M., t Hoen P.A., van Engelen B.G., et al. A decline in PABPN1 induces progressive muscle weakness in oculopharyngeal muscle dystrophy and in muscle aging. Aging (Albany NY) 2013;5:412–426. doi: 10.18632/aging.100567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abu-Baker A., Messaed C., Laganiere J., Gaspar C., Brais B., Rouleau G.A. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2003;12:2609–2623. doi: 10.1093/hmg/ddg293. [DOI] [PubMed] [Google Scholar]
- 78.Bao Y.P., Cook L.J., O'Donovan D., Uyama E., Rubinsztein D.C. Mammalian, yeast, bacterial, and chemical chaperones reduce aggregate formation and death in a cell model of oculopharyngeal muscular dystrophy. J. Biol. Chem. 2002;277:12263–12269. doi: 10.1074/jbc.M109633200. [DOI] [PubMed] [Google Scholar]
- 79.Feany M.B., Bender W.W. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 80.Chang S.H., Chang W.L., Lu C.C., Tarn W.Y. Alanine repeats influence protein localization in splicing speckles and paraspeckles. Nucleic Acids Res. 2014;42:13788–13798. doi: 10.1093/nar/gku1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.